

Abstract
Much progress has been made in the identification of specific human gene variants that contribute to enhanced susceptibility or resistance to viral diseases. Herein we review multiple discoveries made with genome-wide or candidate gene approaches that have revealed significant insights into virus–host interactions. Genetic factors that have been identified include genes encoding virus receptors, receptor-modifying enzymes, and a wide variety of innate and adaptive immunity-related proteins. We discuss a range of pathogenic viruses, including influenza virus, respiratory syncytial virus, human immunodeficiency virus, human T cell leukemia virus, human papilloma virus, hepatitis B and C viruses, herpes simplex virus, norovirus, rotavirus, parvovirus, and Epstein-Barr virus. Understanding the genetic underpinnings that affect infectious disease outcomes should allow tailored treatment and prevention approaches in the future.
Keywords: virus, infection, single nucleotide polymorphism, SNP, genome-wide association study, GWAS
INTRODUCTION
Viruses co-opt numerous cellular pathways to complete their replication cycles, providing myriad points of contact between virus components and cellular proteins. Viruses also often encode proteins that subvert human immune mechanisms. Thus, pathogenic viruses can provide selective pressures for evolutionary adaptation of multiple features of human biology (101). Virus–human interactions have driven as much as 30% of human genome evolution since divergence from chimpanzees (38), and genetic variation among humans produces a wide variety of responses to viral infections. One of the earliest demonstrations of this genetic effect was provided by the examination of poliovirus infection in twins, in which at least one twin was diagnosed with paralytic poliomyelitis (52). Paralytic polio disease in the second twin was significantly more likely among identical versus fraternal twins, indicating a genetic influence on this infection outcome. Further, a landmark study comparing causes of premature death among adoptees with the causes of death of their biological and adoptive parents provided strong evidence for heritable factors in infectious disease mortality (135). In this review, we summarize a variety of notable examples of specific genes and genetic variants linked to particular outcomes of viral infections (Table 1).
Table 1. List of genes in which variants have been associated with particular disease outcomes in specific virus infections.
| Human gene | Variant-associated disease manifestation | Gene functional category | Reference(s) |
|---|---|---|---|
| Influenza virus | |||
| IFITM3 | Severe influenza | Antiviral restriction factor | 39, 162, 177 |
| IRF7 | Severe influenza | Transcription factor | 27 |
| CPTII | Influenza-associated encephalopathy | Cell homeostasis | 21, 94, 171 |
| SFPA/B | Severe influenza | Cell homeostasis | 53, 155 |
| RSV | |||
| SFPA/D | Bronchiolitis | Cell homeostasis | 83, 92, 153 |
| VDR | Bronchiolitis | Transcription factor | 68, 80, 102, 141 |
| IL8 | Bronchiolitis | Cytokine | 61 |
| IL4 | Bronchiolitis | Cytokine | 26, 56, 173 |
| IL4RA | Bronchiolitis | Cytokine | 56, 149 |
| IL13 | Need for mechanical ventilation | Cytokine | 124 |
| IL10 | Need for mechanical ventilation | Cytokine | 44, 168 |
| HIV | |||
| CCR5 | Resistance to infection, slow disease progression | Virus entry coreceptor | 33, 90, 129 |
| HLAB57 | Low viral load and slow T cell decline | Antigen presentation | 2, 40, 74, 103 |
| KIR3DS1 | Slow disease progression | Adaptive immune cell development | 98, 151 |
| TRIM5A | Accelerated disease progression | Antiviral restriction factor | 136 |
| APOBEC3G | Accelerated disease progression | Antiviral restriction factor | 3 |
| IFITM3 | Accelerated disease progression | Antiviral restriction factor | 176 |
| HTLV-1 | |||
| EPC1 | Aggressive type adult T cell lymphoma | Cell homeostasis; transcription factor | 106 |
| TNF | Adult T cell lymphoma | Cytokine | 157 |
| IL13 | Adult T cell lymphoma | Cytokine | 161 |
| VCAM1 | Adult T cell lymphoma | Adaptive immune cell development | 161 |
| MMP9 | HTLV-1-associated myelopathy/tropical spastic paraparesis | Cell homeostasis | 78 |
| IL18 | High viral load | Cytokine | 127 |
| IFNG | High viral load | Cytokine | 127 |
| IL28B (IFNL3) | High viral load | Cytokine | 5 |
| IL10 | High viral load | Cytokine | 128 |
| HPV | |||
| TNF | Increased cervical cancer risk | Cytokine | 7, 34, 35, 77, 79 |
| P53 | Increased HPV-associated cancer risk | Cell homeostasis | 142 |
| EVER1/2 | Epidermodysplasia verruciformis in beta-HPV infections | Cell homeostasis | 113, 125, 150 |
| HCV | |||
| IL28B (IFNL3) | Spontaneous clearance and improved IFN treatment response | Cytokine | 8, 43, 57, 58, 100, 108, 126, 145, 148, 152, 159 |
| IFNL4 | Spontaneous clearance and improved IFN treatment response | Cytokine | 75, 108, 109, 121, 151 |
| HBV | |||
| INTS10 | Chronic infection | Cell homeostasis | 87 |
| STAT4 | Hepatocellular carcinoma | Transcription factor | 69, 70, 71, 76 |
| NTCP | Resistance to chronic infection | Virus entry receptor | 111, 116 |
| HSV | |||
| STAT1 | Multiple infections including herpes simplex encephalitis | Transcription factor | 20, 37 |
| NEMO | Multiple infections including herpes simplex encephalitis | Signaling molecule | 122 |
| UNC93B | Herpes simplex encephalitis | Virus sensor | 18, 147 |
| TLR3 | Herpes simplex encephalitis | Virus sensor | 175 |
| TRAF3 | Herpes simplex encephalitis | Signaling molecule | 118 |
| TRIF | Herpes simplex encephalitis | Signaling molecule | 130 |
| TBK1 | Herpes simplex encephalitis | Signaling molecule | 51 |
| IRF3 | Herpes simplex encephalitis | Transcription factor | 4 |
| TBX21 | HSV-2 susceptibility | Transcription factor; adaptive immune cell development | 146 |
| CSSG1 | Frequent cold sore outbreaks | Unknown function | 55, 81 |
| Norovirus | |||
| FUT2 | Resistance to infection | Virus entry receptor-modifying enzyme | 31, 41, 88, 89, 93, 96, 158 |
| Rotavirus | |||
| FUT2 | Resistance to infection | Virus entry receptor-modifying enzyme | 114 |
| Parvovirus | |||
| B3GALNT1 | Resistance to infection | Virus entry receptor-modifying enzyme | 48 |
| A4GALT | Resistance to infection | Virus entry receptor-modifying enzyme | 138 |
| EBV | |||
| IL10 | Resistance to infection | Cytokine | 49, 50 |
| IL1B | Resistance to infection | Cytokine | 62 |
| MAGT1 | Chronic infection | Cell homeostasis | 19, 86 |
| SH2D1A | Extreme sensitivity to EBV, fatal mononucleosis | Adaptive immune cell development | 143 |
| ITK | Extreme sensitivity to EBV, fatal lymphoproliferation | Adaptive immune cell development | 60, 95, 140 |
Abbreviations: EBV, Epstein-Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papilloma virus; HSV, herpes simplex virus; HTLV-1, human T cell leukemia virus type 1; IFN, interferon; RSV, respiratory syncytial virus.
Genes discussed herein that increase or decrease susceptibility to viral diseases may be grouped into several broad functional classifications (Table 1 and Figure 1). For example, gene variations associated with resistance to particular infections often involve virus entry receptors, coreceptors, or receptor-modifying enzymes. Likewise, polymorphisms leading to over- or under-production of specific cytokines can also influence viral disease severity. Genetic defects in other aspects of cellular innate and adaptive immune responses to viral infections, such as virus sensing, signaling in response to viruses, activity of antiviral restriction factors, or proper initiation of T cell responses, have also been associated with enhanced severity of numerous viral infections. Recognition of such classifications can provide a framework for identifying new biologically plausible susceptibility or resistance loci, though additional classifications are likely to be added as new genetic determinants of viral diseases continue to be discovered.
Figure 1.
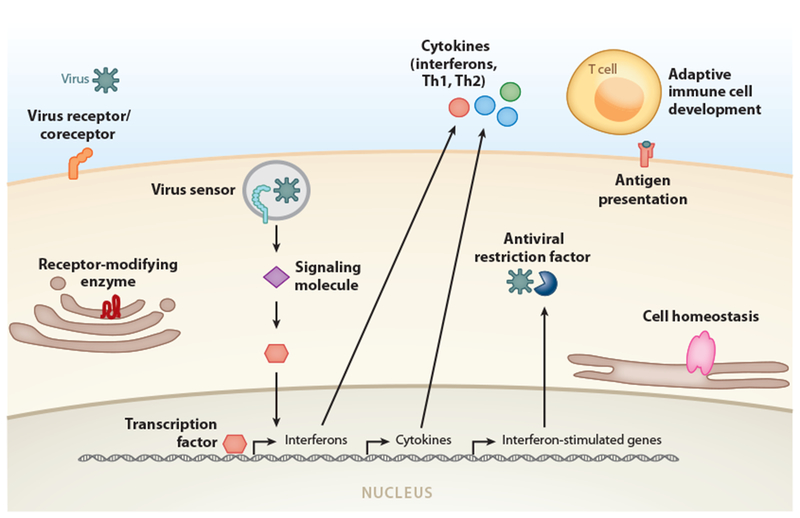
General categories of genes in which variants have been associated with susceptibility or resistance to specific viral diseases. Human genetic determinants of viral diseases can be broadly categorized based on the distinct cellular functions of their associated proteins (labeled in bold text). Abbreviation: Th, T helper.
Several approaches have been applied to identify genetic factors linked to virus infection susceptibility or disease outcome. These studies typically require the identification of individuals with unusual responses to infections, such as an abnormal severity of illness, rare complications, or an unusually rapid or slow progression of disease. The study of candidate genes is one strategy that has proven successful for identifying disease susceptibility loci among such individuals. Candidate genes are usually chosen based on biological plausibility established by in vitro assays or larger genetic screens. Many studies have focused on immunity-related genes, such as human leukocyte antigen (HLA) genes or genes associated with induction or effector functions of antiviral interferons (IFNs). Indeed, such studies are duly warranted given the prominent role of immunity genes in defending against viruses, and that many immunity-related genes are, in humans, highly polymorphic (101). Alternatively, extensive use of unbiased whole-genome approaches, such as genome-wide association studies (GWASs), has identified potential susceptibility genes among patient cohorts afflicted with specific disease manifestations of viral infections. GWASs typically require large groups of correctly diagnosed study participants in order for rare disease-associated polymorphisms to be identified with statistical significance. Similarly, a more recently adopted approach utilizes whole exome sequencing (WES) to pinpoint gene polymorphisms that underlie specific viral disease phenotypes. Application of WES to individual patients can identify polymorphisms previously implicated in an observed infection phenotype while also allowing for variant discovery studies when performed in aggregate (179), although WES does not allow for discovery of noncoding DNA variants. We discuss multiple examples in which important information has been gained from both candidate gene studies and genomic approaches.
For nearly all of the viral infections we consider below, specific HLA alleles have been implicated in susceptibility to, or protection from, severe infection, likely owing to the ability of distinct HLA variants to present unique peptide repertoires to T cells. Because the role of HLA variation in infectious diseases has been reviewed previously (9, 156), we have opted to discuss specific HLAs only when the link to a viral infection outcome is exceptionally well-supported or significant. Likewise, some of the most important infections in human history, such as poliovirus and smallpox virus, are absent from our discussion of specific genetic susceptibility factors because these viruses were largely eliminated from the human population by vaccines prior to the modern genomic era. Nonetheless, our knowledge of human genetic factors involved in viral infections continues to grow, and may provide the information necessary to treat or eradicate additional infectious diseases in the human population in the future.
INFLUENZA VIRUS
Influenza virus circulates in a seasonal pattern, and as much as 20% of the human population is infected in any given year (104). Although most people experience moderate respiratory symptoms and recover within one week, a small percentage is afflicted with severe respiratory distress or other rare complications.
The emergence of novel influenza viruses in recent years has allowed the identification of specific genetic deficiencies of the innate immune system that were perhaps previously masked in these patients by a functional adaptive immune response against seasonal strains. IFN-induced transmembrane protein 3 (1F1TM3) arose as a candidate gene for control of influenza virus infection based on previous studies indicating that IFITM3 mediates a majority of the antiviral action of IFNs against influenza virus in cells (13), and that 1fitm3 is essential for limiting infection severity in mice (39). In two independent studies performed on British and Chinese subjects, respectively, 1F1TM3 was sequenced in individuals hospitalized for severe infection with the newly emergent 2009 pandemic H1N1 influenza A virus (39, 177). Both studies found a significant increase in homozygosity of an 1F1TM3 single-nucleotide polymorphism (SNP), rs12252-C, in severely ill patients versus matched control populations (39, 177). A similar study that examined Chinese patients infected with an emergent H7N9 virus of avian origin also correlated homozygosity of rs12252-C with faster disease progression and fatality (162). This 1F1TM3 polymorphism is predicted to cause alternative splicing of the transcript, resulting in truncation and altered localization of IFITM3, though the existence of a shortened form of IFITM3 remains to be conclusively demonstrated (39). Recent work in vitro suggests that although this alteration of IFITM3 decreases antiviral activity against influenza virus, its activity against retroviruses is increased (29), thus revealing a potential selective advantage for the SNP that may explain its high prevalence in certain human populations (177). Additionally, given that IFITM3 antiviral activity is highly regulated by at least four posttranslational modifications, polymorphisms in factors that install or remove these modifications and impact influenza virus susceptibility may be uncovered in the future (22–24, 117, 172).
Although the 1F1TM3 SNP is the most reproducibly identified genetic factor associated with severe influenza, it accounts for only a fraction of severe infections (39). Importantly, polymorphisms in other IFN-related genes also affect influenza virus infection outcomes. Indeed, a study of a French child with a near-fatal infection by the 2009 pandemic H1N1 virus utilized WES to identify distinct rare mutations in each copy of the patient’s IFN regulatory factor 7 (1RF7) gene, which encodes a critical transcription factor involved in type I IFN production (27). Remarkably, both mutations decreased IRF7 protein function, and cells from this patient supported aberrantly high virus replication that was rescued by complementation with wild-type IRF7 or by treatment with IFN (27). Thus, inactivation of IFN responsiveness by a variety of mechanisms can result in negative influenza virus infection outcomes.
Influenza-associated encephalopathy (IAE) is a rare neurological complication of infection that does not usually involve direct infection of neural tissues (139). IAE patients often display abnormal acylcarnitine fatty acid chain lengths, which suggest a potential aberration in lipid metabolism (21). A candidate gene approach in Japanese and Chinese cohorts demonstrated that IAE is reproducibly associated with several specific polymorphisms in the carnitine palmitoyltransferase II (CPT11) gene (21, 94, 171). The amino acid changes within CPTII result in decreased enzymatic activity at 41°C, consistent with a buildup of long chain fatty acids in the serum of affected patients upon onset of fever (171). Thus, it appears that some cases of IAE result from an underlying metabolic abnormality that is exacerbated by infection.
In addition to the genes discussed above, many other potential influenza virus susceptibility genes have been identified using SNP arrays, though, in general, results have not been reproducible between studies. One notable finding from these studies is that SNPs in genes encoding lung surfactant proteins (SFTPs), including SFTPA and SFTPB, have been associated with severe influenza virus infections (53, 155), as well as other respiratory infections as discussed below. SFTPs are secreted into the lung alveolar space, and may have direct antiviral or immunomodulatory activities (47), although additional studies are needed to fully validate the importance of SFTP variations in human influenza virus infections.
RESPIRATORY SYNCYTIAL VIRUS
Respiratory syncytial virus (RSV) infection leads to a wide spectrum of clinical outcomes, from a mild cold to severe bronchiolitis, pneumonia, or even asthma. Nearly all children have been infected with RSV at least once by two years of age. Approximately 1–2% of infected children develop disease that requires hospitalization (105, 144). Genetic studies of otherwise healthy infants and children have identified SNPs in genes involved in immune defense that are overrepresented in patients hospitalized with RSV compared to either healthy controls or patients with milder RSV disease. At present, genetic susceptibility to RSV is best characterized as a complex trait in which multiple loci across the genome likely contribute to disease severity (25).
Similar to influenza virus, lung SFTPs can impact RSV infections by directly limiting the infection of lung epithelia by RSV and by regulating the immune response against the virus (47). Polymorphisms in SFTPA2 (G223L) and SFTPD (M11T) were shown to be associated with severity of RSV infection in cohorts of patients from Finland and the United States (83, 92, 153). Several studies have also demonstrated a significant association between the FokI start codon polymorphism of the vitamin D receptor gene (VDR) (which codes for an intracellular receptor and transcription factor) and severity of RSV bronchiolitis (68, 80, 102). The FokI polymorphism exacerbates RSV pathogenesis by coding for a VDR that fails to restrain STAT1-mediated antiviral cytokine responses, leading to increased immunopathology (141). Similarly, a promoter polymorphism in the IL8 gene (−251A), encoding the neutrophil-recruiting chemoattractant IL-8, leads to increased IL-8 production and is associated with RSV bronchiolitis, particularly in infants without other known risk factors (61). TLR4 and CD14 are involved in the innate immune sensing of RSV infection and induction of inflammatory mediators and were thus among the first proteins to be predicted to be associated with disease severity. Although some investigations observed correlations between CD14 and TLR4 polymorphisms and RSV bronchiolitis, recent meta-analysis of multiple studies indicates a lack of association (64, 91, 123, 178).
In adaptive immunity to RSV, the balance between T helper 1 (Th1) and Th2 immune responses plays a critical role in pathology during infection, as Th2-dominated responses associate with disease progression. Multiple studies have shown that the RSV G protein triggers the expression of the Th2 cytokines IL-13, IL-5, and IL-4 in mice and human cells, promoting eosinophilia and Th2 immunopathology (10, 67, 73). Genetic association studies in cohorts of children from China, Korea, Chile, and the Netherlands showed that gain of function variants in the IL4 promoter (−590T and −589T) (26, 56, 173) or in the IL-4 receptor alpha chain gene (Q551R) (56, 149) were associated with higher susceptibility to RSV. Likewise, a large study in a German cohort found an association between the IL-13 promoter variant −1112T and severity of RSV disease (124). In two additional studies, variants that impact the production of the Th2-promoting cytokine IL-10 (−1117G and −3585A) were found to increase the risk of needing mechanical ventilation during RSV infection (44, 168). Overall, susceptibility to severe RSV infections appears to involve multifactorial immunity gene networks operating at multiple levels within the host.
HUMAN IMMUNODEFICIENCY VIRUS
More than 30 years after the discovery of the human immunodeficiency virus (HIV) and acquired immune deficiency syndrome (AIDS), research clearly indicates that individuals have significant variability in their susceptibility to HIV type 1 (HIV-1) infection, in their viral control, and in their progression to AIDS. A small subset of HIV-1-infected individuals, termed HIV-1 controllers, do not exhibit viremia even in the absence of antiretroviral therapy and do not progress to AIDS. Multiple genetic studies of HIV-1 controllers have identified a homozygous 32-bp deletion in the CCR5 gene, encoding a nonfunctional HIV-1 coreceptor (33, 90, 129). This genetic variant, termed CCR5Δ32, is the only genetic factor that has been consistently associated with initial resistance to HIV-1, highlighting the important role of CCR5 in HIV-1 infection in vivo. Though additional human gene polymorphisms have been linked with initial HIV-1 infection susceptibility, including genes encoding CCR5 ligands, they occur at very low frequencies, they occur only in specific ethnicities, and their functional contributions to HIV-1 infection in patient populations are largely unconfirmed (134).
Host genetic determinants also affect the rate at which viremia is controlled, and the severity of disease progression. An early study of the role of HLAs in HIV-1 infection found that homozygosity of any HLA class I allele was associated with more rapid progression to AIDS and death and also that specific HLAs could be particularly detrimental (17). Conversely, several GWASs and additional candidate gene studies examining HLAs have converged on a strong and consistent protective link between HLA-B* 57, decreased HIV-1 viral load, and reduced CD4 T cell decline; carriers of the allele also appear asymptomatic of acute infection (2, 40, 74, 103). Mechanistically, these protective effects are elicited through HLA-B*57 presentation of immune epitopes from HIV-1 Gag to cytotoxic T cells (65).
Certain haplotypes and copy number variants of the killer cell immunoglobulin-like receptor (KIR) genes, which are expressed on natural killer cells and some CD8 T cells, affect HIV-1 disease progression. Despite an incomplete understanding of the mechanisms involved, the effect is multifaceted and there seems to be significant interplay with HLA ligands to mount effective viral control (1, 11, 115). For example, KIR3DS1 helps prevent progression to AIDS, but this occurs only in those who also have a particular HLA-B allele (98). The ongoing work to dissect these interactions highlights the challenges in identifying and understanding functional networks involved in viral disease outcomes.
Polymorphisms in a variety of host restriction factors thwart HIV-1 replication and affect disease progression in HIV-1-infected individuals. Examples include variations in genes encoding the IFN-inducible proteins TRIM5\g=a\ (136), APOBEC3G (3), and IFITM3 (176). SAMHD1 is a newly discovered HIV-1 restriction factor that can be counteracted by Vpx protein from HIV-2 and certain simian immunodeficiency viruses (59, 82, 137). Although polymorphisms in SAMHD1 are not associated with natural control of HIV-1 infection in European and African-American cohorts (30), it remains unknown whether SAMHD1 polymorphisms are associated with HIV-2 infection in humans
HUMAN T CELL LEUKEMIA VIRUS TYPE 1
Human T cell leukemia virus type 1 (HTLV-1) is an oncogenic retrovirus that infects and transforms CD4 T cells. Although this viral infection is widespread across the world with as many as 20 million infected individuals, only 5–10% will develop any HTLV-1-related disease (66). The two most prominent diseases associated with HTLV-1 are adult T cell leukemia/lymphoma (ATL) and HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), a slow, progressive, chronic disease of the spinal cord (45, 120). A long clinical latency period of several decades and relatively low disease penetrance in infected individuals indicate that host genetic variation plays an important role in HTLV-1 disease.
ATL typically occurs in individuals who acquire HTLV-1 early in life, often as the result of breastfeeding (16). A genome-wide linkage analysis mapped a major susceptibility locus for predisposition to HTLV-1 infection in African breastfed children to chromosome 6q27 (119), though the causative locus has not been determined. Recently, spectral karyotyping in patients with aggressive type ATL—characterized by less than 10 months survival—identified a common breakpoint cluster region on chromosome 10 (106). The chromosomal breakpoints clustered within the EPC1 gene locus, which codes for a protein involved in histone acetylation and gene transcriptional regulation. Overexpression of EPC1 accelerated leukemia cell growth in vitro, suggesting that altered levels of EPC1 may contribute to leukemogenesis in patients with aggressive-type ATL (106).
Specific polymorphisms that influence ATL and HAM/TSP development have also been identified, primarily using candidate-based gene approaches. A study from Japan identified a SNP in the TNF promoter region (−857T) that was enriched in ATL patients compared to healthy HTLV-1 carriers (157). Conversely, a Caribbean study that examined SNPs in 38 gene candidates found that SNPs in the coding regions of IL13 (A98G) and VCAM1 (G149A)—both of which are known to be upregulated in HTLV-1-infected T cells—were associated with decreased risk for ATL development (161). In HAM/TSP patients, expansions of a CA repeat polymorphism in the MMP9 gene correlate with development of disease (78), and individual SNPs in the immunity-related cytokine genes IL18, IFNG, IL28B, and IL10 have each been associated with higher proviral load (5, 127, 128). Such findings further highlight the genetic complexity underlying HTLV-1-mediated disease.
HUMAN PAPILLOMA VIRUS
Human papilloma virus (HPV) is the most common sexually transmitted virus in the United States (36). HPV has been detected in as much as 40% of the sexually active population, and most individuals will become infected at some point in their lifetime (36). Although most infected persons show no symptoms, HPV can cause warts and a variety of cancers. The virus is the leading cause of cervical cancer, with nearly 100% of cases being linked to persistent HPV infection (160). Although the vast majority of the population is exposed to HPV, symptoms are rare and cervical cancer rarer still. Thus, it is likely that host genetic factors contribute significantly to the variation seen in outcomes following HPV infection.
Several studies suggest that polymorphisms in the TNF gene can alter the course of HPV infections. TNF is released in abundance following infection and plays an integral role in the immune response to HPV (34). A deficient response allows persistence of HPV, whereas excess TNF promotes expression of HPV oncoproteins E6 and E7 that constitutively progress the cell cycle in cervical keratinocytes (34, 42). As such, rs1800629-A, a polymorphism in the TNF promoter region that increases TNF production, has been reproducibly observed to confer an increased risk of cervical cancer development (7, 35, 77, 79).
One of the best-established links between HPV infection and cervical cancer is the interaction of the tumor suppressor p53 with the oncogenic E6 protein of HPV. E6 binds to p53 and initiates its degradation, leading to unchecked cellular proliferation (132, 164). Interestingly, a P72R variant of p53 is more susceptible to E6-mediated degradation (142). Homozygosity for p53-P72R is correlated with significantly increased risk of HPV-associated cancer compared to heterozygosity or homozygosity of the reference allele (72P) (142).
Host genetic variation also plays a role in the rarest outcome of HPV infection, epidermodysplasia verruciformis, a disease characterized by massive wart growth primarily on the head and extremities, caused by beta-HPV infection (85, 113). Beta-HPV is considered harmless in the general population because, unlike other HPV genera, viruses in this genus are unable to disrupt cellular zinc homeostasis in order to replicate (85). Zinc homeostasis is regulated by epidermodysplasia verruciformis endoplasmic reticulum (EVER) proteins, and mutations in EVER1 and EVER2 have been consistently linked to epidermodysplasia verruciformis and progression to cancer (113, 125, 150). Defective EVER proteins fail to complex with other zinc transporter proteins, disrupting the intracellular distribution of Zn2+ ions, thus creating an environment favorable to beta-HPV replication (85).
HEPATITIS C VIRUS
Hepatitis C virus (HCV) is a global health problem that affects more than 184 million people worldwide (154). HCV infection has two main outcomes. Following viral exposure, approximately 25% of people spontaneously clear the virus. The remaining 75% of those infected progress to chronic infection, which can result in cirrhosis, hepatocellular carcinoma (HCC), and extrahepatic complications (154). Spontaneous viral clearance is mediated in part by antiviral IFN responses. Indeed, exogenous IFN-α was the mainstay of chronic HCV treatment until the recent introduction of highly effective direct-acting antiviral medications. Although IFN therapies can be effective in achieving a sustained antiviral response, they fail to eliminate HCV in a significant proportion of patients (154).
In attempts to identify genetic factors that contribute to differential infection and therapeutic outcomes, several large independent GWAS efforts revealed an association between SNPs proximal to the IL28B locus (rs8099917 and rs12979860) and response to pegylated IFN plus ribavirin treatment (43, 145, 148). Of note, these SNPs are in strong linkage disequilibrium. In subsequent studies, they were found to be associated with spontaneous clearance of HCV (126, 152), and rs12979860 was associated with the control of viremia that often occurs following pregnancy (58). The IL28B gene, recently renamed IFNL3, encodes a type III IFN that elicits the expression of antiviral genes. Although SNPs proximal to a gene with clear antiviral function suggest a potential mechanistic connection to HCV clearance, both rs8099917 and rs12979860 SNPs lie outside of the IFNL3 coding sequence.
Hepatocyte RNA sequencing studies identified an additional genetic variant, rs368234815 (originally designated ss469415590), adjacent to the IFNL3 locus. This variant is in strong linkage disequilibrium with rs12979860 and has similar associated effects on HCV infection and treatment outcomes (121). The rs368234815 variant resides within a previously unidentified type III IFN gene, which was designated IFNL4. This polymorphic site has two variant alleles: ΔG [encoding an intact IFNL4 open reading frame (ORF)] and TT (an insertion frameshift variant that disrupts the IFNL4 ORF). Unexpectedly, the ΔG variant, predicted to encode full-length IFNL4, is associated with unfavorable clinical outcomes, including decreased viral clearance and decreased response to IFN therapy (109). Furthermore, patients with an intact IFNL4 ORF encoding a functionally impaired IFNL4 S70 variant (rs117648444) exhibit better clinical outcomes than patients with a proline at position 70 encoding fully active IFNL4 (151).
A mechanistic understanding of how IFNL3/4 variants affect HCV pathogenesis and clearance remains unclear. Paradoxically, clinically unfavorable genotypes are associated with higher IFN-stimulated gene expression in the liver during chronic HCV infection (57, 159). However, this expression profile, perhaps established by IFNL3 or IFNL4 signaling, could render cells refractory to subsequent IFN-α stimulation (131). Although data support IFNL4 rs368234815 as the strongest predictor of spontaneous clearance and positive treatment outcomes (8, 108, 121), evidence for regulatory effects of additional IFNL3 noncoding variants (100) presents challenges in ascribing a phenotype to a single site. Further mechanistic clarification of these effects remains an area of active research.
Characterization of the IFNL4 locus has raised additional questions regarding its evolutionary history. The ΔG variant (intact ORF) has been selected against in non-African human populations (75). It is possible that intact IFNL4 emerged as a host defense factor to combat other ancient viral pathogens, but was later negatively selected in shifting host–pathogen relationships. At present, many unresolved questions are driving research on this intriguing example of virus–host coevolution.
HEPATITIS B VIRUS
It is estimated that hepatitis B virus (HBV) has infected two billion people worldwide, and although most infections are effectively controlled and cleared by the immune response, as many as 250 million people remain chronically infected (84, 133). HBV persistence can lead to immune-mediated damage to the liver, a major risk factor for cirrhosis and HCC (170). Efforts to identify host susceptibility factors for chronic HBV infection and HCC have been focused on areas with the highest infection rates, such as Southeast Asia or the East Pacific (32).
GWASs and candidate gene studies aimed at identifying HBV susceptibility loci have most frequently identified genetic variants in the HLA class I and II genes (reviewed in 99), although GWASs have also uncovered unexpected genes that influence HBV infections. A study comparing individuals who have experienced spontaneous recovery versus chronic infection identified a novel susceptibility locus for persistent HBV at chromosome 8p21.3 (87). Expression quantitative trait loci associations from liver tissues of patients with persistent infection suggested a functional role for the INTS10 gene from this region. Researchers found a two-fold decrease of INTS10 in the plasma of patients with persistent HBV and also demonstrated that INTS10 is capable of suppressing HBV replication in vitro. This antiviral function was dependent on IRF3, suggesting that INTS10 augments the type IIFN response.
Another noteworthy GWAS effort associated increased risk of HBV-related HCC with a variant in the third intron of STAT4 (70). Further, carriers of the high-risk allele had lower levels of STAT4 in HCC and nontumor tissues compared to other genotypes, and lower STAT4 expression in tumor compared to adjacent normal tissue. This link between STAT4 variation and HBV infection severity has been verified in additional cohorts (69, 71, 76), although the mechanistic role of STAT4 in controlling infection remains under investigation.
In 2012, identification of the putative receptor for HBV, the sodium taurocholate cotransporting polypeptide (NTCP), revealed a new gene candidate with a potential role in modulating human responses to infection (169). Indeed, an NTCP variant (S267F) that is primarily found in Asian populations (111) was associated with resistance to chronic HBV infection (116). Structural modeling suggested that this switch from a hydrophilic to hydrophobic residue is positioned to affect NTCP-ligand interactions and therefore, potentially, HBV entry into the cell.
HERPES SIMPLEX VIRUS
Herpes simplex virus (HSV) types 1 and 2 are double-stranded DNA viruses in the α-herpes virus family and are widespread throughout the world (12, 167). These viruses cause lifelong infection in the sensory nerve ganglia, producing oral or genital lesions as the primary symptom of infection (167). Although infection is currently incurable, balance is generally attained between the virus and host, with lesions occurring only as a result of local triggers. However, severe disease can occur in immunocompromised individuals, and some individuals are more susceptible to frequent HSV lesion outbreaks. Studies have now linked genetic susceptibility loci to recurrent HSV lesions, or to the rare HSV-1 infection complication known as herpes simplex encephalitis (HSE).
HSE affects a small number of HSV-1 patients but is the most common form of viral encephalitis in Western countries. Identification of gene mutations in IFN-related pathways has revealed a critical role for these cytokines in controlling HSV and limiting its subsequent pathology in humans. STAT1, a molecule essential for IFN signaling, was first correlated with HSE pathology in 2003 when the STAT1 genes were sequenced in two unrelated infants exhibiting severe viral and bacterial disease (37). Both died from viral disease, one specifically from HSE, and both were identified as having distinct homozygous mutations in STAT1 (L600P and 1757–1758delAG) that made cells unresponsive to stimulation by IFN-α/β or -γ (37). This work provided an initial link between a defective IFN response and HSE. A follow-up report examined two siblings with severe and recurrent intracellular bacterial and viral infections, including recurrent HSV infections in one of the siblings. Again, examination of STAT1 as a candidate gene revealed a homozygous mutation (P696S) that caused impaired splicing of STAT1 mRNA and reduced expression, thus diminishing IFN responsiveness in the patients’ cells (20). Another study identified a child with HSE who possessed a frameshift insertion creating a premature stop codon in NEMO (exon 2,110_111insC), a gene which has been shown to be essential for NF-κB activation and for subsequent IFN-β production (6, 122). Fibroblasts and blood cells from the child exhibited impaired production of IFNs in response to both TLR3 ligation and direct viral infection, and this likely contributed to the patient’s death by HSE (6, 122). Together, these studies indicate that complete or partial loss of the IFN response leads to increased susceptibility to viral diseases including HSV infections and HSE.
Although STAT1 and NEMO impairments result in multiple severe infectious diseases, including HSE, sporadic HSE can also occur in otherwise healthy individuals who are not afflicted with increased susceptibility to other infections. The first genetic link to HSE in otherwise immunocompetent children was identified in 2006 (18). Stimulation of HSE patient blood cells with HSV-1 or endosomal TLR ligands showed impairments in IFN production, and this finding suggested defects in the patients’ TLR responses. Through candidate gene sequencing, it was determined that the two children in this study inherited autosomal recessive mutations resulting in a loss of expression of the protein UNC-93B (exon 8, del1034–1037; exon 6, 781G>A) (18). UNC-93B is required for signaling of TLRs 3, 7, and 9 (147), and has been implicated in the control of HSV-1 through promoting production of IFN-α/β in a TLR3-dependent manner (18). Indeed a dominant negative form of TLR3 (P554S) has now also been linked to HSE in otherwise healthy children (175), as have mutations in the TLR3-associated signaling molecules TRAF3 (R118W) (118), TRIF (R141X; S186L) (130) and TBK1 (D50A; G159A) (51), as well as the transcription factor IRF3 (R285Q) (4). These genetic studies revealed that although TLR3 is expressed on multiple cell types and recognizes a common virus product, double-stranded RNA, surprisingly, it appears to be essential only for control of neurotropic virus infections in humans.
Additional gene polymorphisms likely serve as risk factors for HSV infection and pathology, and these continue to be explored. One study investigated SNPs in the human TBX21 gene, which encodes a transcription factor known to influence HSV-2 adaptive immune responses (146). Investigators observed that a SNP (rs17244587-A) in the 3’ untranslated region of TBX21 was a risk factor for susceptibility to HSV-2, and that a homozygous genotype was found only in HSV-2-positive individuals. Other groups have attempted to address the frequency of oral lesions and what causes certain individuals to have repeated outbreaks. A genetic linkage analysis of over 350 HSV-1 infected individuals identified a region on chromosome 21 associated with increased frequency of cold sore outbreaks (55). This team then further mapped the susceptibility locus to an uncharacterized gene, which they subsequently named cold sore susceptibility gene-1 (CSSG1) (81). Follow-up studies on the role of this gene have not yet been published.
NOROVIRUS AND ROTAVIRUS
Norovirus, also referred to as Norwalk virus, is one of the most common causes of acute gastroenteritis, yet studies in human subjects have shown that a significant portion of the population is innately resistant to infection, independent of the presence of anti-norovirus serum antibodies (72, 112). Norovirus binds to carbohydrates of the histo-blood group family on intestinal epithelial cells (97). Indeed, cellular binding is dependent upon the presence of α1,2-linked fucose on the surface of cells. This requires a functional α1,2-fucosyltransferase gene (FUT2), which encodes a Golgi enzyme that transfers a terminal fucose onto glycoproteins and glycolipids (97). Approximately 20% of Europeans are homozygous for an inactivating missense mutation in FUT2 (428G>A), and a nonsense mutation (385A>T) is also present in Asian populations (41). In a seminal study, human volunteers were infected with norovirus and assessed for mutation of FUT2. Even at high doses of the virus, none of the individuals with defective FUT2 genes showed clinical signs of infection, developed a norovirus antibody response, or had detectable virus in stool samples, thus decisively demonstrating the role of FUT2 in controlling susceptibility to norovirus infection (88). The requirement for active FUT2 in norovirus infections has been confirmed by several subsequent studies on additional cohorts (31, 89, 93, 158). However, the protective effects of altered FUT2 alleles may be norovirus strain specific (46), and additional related factors, such as ABO blood type, also influence susceptibility (63, 96, 107). In addition, FUT2 functionality has been linked to infection with rotavirus, another enteric pathogen that causes diarrheal disease in children, and that utilizes cell surface carbohydrates for attachment (114). These results provide a basis for identifying individuals resistant to infection by these viruses and suggest that the development of purified fucose-containing oligosaccharides may be effective as anti-infectives (163).
PARVOVIRUS
Although parvoviruses generally do not cause disease in humans, the B19 strain typically causes a mild skin rash in children, and can cause more serious anemia in those who are immunocompromised or in those already genetically predisposed to hemolytic disorders. Infection is largely restricted to erythroid cells, and the virus uses the blood group P antigen, a glycosphingolipid globoside, as an entry receptor (14). In one of the earliest mechanistic examples of specific resistance to infection, it was shown that rare individuals lacking P antigen displayed no serological evidence of previous infections with B19, whereas a high percentage of the controls were positive for anti-B19 antibodies (15). Likewise, cells from P-negative donors could not be infected, even at extreme doses (15). To date, P-negative phenotypes have been attributed to several rare polymorphisms in two genes, B3GALNT1 and A4GALT, which encode glycosyltransferases involved in distinct steps of P antigen biosynthesis (48, 138, 165, 166).
EPSTEIN-BARR VIRUS
Epstein-Barr virus (EBV) infects over 90% of the global population and establishes a lifelong latent infection in B cells (28). Although disease manifestation is often mild or asymptomatic, EBV can cause acute infectious mononucleosis, chronic infections, and a variety of cancers, namely lymphomas (28). Host genetic polymorphisms influence the initial immune response to the virus as well as viral reactivation and latency. Indeed, these factors account for much of the observed variation in EBV-related disease outcomes.
Cytokine genes that affect the inflammatory response have been implicated in controlling EBV infections. High levels of IL-10 production, for example, are protective against primary and chronic EBV infection despite IL-10 being an immunosuppressive anti-inflammatory cytokine (50). Three polymorphic sites in the IL10 promoter region (rs1800871, rs1800872, and rs1800896) cause varying levels of IL-10 production. The high production allele, in which all three SNPs are present, is significantly linked to EBV resistance (49, 50). In a group of adults, EBV-seronegative individuals were nearly twice as likely to have the high production allele compared to seropositive individuals (50). Moreover, in a cohort of children, high IL-10 production was significantly correlated with seronegativity and effectively postponed, but did not prevent, primary EBV infection (49). A polymorphism in IL1B (rs16944), which results in a weaker inflammatory response, is also associated with EBV resistance (62). These studies collectively suggest that a reduced inflammatory response may be beneficial in controlling EBV.
There are also several rare genetic abnormalities affecting lymphocyte function that lead to increased susceptibility to EBV. Mutations observed in the magnesium transporter gene MAGT1 (10-bp deletion at the exon 7 splicing site; nonsense mutation in exon 3) lead to impaired lymphocyte responses following infection (86). Under normal conditions, there is a rapid influx of Mg2+ ions following antigen stimulation, which promotes expression of the natural killer activating receptor NKG2D in natural killer and CD8 T cells (19, 86). However, MAGT1 mutations reduce the level of free Mg2+, markedly reducing ion influx and causing reduced NK and T cell responses (86). Dysfunctional MAGT1 is a hallmark of X-linked immunodeficiency with Mg2+ defect, Epstein-Barr virus infection, and neoplasia (XMEN) disease (19). Individuals with XMEN disease suffer from rampant EBV infections and often develop lymphomas at young ages due to their inadequate immune responses against the virus (19). Likewise, individuals with X-linked lym-phoproliferative disease type 1 (XLP1) suffer from extreme sensitivity to EBV and often develop fatal mononucleosis (54, 110). XLP1 is characterized by mutations in SH2D1A, which encodes the signaling lymphocytic activation molecule-associated protein (143). This protein is crucial for directing CD8 T cells to virus-infected B cells, making it especially important in EBV infections (54, 110). IL-2-inducible T cell kinase (ITK) deficiency manifests very similarly to XMEN and XLP1 but is distinguished from these diseases because of its inheritance pattern (60). ITK is essential for the development of natural killer T cells (NKTs), and both nonsense mutations and deletions in the ITK gene effectively inhibit NKT development (60, 95, 140). NKTs may prevent viral reactivation following latent EBV infection (140). Overall, there are various mutations underlying EBV susceptibility that each lead to defects in cytotoxic immune cell killing mechanisms, underscoring the important role of this adaptive immune function in controlling EBV.
CONCLUDING REMARKS
Genetic studies of virus susceptibility in humans have contributed to a better understanding of the essential elements necessary for virus resistance and control, although in many instances causative mechanisms for effects of specific gene variants are lacking. Studies focused on HIV, HBV, norovirus, rotavirus, and parvovirus have shown that variations in genes encoding virus receptors or receptor-modifying enzymes render humans highly resistant to infection (15, 33, 88, 90, 114, 116, 129), and suggest that blockade of receptor binding or inhibition of virus entry pathways may represent an effective antiviral paradigm for prevention or treatment of infection.
In other instances, genetic studies have provided unanticipated results. One notable example is the apparent lack of increased susceptibility to infectious diseases other than HSE in individuals lacking functional UNC-93B andTLR3 (18, 175). Unlike the role of TLR3 in detecting numerous pathogens in cell lines and in mice, it appears that human TLR3 and its signaling pathways play a unique role in neuronal defense against HSV-1, but are not essential for defense against other commonly encountered viruses (174). Other innate immune factors, such as IRF7 and IFITM3, appear to be essential for virus control primarily upon encountering a new virus for which individuals lack adaptive immune memory. Thus, immune defects caused by variation in these genes were unmasked in seemingly healthy individuals upon infection with emergent influenza viruses (27, 39, 162, 177).
Still other factors identified by genetic methods are the subjects of ongoing research to clarify their roles in viral pathogenesis. IFNL4, as an inducer of antiviral gene expression, might be expected to contribute to antiviral defense, yet an inactivating variant of the 1FNL4 gene is associated with better clearance of HCV infection, and this paradox remains to be fully explained mechanistically (8, 108, 121). Nonetheless, this link between HCV and 1FNL genes represents a unique example of a genetic association that was rapidly translated for use in the clinic in designing HCV treatment regimens. We anticipate that the ongoing accumulation of knowledge on the genetics of infectious diseases (Table 1 and Figure 1) will increasingly be used to identify individuals who are particularly susceptible or resistant to specific viral diseases and to enable informed and personalized vaccination and treatment approaches in the years ahead.
ACKNOWLEDGMENTS
J.S.Y. is supported by National Institutes of Health (NIH) grant AI130110. A.D.K. is supported by NIH training grant GM068412. J.A.D. is supported by the Pelotonia Fellowship Program of The Ohio State University Comprehensive Cancer Center. L.B. is supported by grant UL1TR001866 from the National Center for Advancing Translational Sciences Clinical and Translational Science Award program of the NIH. T.M.M. is supported by the Gilliam Fellowship Program of the Howard Hughes Medical Institute. L.S.S. is supported by NIH grants AI059639 and AG051428. P.L.G. is supported by NIH grant CA100730. C.B.L. is supported by NIH grant AI083284. B.R.R. is supported by NIH grants AI111825 and OD012142. L.W. is supported by NIH grants AI104483, AI120209, AI127667, and CA181997.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, et al. 2011. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature 476:96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, et al. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581–91 [DOI] [PubMed] [Google Scholar]
- 3.An P, Bleiber G, Duggal P, Nelson G, May M, et al. 2004. APOBEC3G genetic variants and their influence on the progression to AIDS. J. Virol. 78:11070–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, et al. 2015. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 212:1371–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assone T, de Souza FV, Gaester KO, Fonseca LA, Luiz Odo C, et al. 2014. IL28B gene polymorphism SNP rs8099917 genotype GG is associated with HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) in HTLV-1 carriers. PLOS Negl. Trop. Dis. 8:e3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Audry M, Ciancanelli M, Yang K, Cobat A, Chang H-H, et al. 2011. NEMO is a key component of NF-κB- and IRF-3-dependent TLR3-mediated immunity to herpes simplex virus. J. Allergy Clin. Immunol. 128:610–17.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Badano I, Stietz SM, Schurr TG, Picconi AM, Fekete D, et al. 2012. Analysis of TNFα promoter SNPs and the risk of cervical cancer in urban populations of Posadas (Misiones, Argentina). J. Clin. Virol 53:54–59 [DOI] [PubMed] [Google Scholar]
- 8.Bibert S, Roger T, Calandra T, Bochud M, Cerny A, et al. 2013. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J. Exp. Med. 210:1109–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackwell JM, Jamieson SE, Burgner D. 2009. HLA and infectious diseases. Clin. Microbiol. Rev. 22:370–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyoglu-Barnum S, Chirkova T, Todd SO, Barnum TR, Gaston KA, et al. 2014. Prophylaxis with a respiratory syncytial virus (RSV) anti-G protein monoclonal antibody shifts the adaptive immune response to RSV rA2-line19F infection from Th2 to Th1 in BALB/c mice. J. Virol. 88:10569–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brackenridge S, Evans EJ, Toebes M, Goonetilleke N, Liu MK, et al. 2011. An early HIV mutation within an HLA-B*57-restricted T cell epitope abrogates binding to the killer inhibitory receptor 3DL1. J. Virol. 85:5415–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradley H, Markowitz LE, Gibson T, McQuillan GM. 2014. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J. Infect. Dis. 209:325–33 [DOI] [PubMed] [Google Scholar]
- 13.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, et al. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown KE, Anderson SM, Young NS. 1993. Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science 262:114–17 [DOI] [PubMed] [Google Scholar]
- 15.Brown KE, Hibbs JR, Gallinella G, Anderson SM, Lehman ED, et al. 1994. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). New Engl. J. Med. 330:1192–96 [DOI] [PubMed] [Google Scholar]
- 16.Carneiro-Proietti AB, Amaranto-Damasio MS, Leal-Horiguchi CF, Bastos RH, Seabra-Freitas G, et al. 2014. Mother-to-child transmission of human T-cell lymphotropic viruses-1/2: what we know, and what are the gaps in understanding and preventing this route of infection. J. Pediatr. Infect. Dis. Soc. 3(Suppl. 1):S24–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, et al. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 283:1748–52 [DOI] [PubMed] [Google Scholar]
- 18.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, et al. 2006. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–12 [DOI] [PubMed] [Google Scholar]
- 19.Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, et al. 2013. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 341:186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, et al. 2009. A partial form of recessive STAT1 deficiency in humans. J. Clin. Investig. 119:1502–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Mizuguchi H, Yao D, Ide M, Kuroda Y, et al. 2005. Thermolabile phenotype of carnitine palmitoyltransferase II variations as a predisposing factor for influenza-associated encephalopathy. FEBS Lett. 579:2040–44 [DOI] [PubMed] [Google Scholar]
- 22.Chesarino NM, McMichael TM, Hach JC, Yount JS. 2014. Phosphorylation of the antiviral protein interferon-inducible transmembrane protein 3 (IFITM3) dually regulates its endocytosis and ubiquitination. J. Biol. Chem. 289:11986–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chesarino NM, McMichael TM, Yount JS. 2014. Regulation of the trafficking and antiviral activity of IFITM3 by post-translational modifications. Future Microbiol. 9:1151–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chesarino NM, McMichael TM, Yount JS. 2015. E3 ubiquitin ligase NEDD4 promotes influenza virus infection by decreasing levels of the antiviral protein IFITM3. PLOS Pathog. 11:e1005095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi EH, Lee HJ, Chanock SJ. 2013. Human genetics and respiratory syncytial virus disease: current findings and future approaches. Curr. Top. Microbiol. Immunol. 372:121–37 [DOI] [PubMed] [Google Scholar]
- 26.Choi EH, Lee HJ, Yoo T, Chanock SJ. 2002. A common haplotype of interleukin-4 gene IL4 is associated with severe respiratory syncytial virus disease in Korean children. J. Infect. Dis. 186:1207–11 [DOI] [PubMed] [Google Scholar]
- 27.Ciancanelli MJ, Huang SXL, Luthra P, Garner H, Itan Y, et al. 2015. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348:448–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen JI. 2000. Epstein-Barr virus infection. N. Engl. J. Med. 343:481–92 [DOI] [PubMed] [Google Scholar]
- 29.Compton AA, Roy N, Porrot F, Billet A, Casartelli N, et al. 2016. Natural mutations in 1F1TM3 modulate post-translational regulation and toggle antiviral specificity. EMBO Rep. 17:1657–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coon S, Wang D, Wu L. 2012. Polymorphisms of the SAMHD1 gene are not associated with the infection and natural control of HIV type 1 in Europeans and African-Americans. AIDS Res. Hum. Retroviruses 28:1565–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Currier RL, Payne DC, Staat MA, Selvarangan R, Shirley SH, et al. 2015. Innate susceptibility to norovirus infections influenced by FUT2 genotype in a United States pediatric population. Clin. Infect. Dis. 60:1631–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Custer B, Sullivan SD, Hazlet TK, Iloeje U, Veenstra DL, Kowdley KV. 2004. Global epidemiology of hepatitis B virus. J. Clin. Gastroenterol. 38:S158–68 [DOI] [PubMed] [Google Scholar]
- 33.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, et al. 1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273:1856–62 [DOI] [PubMed] [Google Scholar]
- 34.Deshpande A, Nolan JP, White PS, Valdez YE, Hunt WC, et al. 2005. TNF-α promoter polymorphisms and susceptibility to human papillomavirus 16-associated cervical cancer. J. Infect. Dis 191:969–76 [DOI] [PubMed] [Google Scholar]
- 35.Duarte I, Santos A, Sousa H, Catarino R, Pinto D, et al. 2005. G-308A TNF-α polymorphism is associated with an increased risk of invasive cervical cancer. Biochem. Biophys. Res. Commun. 334:588–92 [DOI] [PubMed] [Google Scholar]
- 36.Dunne EF, Unger ER, Sternberg M, McQuillan G, Swan DC, et al. 2007. Prevalence of HPV infection among females in the United States. JAMA 297:813–19 [DOI] [PubMed] [Google Scholar]
- 37.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, et al. 2003. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33:388–91 [DOI] [PubMed] [Google Scholar]
- 38.Enard D, Cai L, Gwennap C, Petrov DA. 2016. Viruses are a dominant driver of protein adaptation in mammals. eLife 5:e12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Everitt AR, Clare S, Pertel T, John SP, Wash RS, et al. 2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484:519–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, et al. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferrer-Admetlla A, Sikora M, Laayouni H, Esteve A, Roubinet F, et al. 2009. A natural history of FUT2 polymorphism in humans. Mol. Biol. Evol. 26:1993–2003 [DOI] [PubMed] [Google Scholar]
- 42.Gaiotti D, Chung J, Iglesias M, Nees M, Baker PD, et al. 2000. Tumor necrosis factor-α promotes human papillomavirus (HPV) E6/E7 RNA expression and cyclin-dependent kinase activity in HPV-immortalized keratinocytes by a ras-dependent pathway. Mol. Carcinog. 27:97–109 [DOI] [PubMed] [Google Scholar]
- 43.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, et al. 2009. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401 [DOI] [PubMed] [Google Scholar]
- 44.Gentile DA, Doyle WJ, Zeevi A, Piltcher O, Skoner DP. 2003. Cytokine gene polymorphisms moderate responses to respiratory syncytial virus in adults. Hum. Immunol. 64:93–98 [DOI] [PubMed] [Google Scholar]
- 45.Gessain A, Barin F, Vernant JC, Gout O, Maurs L, et al. 1985. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 2:407–10 [DOI] [PubMed] [Google Scholar]
- 46.Halperin T, Vennema H, Koopmans M, Kahila Bar-Gal G, Kayouf R, et al. 2008. No association between histo-blood group antigens and susceptibility to clinical infections with genogroup II norovirus. J. 1nfect. Dis. 197:63–65 [DOI] [PubMed] [Google Scholar]
- 47.Hartshorn KL. 2010. Role of surfactant protein A and D (SP-A and SP-D) in human antiviral host defense. Front. Biosci. (Schol. Ed.) 2:527–46 [DOI] [PubMed] [Google Scholar]
- 48.Hellberg A, Poole J, Olsson ML. 2002. Molecular basis of the globoside-deficient Pk blood group phenotype. Identification of four inactivating mutations in the UDP-N-acetylgalactosamine: globotriaosylceramide 3-β-N-acetylgalactosaminyltransferase gene. J. Biol. Chem. 277:29455–59 [DOI] [PubMed] [Google Scholar]
- 49.Helminen ME, Kilpinen S, Virta M, Hurme M. 2001. Susceptibility to primary Epstein-Barr virus infection is associated with interleukin-10 gene promoter polymorphism. J. Infect. Dis. 184:777–80 [DOI] [PubMed] [Google Scholar]
- 50.Helminen ME, Lahdenpohja N, Hurme M. 1999. Polymorphism of the interleukin-10 gene is associated with susceptibility to Epstein-Barr virus infection. J. 1nfect. Dis. 180:496–99 [DOI] [PubMed] [Google Scholar]
- 51.Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, et al. 2012. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 209:1567–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herndon CN, Jennings RG. 1951. A twin-family study of susceptibility to poliomyelitis. Am. J. Hum.Genet. 3:17–46 [PMC free article] [PubMed] [Google Scholar]
- 53.Herrera-Ramos E, López-Rodríguez M, Ruiz-Hernández JJ, Horcajada JP, Borderías L, et al. 2014. Surfactant protein A genetic variants associate with severe respiratory insufficiency in pandemic influenza A virus infection. Crit. Care 18:R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, et al. 2010. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood 116:3249–57 [DOI] [PubMed] [Google Scholar]
- 55.Hobbs MR, Jones BB, Otterud BE, Leppert M, Kriesel JD. 2008. Identification of a herpes simplex labialis susceptibility region on human chromosome 21. J. Infect. Dis. 197:340–46 [DOI] [PubMed] [Google Scholar]
- 56.Hoebee B, Rietveld E, Bont L, Oosten M, Hodemaekers HM, et al. 2003Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor α polymorphisms. J. Infect. Dis. 187:2–11 [DOI] [PubMed] [Google Scholar]
- 57.Honda M, Sakai A, Yamashita T, Nakamoto Y, Mizukoshi E, et al. 2010. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology 139:499–509 [DOI] [PubMed] [Google Scholar]
- 58.Honegger JR, Tedesco D, Kohout JA, Prasad MR, Price AA, et al. 2016. Influence of IFNL3 and HLA-DPB1 genotype on postpartum control of hepatitis C virus replication and T-cell recovery. PNAS 113:10684–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, et al. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474:658–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huck K, Feyen O, Niehues T, Ruschendorf F, Hübner N, et al. 2009. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Investig 119:1350–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hull J, Thomson A, Kwiatkowski D. 2000. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax 55:1023–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hurme M, Helminen ME. 1998. Polymorphism of the IL-1 gene complex in Epstein-Barr virus seronegative and seropositive adult blood donors. Scand. J. Immunol 48:219–22 [DOI] [PubMed] [Google Scholar]
- 63.Hutson AM, Atmar RL, Graham DY, Estes MK. 2002. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis 185:1335–37 [DOI] [PubMed] [Google Scholar]
- 64.Inoue Y, Shimojo N, Suzuki Y, Campos Alberto EJ, Yamaide A, et al. 2007. CD14–550 C/T, which is related to the serum level of soluble CD14, is associated with the development of respiratory syncytial virus bronchiolitis in the Japanese population. J. Infect. Dis 195:1618–24 [DOI] [PubMed] [Google Scholar]
- 65.International HIV Controllers Study Writing Team, Pereyra F, Jia X, McLaren PJ, Telenti A, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishitsuka K, Tamura K. 2014. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 15:e517–26 [DOI] [PubMed] [Google Scholar]
- 67.Jackson M, Scott R. 1996. Different patterns of cytokine induction in cultures of respiratory syncytial (RS) virus-specific human TH cell lines following stimulation with RS virus and RS virus proteins. J. Med. Virol. 49:161–69 [DOI] [PubMed] [Google Scholar]
- 68.Janssen R, Bont L, Siezen CL, Hodemaekers HM, Ermers MJ, et al. 2007. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J. Infect. Dis 196:826–34 [DOI] [PubMed] [Google Scholar]
- 69.Jiang D-K, Ma X-P, Wu X, Peng L, Yin J, et al. 2015. Genetic variations in STAT4, C2, HLA-DRB1 and HLA-DQ associated with risk of hepatitis B virus-related liver cirrhosis. Sci. Rep 5:16278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang D-K, Sun J, Cao G, Liu Y, Lin D, et al. 2013. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus–related hepatocellular carcinoma. Nat. Genet 45:72–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang X, Su K, Tao J, Fan R, Xu Y, et al. 2016. Association of STAT4 polymorphisms with hepatitis B virus infection and clearance in Chinese Han population. Amino Acids 48:2589–98 [DOI] [PubMed] [Google Scholar]
- 72.Johnson PC, Mathewson JJ, DuPont HL, Greenberg HB. 1990. Multiple-challenge study of host susceptibility to Norwalk gastroenteritis in US adults. J. Infect. Dis 161:18–21 [DOI] [PubMed] [Google Scholar]
- 73.Johnson TR, Parker RA, Johnson JE, Graham BS. 2003. IL-13 is sufficient for respiratory syncytial virus G glycoprotein-induced eosinophilia after respiratory syncytial virus challenge. J. Immunol 170:2037–45 [DOI] [PubMed] [Google Scholar]
- 74.Kaslow RA, Carrington M, Apple R, Park L, Munoz A, et al. 1996. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat. Med 2:405–11 [DOI] [PubMed] [Google Scholar]
- 75.Key FM, Peter B, Dennis MY, Huerta-Sánchez E, Tang W, et al. 2014. Selection on a variant associated with improved viral clearance drives local, adaptive pseudogenization of interferon lambda 4 (IFNL4). PLOS Genet. 10:e1004681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim LH, Cheong HS, Namgoong S, Kim JO, Kim J-H, et al. 2015. Replication of genome wide association studies on hepatocellular carcinoma susceptibility loci of STAT4 and HLA-DQ in a Korean population. Infect. Genet. Evol 33:72–76 [DOI] [PubMed] [Google Scholar]
- 77.Kirkpatrick A, Bidwell J, van den Brule AJC, Meijer CJLM, Pawade J, Glew S. 2004. TNFα polymorphism frequencies in HPV-associated cervical dysplasia. Gynecol. Oncol 92:675–79 [DOI] [PubMed] [Google Scholar]
- 78.Kodama D, Saito M, Matsumoto W, Sabouri AH, Izumo S, et al. 2004. Longer dinucleotide repeat polymorphism in matrix metalloproteinase-9 (MMP-9) gene promoter which correlates with higher HTLV-I Tax mediated transcriptional activity influences the risk of HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP). J. Neuroimmunol 156:188–94 [DOI] [PubMed] [Google Scholar]
- 79.Kohaar I, Thakur N, Salhan S, Batra S, Singh V, et al. 2007. TNFα-308G/A polymorphism as a risk factor for HPV associated cervical cancer in Indian population. Cell Oncol. 29:249–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kresfelder TL, Janssen R, Bont L, Pretorius M, Venter M 2011. Confirmation of an association between single nucleotide polymorphisms in the VDR gene with respiratory syncytial virus related disease in South African children. J. Med. Virol. 83:1834–40 [DOI] [PubMed] [Google Scholar]
- 81.Kriesel JD, Jones BB, Matsunami N, Patel MK, Pierre CA St., et al. 2011. C21orf91 genotypes correlate with herpes simplex labialis (cold sore) frequency: description of a cold sore susceptibility gene. J. Infect. Dis 204:1654–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, et al. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474:654–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lahti M, Lofgren J, Marttila R, Renko M, Klaavuniemi T, et al. 2002. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr. Res 51:696–99 [DOI] [PubMed] [Google Scholar]
- 84.Lavanchy D 2004. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepatol 11:97–107 [DOI] [PubMed] [Google Scholar]
- 85.Lazarczyk M, Pons C, Mendoza JA, Cassonnet P, Jacob Y, Favre M. 2008. Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J. Exp. Med 205:35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, et al. 2011. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475:471–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y, Si L, Zhai Y, Hu Y, Hu Z, et al. 2016. Genome-wide association study identifies 8p21.3 associated with persistent hepatitis B virus infection among Chinese. Nat. Commun 7:11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, et al. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med 9:548–53 [DOI] [PubMed] [Google Scholar]
- 89.Liu P, Wang X, Lee J-C, Teunis P, Hu S, et al. 2014. Genetic susceptibility to norovirus GII.3 and GII.4 infections in Chinese pediatric diarrheal disease. Pediatr. Infect. Dis. J 33:e305–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, et al. 1996. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86:367–77 [DOI] [PubMed] [Google Scholar]
- 91.Löfgren J, Marttila R, Renko M, Rämet M, Hallman M. 2010. Toll-like receptor 4 Asp299Gly polymorphism in respiratory syncytial virus epidemics. Pediatr. Pulmonol 45:687–92 [DOI] [PubMed] [Google Scholar]
- 92.Löfgren J, Rämet M, Renko M, Marttila R, Hallman M. 2002. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J. Infect. Dis 185:283–89 [DOI] [PubMed] [Google Scholar]
- 93.Lopman BA, Trivedi T, Vicuna Y, Costantini V, Collins N, et al. 2015. Norovirus infection and disease in an Ecuadorian birth cohort: association of certain norovirus genotypes with host FUT2 secretor status. J. Infect. Dis 211:1813–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mak CM, Lam C-W, Fong N-C, Siu W-K, Lee H-C, et al. 2011. Fatal viral infection-associated encephalopathy in two Chinese boys: a genetically determined risk factor of thermolabile carnitine palmitoyltransferase II variants. J. Hum. Genet 56:617–21 [DOI] [PubMed] [Google Scholar]
- 95.Mansouri D, Mahdaviani SA, Khalilzadeh S, Mohajerani SA, Hasanzad M, et al. 2012. IL-2-inducible T-cell kinase deficiency with pulmonary manifestations due to disseminated Epstein-Barr virus infection. Int. Arch. Allergy Immunol 158:418–22 [DOI] [PubMed] [Google Scholar]
- 96.Marionneau S, Airaud F, Bovin NV, Le Pendu J, Ruvoën-Clouet N. 2005. Influence of the combined ABO, FUT2 and FUT3 polymorphism on susceptibility to Norwalk virus attachment. J. Infect. Dis 192:1071–77 [DOI] [PubMed] [Google Scholar]
- 97.Marionneau S, Ruvoën N, Le Moullac-Vaidye B, Clement M, Cailleau-Thomas A, et al. 2002. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology 122:1967–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martin MP, Gao X, Lee JH, Nelson GW, Detels R, et al. 2002. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat. Genet 31:429–34 [DOI] [PubMed] [Google Scholar]
- 99.Matsuura K, Isogawa M, Tanaka Y. 2016. Host genetic variants influencing the clinical course of hepatitis B virus infection. J. Med. Virol 88:371–79 [DOI] [PubMed] [Google Scholar]
- 100.McFarland AP, Horner SM, Jarret A, Joslyn RC, Bindewald E, et al. 2014. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat. Immunol 15:72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McLaughlin RN Jr., Malik HS. 2017. Genetic conflicts: the usual suspects and beyond. J. Exp. Biol 220:6–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McNally JD, Sampson M, Matheson LA, Hutton B, Little J. 2014. Vitamin D receptor (VDR) polymorphisms and severe RSV bronchiolitis: a systematic review and meta-analysis. Pediatr. Pulmonol 49:790–99 [DOI] [PubMed] [Google Scholar]
- 103.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, et al. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. PNAS 97:2709–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, et al. 2007. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25:5086–96 [DOI] [PubMed] [Google Scholar]
- 105.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, et al. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nakahata S, Saito Y, Hamasaki M, Hidaka T, Arai Y, et al. 2009. Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer 48:768–76 [DOI] [PubMed] [Google Scholar]
- 107.Nordgren J, Nitiema LW, Ouermi D, Simpore J, Svensson L. 2013. Host genetic factors affect susceptibility to norovirus infections in Burkina Faso . PLOS ONE 8:e69557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.O’Brien TR, Pfeiffer RM, Paquin A, Lang Kuhs KA, Chen S, et al. 2015. Comparison of functional variants in IFNL4 and IFNL3 for association with HCV clearance. J. Hepatol 63:1103–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Brien TR, Prokunina-Olsson L, Donnelly RP. 2014. IFN-A4: the paradoxical new member of the interferon lambda family. J. Interferon. Cytokine Res 34:829–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Palendira U, Low C, Chan A, Hislop AD, Ho E, et al. 2011. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLOS Biol. 9:e1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pan W, Song IS, Shin HJ, Kim MH, Choi YL, et al. 2011. Genetic polymorphisms in Na+-taurocholate co-transporting polypeptide (NTCP) and ileal apical sodium-dependent bile acid transporter (ASBT) and ethnic comparisons of functional variants of NTCP among Asian populations. Xenobiotica 41:501–10 [DOI] [PubMed] [Google Scholar]
- 112.Parrino TA, Schreiber DS, Trier JS, Kapikian AZ, Blacklow NR. 1977. Clinical immunity in acute gastroenteritis caused by Norwalk agent. N. Engl. J. Med 297:86–89 [DOI] [PubMed] [Google Scholar]
- 113.Patel AS, Karagas MR, Pawlita M, Waterboer T, Nelson HH. 2008. Cutaneous human papillomavirus infection, the EVER2 gene and incidence of squamous cell carcinoma: a case-control study. Int. J. Cancer 122:2377–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Payne DC, Currier RL, Staat MA, Sahni LC, Selvarangan R, et al. 2015. Epidemiologic association between FUT2 secretor status and severe rotavirus gastroenteritis in children in the United States. JAMA Pediatr. 169:1040–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pelak K, Need AC, Fellay J, Shianna KV, Feng S, et al. 2011. Copy number variation of KIR genes influences HIV-1 control. PLOS Biol. 9:e1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peng L, Zhao Q, Li Q, Li M, Li C, et al. 2015. The p.Ser267Phe variant in SLC10A1 is associated with resistance to chronic hepatitis B. Hepatology 61:1251–60 [DOI] [PubMed] [Google Scholar]
- 117.Percher A, Ramakrishnan S, Thinon E, Yuan X, Yount JS, Hang HC. 2016. Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. PNAS 113:4302–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pérez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, et al. 2010. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33:400–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Plancoulaine S, Gessain A, Tortevoye P, Boland-Auge A, Vasilescu A, et al. 2006. A major susceptibility locus for HTLV-1 infection in childhood maps to chromosome 6q27. Hum. Mol. Genet 15:3306–12 [DOI] [PubMed] [Google Scholar]
- 120.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. PNAS 77:7415–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, et al. 2013. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet 45:164–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puel A, Reichenbach J, Bustamante J, Ku CL, Feinberg J, et al. 2006. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am. J. Hum. Genet 78:691–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Puthothu B, Forster J, Heinzmann A, Krueger M. 2006. TLR-4 and CD14 polymorphisms in respiratory syncytial virus associated disease. Dis. Markers 22:303–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Puthothu B, Krueger M, Forster J, Heinzmann A 2006. Association between severe respiratory syncytial virus infection and IL13/IL4 haplotypes. J. Infect. Dis 193:438–41 [DOI] [PubMed] [Google Scholar]
- 125.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. 2002. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet 32:579–81 [DOI] [PubMed] [Google Scholar]
- 126.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, et al. 2010. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology 138:1338–45.e7 [DOI] [PubMed] [Google Scholar]
- 127.Rocha-Júnior MC, Haddad R, Cilião Alves DC, de Deus Wagatsuma VM, Mendes-Junior CT, et al. 2012. Interleukin-18 and interferon-gamma polymorphisms are implicated on proviral load and susceptibility to human T-lymphotropic virus type 1 infection. Tissue Antigens 80:143–50 [DOI] [PubMed] [Google Scholar]
- 128.Sabouri AH, Saito M, Lloyd AL, Vine AM, Witkover AW, et al. 2004. Polymorphismin theinterleukin-10 promoter affects both provirus load and the risk of human T lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis. J. Infect. Dis. 190:1279–85 [DOI] [PubMed] [Google Scholar]
- 129.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, et al. 1996. Resistance to HIV-1 infection in caucasianindividuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722–25 [DOI] [PubMed] [Google Scholar]
- 130.Sancho-Shimizu V, Pérez de Diego R, Lorenzo L, Halwani R, Alangari A, et al. 2011. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig 121:4889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, et al. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. PNAS 105:7034–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–36 [DOI] [PubMed] [Google Scholar]
- 133.Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. 2015. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386:1546–55 [DOI] [PubMed] [Google Scholar]
- 134.Shea PR, Shianna KV, Carrington M, Goldstein DB. 2013. Host genetics of HIV acquisition and viral control. Annu. Rev. Med 64:203–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sorensen TI, Nielsen GG, Andersen PK, Teasdale TW. 1988. Genetic and environmental influences on premature death in adult adoptees. N. Engl. J. Med 318:727–32 [DOI] [PubMed] [Google Scholar]
- 136.Speelmon EC, Livingston-Rosanoff D, Li SS, Vu Q, Bui J, et al. 2006. Genetic association of the antiviral restriction factor TRIM5 α with human immunodeficiency virus type 1 infection. J. Virol 80:2463–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gelais C St., Wu L.2011. SAMHD1: a new insight into HIV-1 restriction in myeloid cells. Retrovirology 8:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Steffensen R, Carlier K, Wiels J, Levery SB, Stroud M, et al. 2000. Cloning and expression of the histo-blood group PkUDP-galactose: Galβ1–4Glcβ1-cer α1,4-galactosyltransferase. Molecular genetic basis of the p phenotype. J. Biol. Chem 275:16723–29 [DOI] [PubMed] [Google Scholar]
- 139.Steininger C, Popow-Kraupp T, Laferl H, Seiser A, Godl I, et al. 2003. Acute encephalopathy associated with influenza A virus infection. Clin. Infect. Dis 36:567–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, et al. 2011. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica 96:472–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stoppelenburg AJ, von Hegedus JH, Huis in’t Veld R, Bont L, Boes M. 2014. Defective control of vitamin D receptor-mediated epithelial STAT1 signalling predisposes to severe respiratory syncytial virus bronchiolitis. J. Pathol 232:57–64 [DOI] [PubMed] [Google Scholar]
- 142.Storey A, Thomas M, Kalita A, Harwood C, Gardiol D, et al. 1998. Role of a p53 polymorphism in the development of human papillomavirus-associated cancer. Nature 393:229–34 [DOI] [PubMed] [Google Scholar]
- 143.Sumegi J, Huang D, Lanyi A, Davis JD, Seemayer TA, et al. 2000. Correlation of mutations of the SH2D1A gene and Epstein-Barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood 96:3118–25 [PubMed] [Google Scholar]
- 144.Sun Y, López CB. 2017. The innate immune response to RSV: advances in our understanding of critical viral and host factors. Vaccine 35:481–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, et al. 2009. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet 41:1100–4 [DOI] [PubMed] [Google Scholar]
- 146.Svensson A, Bergin AM, Lowhagen GB, Tunback P, Bellner L, et al. 2008. A 3′ untranslated region polymorphism in the TBX21 gene encoding T-bet is a risk factor for genital herpes simplex virus type 2 infection in humans. J. Gen. Virol 89:2262–68 [DOI] [PubMed] [Google Scholar]
- 147.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, et al. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol 7:156–64 [DOI] [PubMed] [Google Scholar]
- 148.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, et al. 2009. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat. Genet 41:1105–9 [DOI] [PubMed] [Google Scholar]
- 149.Tapia LI, Ampuero S, Palomino MA, Luchsinger V, Aguilar N, et al. 2013. Respiratory syncytial virus infection and recurrent wheezing in Chilean infants: a genetic background? Infect. Genet. Evol 16:54–61 [DOI] [PubMed] [Google Scholar]
- 150.Tate G, Suzuki T, Kishimoto K, Mitsuya T. 2004. Novel mutations ofEVER1/TMC6 gene in a Japanese patient with epidermodysplasia verruciformis. J. Hum. Genet 49:223–25 [DOI] [PubMed] [Google Scholar]
- 151.Terczyńska-Dyla E, Bibert S, Duong FH, Krol I, Jørgensen S, et al. 2014. Reduced IFNλ4 activity is associated with improved HCV clearance and reduced expression of interferon-stimulated genes. Nat. Commun 5:5699. [DOI] [PubMed] [Google Scholar]
- 152.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, et al. 2009. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461:798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thomas NJ, DiAngelo S, Hess JC, Fan R, Ball MW, et al. 2009. Transmission of surfactant protein variants and haplotypes in children hospitalized with respiratory syncytial virus. Pediatr. Res 66:70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Thrift AP, El-Serag HB, Kanwal F. 2017. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol 14:122–32 [DOI] [PubMed] [Google Scholar]
- 155.To KKW, Zhou J, Song Y-Q, Hung IFN, Ip WCT, et al. 2014. Surfactant protein B gene polymorphism is associated with severe influenza. Chest 145:1237–43 [DOI] [PubMed] [Google Scholar]
- 156.Trowsdale J, Knight JC. 2013. Major histocompatibility complex genomics and human disease. Annu. Rev. Genom. Hum. Genet 14:301–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tsukasaki K, Miller CW, Kubota T, Takeuchi S, Fujimoto T, et al. 2001. Tumor necrosis factor α polymorphism associated with increased susceptibility to development of adult T-cell leukemia/lymphoma in human T-lymphotropic virus type 1 carriers. Cancer Res. 61:3770–74 [PubMed] [Google Scholar]
- 158.Tu L-T, Liu F-P, Huang Y-C, Huang C-G, Yang S, et al. 2017. Genetic susceptibility to norovirus GII.4 Sydney strain infections in Taiwanese children. Pediatr. Infect. Dis. J 36:353–57 [DOI] [PubMed] [Google Scholar]
- 159.Urban TJ, Thompson AJ, Bradrick SS, Fellay J, Schuppan D, et al. 2010. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology 52:1888–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, et al. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol 189:12–19 [DOI] [PubMed] [Google Scholar]
- 161.Wang SS, Carreon JD, Hanchard B, Chanock S, Hisada M. 2009. Common genetic variants and risk for non-Hodgkin lymphoma and adult T-cell lymphoma/leukemia in Jamaica. Int. J. Cancer 125:1479–82 [DOI] [PubMed] [Google Scholar]
- 162.Wang ZF, Zhang AL, Wan YM, Liu XN, Qiu C, et al. 2014. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. PNAS 111:769–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Weichert S, Koromyslova A, Singh BK, Hansman S, Jennewein S, et al. 2016. Structural basis for norovirus inhibition by human milk oligosaccharides. J. Virol 90:4843–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Werness BA, Levine AJ, Howley PM. 1990. Association of human papillomavirus type-16 and type-18 E6 proteins with p53. Science 248:76–79 [DOI] [PubMed] [Google Scholar]
- 165.Westman JS, Benktander J, Storry JR, Peyrard T, Hult AK, et al. 2015. Identification of the molecular and genetic basis of PX2, a glycosphingolipid blood group antigen lacking on globoside-deficient erythrocytes. J. Biol. Chem 290:18505–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Westman JS, Hellberg A, Peyrard T, Hustinx H, Thuresson B, Olsson ML. 2013. P1/P2 genotyping of known and novel null alleles in the P1PK and GLOB histo-blood group systems. Transfusion 53:2928–39 [DOI] [PubMed] [Google Scholar]
- 167.Whitley RJ, Kimberlin DW, Roizman B. 1998. Herpes simplex viruses. Clin. Infect. Dis 26:541–53 [DOI] [PubMed] [Google Scholar]
- 168.Wilson J, Rowlands K, Rockett K, Moore C, Lockhart E, et al. 2005. Genetic variation at the IL10 gene locus is associated with severity of respiratory syncytial virus bronchiolitis. J. Infect. Dis 191:1705–9 [DOI] [PubMed] [Google Scholar]
- 169.Yan H, Zhong G, Xu G, He W, Jing Z, et al. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yang JD, Roberts LR. 2010. Hepatocellular carcinoma: a global view. Nat. Rev. Gastroenterol. Hepatol 7:448–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yao D, Mizuguchi H, Yamaguchi M, Yamada H, Chida J, et al. 2008. Thermal instability of compound variants of carnitine palmitoyltransferase II and impaired mitochondrial fuel utilization in influenza-associated encephalopathy. Hum. Mutat 29:718–27 [DOI] [PubMed] [Google Scholar]
- 172.Yount JS, Moltedo B, Yang Y-Y, Charron G, Moran TM, et al. 2010. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Biol 6:610–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang M, Lu Y, Zhang X, Lu A, Wang L, Chen C. 2016. Interleukin-4 polymorphism is associated with severity of respiratory syncytial virus infection. J. Paediatr. Child Health 52:25–29 [DOI] [PubMed] [Google Scholar]
- 174.Zhang S-Y, Herman M, Ciancanelli MJ, Pérez de Diego R, Sancho-Shimizu V, et al. 2013. TLR3 immunity to infection in mice and humans. Curr. Opin. Immunol 25:19–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang S-Y, Jouanguy E, Ugolini S, Smahi A, Elain G, et al. 2007. TLR3 deficiency in patients with herpes simplex encephalitis. Science 317:1522–27 [DOI] [PubMed] [Google Scholar]
- 176.Zhang Y-H, Makvandi-Nejad S, Qin L, Zhao Y, Zhang T, et al. 2015. Interferon-induced transmembrane protein-3 rs12252-C is associated with rapid progression of acute HIV-1 infection in Chinese MSM cohort. AIDS 29:889–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang Y-H, Zhao Y, Li N, Peng Y-C, Giannoulatou E, et al. 2013. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat. Commun 4:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhou J, Zhang X, Liu S, Wang Z, Chen Q, et al. 2016. Genetic association of TLR4 Asp299Gly, TLR4 Thr399Ile, and CD14 C-159T polymorphisms with the risk of severe RSV infection: a meta-analysis. Influenza Other Respir. Viruses 10:224–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, et al. 2015. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet. Med 17:774–81 [DOI] [PMC free article] [PubMed] [Google Scholar]