

Abstract
The nuclear lamina is a multi-protein lattice composed of A-and B-type lamins and their associated proteins. This protein lattice associates with heterochromatin and integral inner nuclear membrane proteins, providing a link between the genome, nucleoskeleton, and cytoskeleton. In the 1990s, mutations in EMD and LMNA were linked to Emery-Dreifuss muscular dystrophy. Since then, the number of diseases attributed to nuclear lamina defects, including laminopathies and other disorders, has increased to include more than 20 distinct genetic syndromes. Studies of patients and mouse genetic models have indicated the important roles for lamins and their associated proteins in the function of gastrointestinal organs including liver and pancreas. We review the interactions and functions of the lamina in relation to the nuclear envelope and genome, the ways in which its dysfunction is thought to contribute to human disease, and possible avenues for targeted therapies.
Keywords: nucleoskeleton, envelopathies, progeria, myopathy, neuropathy, lipodystrophy, nonalcoholic fatty liver disease
In metazoan cells, a structural and functional link between the genome and the cytoskeleton is required to allow cells to quickly and appropriately respond to mechanical, chemical, inflammatory, and other stimuli. This link is provided by nuclear envelope proteins, which have collective and individual structural and regulatory roles. Nuclear pore complexes allow regulated nuclear translocation of transcription factors and co-regulators1. The LINC complex (linker of the nucleoskeleton and cytoskeleton) tethers the nuclear envelope to cytoplasmic cytoskeletal networks, allowing transmission of mechanical and shear stress to the nucleus2. On the inner surface of the nuclear envelope, large regions of the genome, typically dominated by heterochromatin, are tethered to a multi-protein lattice3, 4 (Fig.1). This complex of proteins, the nuclear lamina, lies beneath the inner nuclear membrane and physically associates with nuclear pore proteins and a variety of transmembrane and integral membrane proteins, and in direct contact with large portions of the genome.
Figure 1.
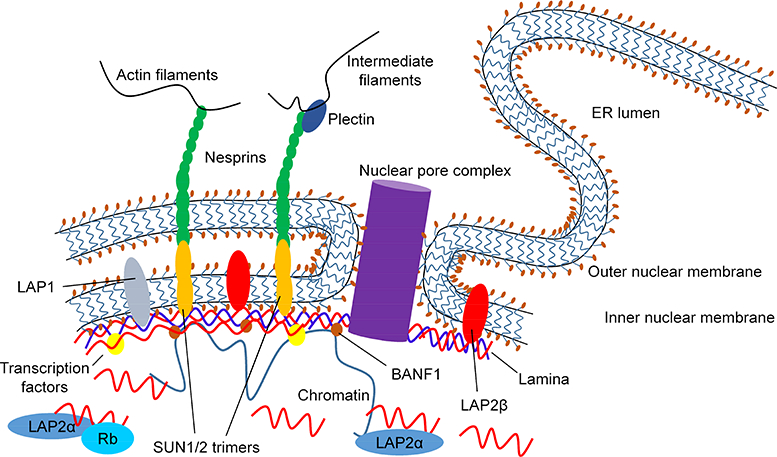
Structure of the nuclear envelope including nuclear pore complex, LINC complex, and the nuclear lamina. Schematic of the nuclear envelope structure including outer and inner nuclear membranes, the lamina, integral membrane proteins, nuclear pore complex, LINC complexes, components of the cytoplasmic cytoskeleton, and chromatin-lamina contacts. The outer nuclear membrane is shown in continuity with the membrane of the endoplasmic reticulum (ER). A portion of LMNA (red) is shown as a soluble nucleoplasmic protein, some of which is bound to LAP2α.
The primary components of the nuclear lamina are lamins, which are type V intermediate filament proteins (IFs)—the most common IFs in the nucleus (although other IFs, such as keratins, are also found at lower levels in the nucleus)5–8. Lamins are encoded by 3 genes that generate the proteins: lamin A/C (LMNA; protein also referred to as LMNA), lamin B1 (LMNB1; protein also referred to as LMNB1), and lamin B2 (LMNB2; protein also referred to as LMNB2)9–11. The B-type lamins are expressed ubiquitously and throughout development, whereas A-type lamins are primarily expressed in differentiated cells12, 13. Together these proteins form a lattice that forms an interface with the inner nuclear membrane, nuclear pore complexes, transcription factors and co-regulatory proteins, and chromatin. Anchoring of the lamina to the inner nuclear membrane is achieved via B-type lamin farnesylation and lamin binding to transmembrane proteins that include lamina-associated polypeptide 1 (LAP1) and LEM-domain containing proteins such as LAP2β, emerin, and MAN1 (also called LEMD3)14, whereas anchoring to the genome is thought to occur via adaptor proteins including barrier to autointegration factor (BANF1), the lamin B receptor (LBR), and direct binding of lamin to chromatin15–18.
Lamin Post-translational Processing and Localization
Lamin C does not require post-translational modification to localize to the inner nuclear membrane. Lamin A, however, requires stepwise post-translational processing at the carboxy terminus via cysteine farnesylation at a cysteine-aliphatic-aliphatic-any amino acid (CAAX) motif, then proteolytic cleavage of the-AAX portion, carboxymethylation of the farnesylcysteine, and final clipping of the 15 carboxy-terminal residues, including the farnesylated cysteine, by the zinc metallopeptidase STE24 (ZMPSTE24)19–22. Although the B-type lamins are permanently farnesylated and found exclusively at the inner nuclear membrane as part of the nuclear lamina, a portion of LMNA is found in the nucleoplasm. Nucleoplasmic LMNA is stabilized by a mammal-specific isoform of thymopoietin (TMPO or LAP2), called LAP2a, but little is known about its function 23, 24
Lamina-associated Proteins
A and B-type lamins form an intricate network of overlapping but independent 3-dimensional protein meshes25 that interact with distinct subsets of the nuclear proteome. Lamin interactors include transmembrane LEM domain proteins such as LAP2β and MAN1, transcription factors such as SREBP1, transcriptional regulators including the RB transcriptional corepressor 1 (RB1), and adaptor proteins such as BANF1 that might facilitate chromatin binding to the lamina15, 17, 26, 27 Two other critical nuclear envelope structures mediate interactions between the nucleus and cytoplasm: nuclear pore complexes and the LINC complex. Nuclear pore complexes allow transcription factors, nuclear receptors, and signaling proteins to shuttle between the nucleus and cytoplasm1. A subset of nuclear pore complex proteins localizes to the nuclear periphery and/or with inactive regions of heterochromatin, whereas others are associated with active areas of euchromatin in the nuclear interior28–30. How this is regulated, and whether lamins or adaptor proteins such as BANF1 are involved, is unclear. Finally, the LINC complex2 forms a key structural and regulatory connection between the nuclear envelope and the cytoskeleton, binding to nuclear lamins and the inner nuclear membrane on one side and actin filaments on the other (Fig.1).
Lamina-heterochromatin Interactions and Lamina-associated Domains
Several landmark studies have demonstrated the physical association between large regions of the genome (typically characterized by heterochromatin) and the nuclear lamina3, 31–34 Some genomic regions—usually gene-poor, transcriptionally inactive regions—are associated with the lamina as part of lamina-associated domains (LADs) in numerous cell types, including pluripotent and terminally differentiated cells. In contrast, other regions of the genome may be found in or out of LADs depending on the cell type, or may move in and out of LADs during the process of cellular differentiation3, 35. For example, genomic regions associated with the lamina and transcriptionally silent in embryonic stem cells were found to dissociate from the lamina and become transcriptionally active during astrocyte differentiation31. Association with or dissociation from the nuclear lamina is therefore an important mechanism of transcriptional regulation during development; differential histone post-translational modification (methylation/acetylation) is likely to be involved in this process. Importantly, few studies have explored how disease-associated lamin variants affect organization of the genome and the LAD landscape in the involved tissues36.
Lamina-related Diseases
Researchers began to realize that alterations in the nuclear lamina can lead to development of disease when genetic mapping and sequencing became widely available in the 1990s. In 1994, mutations in EMD, encoding emerin, an inner nuclear membrane protein, were found to cause X-linked Emery-Dreifuss muscular dystrophy (EDMD)37. Subsequently, autosomal mutations in LMNA were found to cause EDMD38. In the following years, many other monogenic diseases, called laminopathies and envelopathies, were attributed to mutations in lamins or their associated proteins, respectively (Table 1).
Table 1:
Human diseases linked to mutations in genes encoding lamins and associated proteins. Key: A-type lamins | B-type lamins | Lamin associated proteins | Lamin processing proteins
| Disease [references] | Gene (Phenotype MIM number) |
Phenotype | |
|---|---|---|---|
| LMNA (181350, 616516) | |||
| Emery-Dreifuss muscular dystrophy [37,38,S1–3] |
EMD (310300) SYNE1 (612998) SYNE2 (612999) TMEM43 (614302) |
Skeletal myopathy, cardiomyopathy, early contractures, cardiac conduction defects |
|
| Limb girdle muscular dystrophy [46,47] |
LMNA (159001) TOR1AIP1 (617072) |
Progressive limb weakness, late contractures, arrhythmogenic cardiomyopathy | |
| Muscular dystrophy, congenital [39] | LMNA (613205) | Limb and axial muscle weakness and wasting | |
| Dilated cardiomyopathy, type 1A [48,S4] | LMNA (115200) | Cardiac dilation, reduced ejection fraction | |
| Cardiomyopathy, dilated, with hypergonadotropic hypogonadism [S5] | LMNA (212112) | Cardiomyopathy, hypogonadism | |
| Heart-hand syndrome, Slovenian type [50] | LMNA (610140) | Heart conduction defects, cardiomyopathy, abnormal bone development in hands and feet |
|
| Hutchinson-Gilford progeria syndrome [51,53] | LMNA (176670) | Symptoms of premature ageing, alopecia, scleroderma, lipodystrophy, cardiovascular defects | |
| Restrictive dermopathy [56,S6] |
LMNA (275210) ZMPSTE24 (275210) |
Taut facies, intrauterine growth retardation, death within weeks of extrauterine life | |
| Mandibuloacral dysplasia [54,S7] |
LMNA (248370) ZMPSTE24 (608612) |
Mandibular hypoplasia, growth restriction, progressive osteolysis, variable lipodystrophy and progeroid symptoms. | |
| Charcot-Marie-Tooth disease, type 2B1 [59,S8] | LMNA (605588) | Lower limb motor and sensory neuropathy, pes cavus | |
| Familial partial lipodystrophy, type 2 [62–64] | LMNA (151660) | Abnormal distribution of subcutaneous fat with cushingoid appearance, metabolic defects including diabetes mellitus and hypertriglyceridemia | |
| Leukodystrophy, adult-onset, autosomal dominant [61] | LMNB1 (169500) | Multiple-sclerosis-like symptoms, autonomic dysfunction, CNS demyelination | |
| Progressive myoclonic epilepsy-9 [60] | LMNB2 (616540) | Myoclonic epilepsy, brain developmental defects, muscle atrophy | |
| Lipodystrophy, partial, acquired, susceptibility to [65] | LMNB2 (608709) | Loss of subcutaneous fat, metabolic disorder | |
| Nestor-Guillermo progeria syndrome [55] | BANF1 (614008) | Variable lipoatrophy, skeletal and cardiac abnormalities | |
| Greenberg skeletal dysplasia [58] | LBR (215140) | Osteochondroplasia, fetal demise, hydrops | |
| Pelger-Huet anomaly [S9] | LBR (169400) | Skeletal defects, epilepsy, developmental delay, abnormal granulocyte nuclear morphology | |
| Buschke-Ollendorff syndrome [57] | LEMD3 (166700) | Multiple nevi, osteopoikilosis | |
| Spinocerebellar ataxia, autosomal recessive 8 [S1] | SYNE1 (610743) | Ataxia, dysarthria, variable muscle atrophy | |
| Deafness, autosomal recessive 76 [S10] | SYNE4 (615540) | Progressive high-frequency hearing loss | |
| Arrhythmogenic right ventricular dysplasia 5 [49] | TMEM43 (604400) | Arrhythmogenic cardiomyopathy, right ventricular dysplasia, center ventricular enlargement |
|
The laminopathies are characteristically syndromic and frequently have overlapping features. The pleiotropism of lamins and their associated proteins, combined with overlapping phenotypes, reflects their sophisticated regulation and diverse roles in many tissues39. Shared clinical features among laminopathies were initially interpreted as evidence of a single disease process with a spectrum of manifestations40, but careful mapping of mutations revealed clear associations between mutations in distinct regions of the LMNA gene and different diseases41. For example, most patients with type 2 (Dunnigan) familial partial lipodystrophy (FPLD2) carry mutations in exons 7, 8, or 11 (UMD-LMNA mutations database [http://www.umd.be/LMNA]). Most of these mutations change the surface charge of an immunoglobulin-like motif in LMNA, even though the overall integrity of the motif is maintained42, 43. Some patients with FPLD2 have additional findings consistent with limb-girdle muscular dystrophy (LGMD), whereas others with identical mutations are completely asymptomatic44. Intra-familial phenotypic differences, variable expressivity, and incomplete penetrance highlight the importance of undiscovered modifier genes that might serve as therapeutic targets.
Myopathies
Several striated myopathies are caused by mutations in genes encoding lamins or their associated proteins, characterized by dilated cardiomyopathy with variable skeletal muscle involvement, although disorders primarily involving smooth muscle have not been reported. The most common myopathic laminopathy resulting from mutations in LMNA and genes encoding other LAPs is EDMD, characterized by early contractures, progressive weakness, muscle wasting, and cardiomyopathy with conduction defects45. Distal (more so than proximal) muscle involvement and early neck, elbow, and achilles contractures in EDMD differentiate it from LGMD, which generally has more proximal muscle involvement and results from mutations in LMNA or TOR1AIP146, 47 Multiple clinically distinct cardiomyopathies have also been attributed to mutations in lamins and their associated proteins including dilated cardiomyopathy, arrhythmogenic right ventricular dysplasia, and Slovenian type heart-hand syndrome48–50 (Table 1).
Progerias and developmental disorders
Hutchinson-Gilford progeria syndrome (HGPS) is perhaps the most well-recognized laminopathy because of its striking presentation of premature aging51. Classically, it is caused by a de novo point mutation activating a cryptic splice site in LMNA exon 11, generating truncated prelamin A that cannot be cleaved by ZMPSTE24 and remains farnesylated (resulting in a protein product called progerin), though other LMNA mutations can also cause HGPS51–53. Manifestations include alopecia, scleroderma, osteoporosis, lipodystrophy, atherosclerosis, and death within the first 2 decades of life53. Nestor-Guillermo progeria syndrome and progeria-like mandibuloacral dysplasia have also been attributed to mutations in genes encoding lamins or their associated proteins; these share features with other laminopathies including striated muscle defects and lipodystrophy54, 55. Restrictive dermopathy, caused by mutations in LMNA or ZMPSTE24, shares features with the progerias but is defined by tight skin resulting in the fetal hypokinesia sequence, including intrauterine growth retardation56. Other developmental disorders include Greenberg skeletal dysplasia and Buschke-Ollendorff syndrome, caused by mutations in LBR and LEMD3, respectively, with predominant skeletal phenotypes57, 58.
Neuropathies and lipodystrophies
Numerous neuropathies have been linked to mutations in genes encoding lamins, including B-type lamins, and their associated proteins. For example, type 2B1 Charcot-Marie-Tooth disease (caused by LMNA mutation; Table 1) manifests as pes cavus with progressive sensory and motor neuropathy beginning in the lower extremities59. Mutations in LMNB1 or LMNB2 can cause a rare leukodystrophy or myoclonic dystrophy, respectively60, 61.
Mutations in LMNA cause autosomal-dominant FPLD262–64, a fat storage disorder characterized by cushingoid appearance with decreased subcutaneous fat in the trunk and limbs, dyslipidemia, diabetes mellitus, and hepatic steatosis. Mutations in LMNB2 predispose to acquired partial lipodystrophy, which shares many features with FPLD265.
Multifactorial diseases
Classical laminopathies are rare monogenic diseases, but there is mounting evidence that lamins and their associated proteins are also involved in more-common, multi-factorial disease processes. For example, lamin and nuclear pore complex dysfunction leads to neuronal cell death and modulates heterochromatin relaxation in Alzheimer disease66, 67 There are conflicting data on whether defects in LMNA contribute to development of gastrointestinal cancers68, 69 (section 3.1;Table 2); nuclear membrane-targeted gold particles can decrease tumor cell metastatic potential by increasing LMNA expression and nuclear stiffness70. A common variant of LMNA was reported as a risk factor for type 2 diabetes mellitus71, and several LAPs were reported to be necessary for HIV-1 integration into nuclear chromatin in primary macrophages72. Continued advances in imaging and biochemical techniques will likely identify novel interactions within the nuclear lamina that lead to new disease associations.
Table 2:
Changes in lamins and associated proteins in human gastrointestinal cancers. Key: A-type lamins | B-type lamins | Lamin associated proteins
| Cancer | Gene | Finding [references] | Clinical significance |
|---|---|---|---|
| Esophageal adenocarcinoma | LMNA | Upregulation (protein & mRNA) [82] Downregulation (protein) [73] | Unknown |
| LMNB1 | Downregulation (protein) [73] | Unknown | |
| Esophageal squamous cell carcinoma | LMNA | Downregulation (protein) [73] | Unknown |
| LMNB1 | Downregulation (protein) [73] | Unknown | |
| BANF1 | Upregulation (protein & mRNA) [86] | Poor prognosis | |
| Gastric adenocarcinoma | LMNA | Downregulation (protein & mRNA) [73,74] | Poorly differentiated tumors, poor prognosis |
| LMNB1 | Downregulation (protein) [73] | Unknown | |
| TMPO | Upregulated (mRNA) [85] | Unknown | |
| Duodenal and rectal adenocarcinoma | SYNE2 | Upregulation, mislocalization (protein) [89] | Unknown |
| LMNA | Positive staining [69] Low expression (protein) [68] | Poor prognosis Increased risk of recurrence | |
| LMNA/LMNB1 | Downregulation, mislocalization (protein) [73,74] | Unknown | |
| Colorectal adenocarcinoma | TMPO | Upregulation (mRNA) [85] | Unknown |
| SYNE1 | Non-synonymous variants [88] | Unknown | |
| NUP88 | Upregulation (protein) [92 | Unknown | |
| Hepatocellular carcinoma | LMNB1 | Upregulation (protein & plasma mRNA) [83,84] | Larger tumor size and number, more advanced disease; potential biomarker |
| BANF1 | Upregulation (protein & mRNA) [87] | Putative early biomarker of HCC | |
| NUP88 | Upregulation (protein) [90,91] | Poorly differentiated tumors; increased expression during carcinogenesis |
Lamins and Associated Proteins in Gastrointestinal Diseases
Altered expression and/or localization of lamins and lamin-associated proteins has been associated with several gastrointestinal cancers73, 74, although little is known about laminopathies that specifically affect gastrointestinal organs aside from the liver and pancreas75–78. What laminopathies affect the gastrointestinal tract, and what gastrointestinal diseases have been associated with lamina gene variants or altered expression/localization of nuclear lamina proteins?
Cancer
Malignant cells typically have changes in nuclear structure and morphology, such as altered protein composition and irregularly shaped nuclei79–81, that resemble changes in cells that express mutant lamins or lamin-associated proteins. Altered expression and/or localization of lamins and their associated proteins has been reported in different types of gastrointestinal tumors (Table 2)80, 81. For example, levels of LMNA mRNA and protein were increased in esophageal adenocarcinoma specimens, compared to Barrett’s esophagus with high-grade dysplasia82. However, another study showed that esophageal tumors (squamous and adenocarcinoma) had reduced expression of LMNA and LMNB1 compared to control esophageal tissue73. Levels of LMNA and LMNB1 were also reduced in gastric tumor and colon carcinoma and adenoma specimens, and were mislocalized to the cytoplasm in some colon carcinomas and adenomas73, 74 In gastric adenocarcinoma, decreased LMNA expression correlated with poorly differentiated tumors and poor patient outcomes, compared to tumors that expressed normal levels of LMNA74.
Findings from studies of colorectal cancer are contradictory. In a study of archived patient samples, tumor staining for LMNA correlated with decreased overall survival time of patients, compared to samples that were negative for LMNA69. In another study, low LMNA expression was associated with increased recurrence among patients with stage II or III colon cancer68. This discrepancy may be related to the greater homogeneity of this cohort, which did not include patients with stage-I disease or rectal cancer68 and/or the fact that in the former study, tumors with low and high levels of LMNA were grouped together as LMNA positive69. Notably, both studies used archived tissue samples and retrospective registry data; further study is needed to clarify the role of LMNA in colorectal cancer.
LMNB1 was consistently increased in liver tissues from patients with hepatocellular carcinoma (HCC) compared to patients without HCC83, 84 Furthermore, increased levels of LMNB1 associated with larger tumors, increased number of tumor foci, and more advanced disease; LMNB1 mRNA was increased in plasma from patients with HCC compared to individuals without HCC51. LMNB1 might therefore serve as a prognostic factor for patients with early-and late-stage HCC83, 84
Levels of lamin-associated and other nuclear proteins have also been examined in gastrointestinal tumors. For example, levels of LAP2α mRNA were increased in gastric and colon tumor tissues compared to non-tumor tissues85. Levels of BANF1 (protein and mRNA) were increased in esophageal squamous cell carcinoma, and high levels were associated with poor outcomes of patients86. Moreover, BANF1 mRNA and protein were increased in HCC samples compared to non-tumor liver tissues87. Notably, multiple heterozygous non-synonymous mutations in the spectrin repeat containing nuclear envelope protein 1 gene (SYNE1 or Nesp1) were identified in colorectal tumors88, and SYNE1 was increased and mis-localized to the cytoplasm in duodenal and rectal tumors89. Nucleoporin 88 (NUP88), a nuclear pore complex protein, was highly expressed in HCC compared to non-tumor liver tissues, and overexpressed in colorectal tumors compared to non-tumor tissues. Levels of NUP88 increased during carcinogenesis and correlated with poorly differentiated tumors90–92. There is much evidence for alterations in lamins and their associated proteins in gastrointestinal tumors. Further studies are needed to determine the mechanisms of these changes and their potential role in tumor development, but they might be used as biomarkers of tumorigenesis or tumor progression.
Primary Biliary Cholangitis
Negative serologic results for anti-mitochondrial antibodies (AMA) present a challenge in the diagnosis of primary biliary cholangitis (PBC, previously called primary biliary cirrhosis). Antinuclear antibodies are present in sera from 25% of patients with PBC, and anti-LBR antibodies, along with anti-gp210 and anti-nucleoporin p62 autoantibodies, characterize a subset of PBC93–95. These autoantibodies produce a rim-like or membranous pattern that is highly specific for PBC and may represent a useful tool in the diagnosis of AMA-negative PBC96–98. Notably, Reynolds syndrome, characterized by concurrent scleroderma and PBC, was linked to an LBR missense variant in exon 9 (p.R372C), so it could be a previously unrecognized laminopathy99 A systematic analysis of a large cohort of patients with PBC is necessary to validate LBR variants as a common cause of Reynolds syndrome100.
Porphyria-associated liver injury
Porphyrias include 8 metabolic disorders of the heme biosynthetic pathway, each resulting from a specific enzyme defect, and are characterized by accumulation of heme precursors in diverse organs101, 102. Aggregation of LMNA and LMNB1 in the liver, a major site of heme synthesis, is an early marker of porphyria-associated liver injury in mice 102,103. Furthermore, accumulation of porphyrin in HepG2 cells results in light-dependent aggregation of lamins and nuclear shape alterations104, resembling the changes observed in patients with laminopathies. These nuclear alterations might affect transcription, via aggregate sequestration of lamins and other nuclear proteins103,104.
Nonalcoholic fatty liver disease (NAFLD)
Hepatic steatosis, with progression to nonalcoholic steatohepatitis (NASH), is a common clinical feature of laminopathies including FPLD2 and other lipodystrophic syndromes and is considered a complication of these diseases75, 105. However, there is evidence that alterations in lamins and their associated proteins contribute to development of NAFLD and NASH, independent of lipodystrophy syndromes76,77,106. In a cohort of twin and sibling pairs with NAFLD (without lipodystrophy), coding sequence variants in lamina-related genes were identified in 90% of patients with NAFLD vs 36% of subjects without NAFLD76. Among these variants was an insertion in TMPO that causes a frameshift and insertion of a premature stop codon after amino acid 99 in all LAP2 isoforms. When expressed ectopically in human hepatoma cells, truncated LAP2 was mislocalized throughout the nucleus and cytoplasm, unable to bind LMNA, and altered the distribution of endogenous LMNA, LMB1, and LMNB2 in transfected cells. Cells expressing truncated LAP2 had greater lipid droplet accumulation than control cells (transfected with full-length LAP2α) after incubation with oleic acid76. Supporting these findings, hepatocyte-specific deletion of LMNA in mice altered growth hormone signaling via Jak and Stat proteins in hepatocytes and led to steatosis with progression to NASH with fibrosis (section 4.6)77 It appears that patients with laminopathies are at greater risk for NAFLD or NASH due to hepatocyte-specific defects in LMNA; some cases of NAFLD or NASH might be associated with unrecognized laminopathies75–77,107,108.
Insights From Animal Models
Our understanding of lamin function and the etiology of laminopathies has been greatly enhanced by the development of animal models. Mice with loss or gain of function alleles that are orthologous to human disease alleles recapitulate much of the tissue-specific features of human laminopathies. Phenotypic characterization of these mice has enabled the identification of physiologic abnormalities in affected tissues and alterations in signal transduction pathways and the transcriptome that contribute to pathogenesis. Mouse models of laminopathies have been useful in identifying agents that could alleviate disease symptoms and prolong life. What models of laminopathy have been developed and what have we learned about lamin function from these models (Table 3)?
Table 3:
Mouse models of lamin-related disease.
| Mouse model [references] | Disease Relevance/Affected Tissues |
Model design | Model phenotype | |
|---|---|---|---|---|
| LMNASul/Sul[119,124,143,164,811] | DCM, EDMD, FPLD2/ Striated muscle, bone, liver, pancreas, adipose tissue | LMNA exons 8 through top of exon 11 deleted | Reduced growth from 2–3 weeks of age; abnormal gait and posture, muscular dystrophy, decreased bone mass, cardiomyopathy, reduced subcutaneous fat (lethal by 8 weeks of age) | |
| LMNAGT−/− [122] | DCM, EDMD, FPLD2/ Striated muscle, adipose tissue | Gene trap insertion in LMNA intron 2 | Reduced growth, abnormal gait, muscle weakness, cardiomyopathy, reduced subcutaneous fat, lethal by postembryonic day 16–18 (P16-P18) | |
| LMNAflx/flx CMV-Cre [121] | DCM, EDMD, FPLD2/ Striated muscle, adipose tissue | LoxP sites flanking LMNA exon 2, CMV-Cre drives whole-body LMNA deletion | Similar to LMNASul/Sul and LMNAGT−/− models; lethal by P16-P18 | |
| LMNAflx/flx; Zp3-Cre [125] | DCM, EDMD, FPLD2/ Striated muscle, adipose tissue | LoxP sites flanking LMNA exons 10–11, Zp3-Cre drives whole-body LMNA deletion | Phenotype not described; lethal between P13-P18 | |
| LMNAδ9/δ9 [129,145] | HGPS Skin, bone, adipose tissue |
LMNA L530P; alternative splicing generates truncated protein that remains farnesylated. | Subcutaneous fat loss, osteoporosis, abnormal dentition, thin skin, growth retardation, and shortened lifespan | |
| LMNAG609G/G609G [127] | HGPS Skin, bone, adipose tissue | LMNA G609G; cryptic splice site results in loss of Zmpste24 cleavage site and expression of progerin | Subcutaneous fat loss, alopecia, reduced bone density, kyphosis, thymic and splenic atrophy, reduced lifespan | |
| LMNAHG/+ [128] | HGPS Skin, bone, adipose tissue | LMNA introns 10–11, part of exon 11 removed; loss of Zmpste24 cleavage site and expression of progerin | Heterozygotes exhibited slow weight gain, rib fractures, loss of body fat, reduced lifespan | |
| LMNAnHG/+ [128] | HGPS Skin, bone, adipose tissue | Same as LMNAHG/+ except CAAX mutated to SAAX; non-farnesylated progerin expressed | Heterozygotes exhibited slow weight gain, rib fractures, loss of body fat, and shortened lifespan (less severe LMNAnHG/+) | |
| Zmpste24−/− (Pendas) [130] | HGPS Skin, bone, adipose tissue | Zmpste24−/− exons 2–3 deleted | Weight loss, kyphosis, muscle weakness, alopecia; average lifespan of 20 weeks | |
| Zmpste24−/− (Bergo) [19] | HGPS Skin, bone, adipose tissue | Zmpste24−/− exon 8 deleted | Spontaneous fractures, slow weight gain, alopecia, kyphosis, muscle weakness, average lifespan of 6–7 months | |
| LMNALCO/LCO [135] | HGPS | LMNA intron 11, part of exon 11 removed; lamin C but not prelamin A produced | Homozygotes similar to WT | |
| LMNALAO/LAO [134] | HGPS | LMNA introns 10–11, first 24 bp of exon 12 removed; mature lamin A expressed, not prelamin A or lamin C | Homozygotes similar to WT | |
| H222P LMNA [137,139,155,156] | DCM and EDMD Striated muscle | LMNA H222P (causes EDMD in humans) | Kyphosis, shallow breathing, dilated cardiomyopathy, reduced lifespan; no lipodystrophy | |
| N195K LMNA [138] | DCM Cardiac muscle | LMNA N195K (causes DCM in humans) | Cardiac muscle degeneration, dilated heart chambers, conduction defects; lethal by 12–14 weeks of age | |
| LMNAflx/flx; Villin-Cre [146] | Intestinal epithelium | LoxP sites flanking LMNA exons 10–11, Villin-Cre drives LMNA deletion in intestinal epithelial cells | Slight increase in number of intestinal polyps with ApcMin/+ background | |
| LMNAflx/flx; Alb-Cre [77] | FPLD2 and NAFLD Liver | LoxP sites flanking LMNA exons 10–11, Alb-Cre drives LMNA deletion in hepatocytes | Male-specific liver injury and steatosis, with steatohepatitis and fibrosis after high fat diet | |
| LMNAflx/flx; Cela1-CreERT2 [78] | Pancreas | LoxP sites flanking LMNA exons 10–11, Cela1-CreERT2 drives inducible LMNA deletion in acinar cells | ER stress, increased apoptosis and proliferation, chronic pancreatitis, fibrosis | |
| R482Q LMNA transgenic mice [160,161] | FPLD2 Adipose tissue | LMNA (human) R482Q transgene | Weight plateau at 41 weeks, fat pad loss, hepatic steatosis, thermogenesis defects, adipocyte differentiation defects [160]; TGF-β activation and fibrosis in adipose tissue [161] | |
| Lap2α−/− [24,S12–13] | DCM Striated muscle, epidermal/erythroid progenitor cells | Tmpo/Lap2 exon 4 deleted, eliminates Lap2α expression, preserves other isoforms | Systolic dysfunction, cardiac fibrosis in older mice, hyperproliferation of epidermal and erythroid progenitor cells | |
| Lmnb1−/− (Kim) [113] | Lung, diaphragm, brain | Lmnb1 exon 1 deleted | Delayed embryonic growth, lung/diaphragm defects, microencephaly, respiratory failure, perinatal lethality | |
| Lmnb1Δ/Δ [112,114] | Lung, bone, brain | Gene trap cassette in Lmnb1 intron 5; protein lacks NLS and CAAX motif | Abnormal lung development and bone ossification, microencephaly, respiratory failure, perinatal lethality | |
| Lmnb2−/− (Kim) [113] | Diaphragm, brain | Lmnb2 exon 1 deleted | Brain and diaphragm defects, respiratory failure, perinatal lethality | |
| Lmnb2−/− (Coffinier) [112] | Brain | Lmnb2 exon 1 replaced with lacZ reporter | Abnormal layering of cortical neurons in cerebral cortex, perinatal lethality | |
| Lmnb1−/−; Lmnb2−/− (Kim) [113] | Lung, diaphragm, brain | Lmnb1, Lmnb2 loci deleted | Thin diaphragm, microencephaly, delayed embryonic growth, perinatal lethality | |
| Lmnb1f Lmnb2flx/flx; Emx1-Cre [112] | Forebrain | LoxP sites flanking exon 2 of Lmnb1 and Lmnb2, Emx1-Cre drives forebrain-specific deletion | Cortical atrophy, loss of hippocampal structures, perinatal lethality | |
| Lmnb1flx/flx; Lmnb2flx/flx; K14-Cre [117] | Skin | LoxP sites flanking exon 2 of Lmnb1 and Lmnb2, K14-Cre drives keratinocyte-specific deletion | Normal skin and hair, normal keratinocyte proliferation | |
| Lmnb1flx/flx; Lmb2flx/flx; Alb-Cre [116] | Liver | LoxP sites flanking exon 2 of Lmnb1 and Lmnb2, Alb-Cre drives hepatocyte-specific deletion | Similar to WT; liver chemistries and histology normal | |
| Lmnb1B2/B2[115,S14] | Brain | Lmnb1 locus replaced with Lmnb2 | Cortical neuron layering defect, decreased body mass (less severe than Lmnb1−/−) | |
| Lmnb2B1/B1 [115,S14] | Brain | Lmnb2 locus replaced with Lmnb1 | Normal body mass, slightly decreased brain size, cortical neuron layering defect | |
| Lmnb1CS/CS [118] | Lung, brain | Lmnb1 CAAX motif replaced with SAAX | Cortical layering defect, microencephaly, lung defects, perinatal lethality | |
| Lmnb2CS/CS [118] | Brain | Lmnb2 CAAX motif replaced with SAAX | Similar to WT (normal growth, fertility, lifespan) | |
B-type lamins
B-type lamins are expressed ubiquitously from early in embryonic development and regulate basic cellular functions such as senescence, replication, spindle assembly, chromatin organization, transcription, and resistance to oxidative stress109–111. It might be assumed, therefore, that mice lacking B-type lamins would die at an early stage of embryogenesis. In fact, mice with disruptions in Lmnb1 and/or Lmnb2 develop to term but die perinatally, with developmental defects in lung, bone, and brain112–114. Because Lmnb1 and Lmnb2 have 60% homology and similar expression patterns115, functional redundancy could account for the relatively mild phenotype. However, Lmnbl−/− and Lmnb2−/− mice develop to term at the expected Mendelian frequency113. Moreover, combined tissue-specific deletion of Lmnbl and Lmnb2 in keratinocytes and hepatocytes did not produce abnormal phenotypes or visible defects in tissue histology, nuclear shape, or cell proliferation116, 117 These findings indicate that B-type lamins are not required for basic cellular functions but are required for the normal development of a subset of tissue types.
B-type lamins have important roles in brain development, based on defects in neuron migration and layering of forebrain neurons in Lmnb1−/− and Lmnb2−/− mice112. The similar phenotypes indicate that Lmnb1 and Lmnb2 have similar functions in brain. However, neuronal nuclei from Lmnb1−/− brains have a bleb-like structure, whereas those from Lmnb2−/− brains are elongated112. Moreover, differences in brain size and neuron numbers between these 2 lines indicate that Lmnb1 and Lmnb2 have distinct roles112. This finding was supported by studies in which the Lmnb2 locus was replaced with Lmnb1, and vice versa. Lmnb1 at the Lmnb2 locus could not substitute for Lmnb2, and vice versa, resulting in brain phenotypes similar to those of Lmnb1−/− and Lmnb2−/− mice115. In addition, mice that express a nonfarnesylated form of Lmnb2 develop normally, as opposed to those that express nonfarnesylated Lmnb1, which die at birth with severe neurodevelopmental defects. These observations indicate different requirements in farnesylation for lamin function118.
A-type lamins: Lmna loss of function alleles
The first strain of mice with a LMNA loss of function allele (LmnaSul) was generated via deletion of exons 8–11 of Lmna119. However, these mice express a truncated version of Lmna120. LmnaSul/Sul mice have growth retardation at 2 weeks and die by 8 weeks of age. Death has been attributed to cachexia, muscular dystrophy, and cardiomyopathy. In embryonic fibroblasts from these mice, emerin mislocalized to the cytoplasm thereby indicating that LmnaSul/Sul phenocopies EDMD (in which emerin expression is lost or the protein is mislocalized in myocytes)119.
Two other strains of mice with loss of function alleles have been generated. These delete exons 2–12 of Lmna. Mice homozygous for these deletions have a similar phenotype mice homozygous for LmnaSul/Sul, but die by 3 weeks rather than 8 weeks of age, indicating that the truncated LmnaSul gene product is partially functional121, 122
LmnaSul/Sul mice were initially reported to have no adipogenic or metabolic defects123. However, subsequent studies with these mice124, and with 2 separate lines of mice with different loss of function alleles121, 122, reported fat loss, impaired ex vivo adipogenesis, increased lipolysis in white adipose tissue, and impaired thermogenesis in brown adipose tissue in homozygous mice. LmnaSul/Sul mice have a decrease in bone mass that correlates with significant reductions in numbers of osteoblast and osteocyte119.
Studies of mice with a floxed allele that removes exons 10–12 of Lmna showed that LMNA, together with LBR, tethers peripheral heterochromatin to the nuclear envelope. In mice lacking LBR, loss of LMNA causes mislocalization of heterochromatin to the nuclear interior, with concomitant changes in gene expression and myocyte differentiation125. These studies indicate that lamin A/C is important for myocyte, osteocyte, and adipocyte differentiation and function, likely due to its role in regulating gene expression.
Lmna knock-in mice
Mouse models of laminopathies have been generated—these mice carry alleles orthologous to those that cause human laminopathies. Unlike in humans, in mice, most of these laminopathy alleles, especially those that cause muscular dystrophy and cardiomyopathy, are not fully penetrant in the heterozygous state or cause disease only in homozygous mice. Reasons for this recessive mode of inheritance are unclear126. Nevertheless, the homozygous mice develop phenotypes that are similar to human diseases and have provided important insights into the pathogenesis of laminopathies.
Progeroid Mouse Models
Mice that express progerin or a similar LMNA variant develop disorders similar to those of patients with HGPS (Table 3)127,128,129. These mice develop osteoporosis, kyphosis, alopecia, reduced subcutaneous fat, and cardiovascular disease and have a shortened lifespan127–129. Homozygous mice have a more severe phenotype than heterozygous mice, indicating that progerin levels determine the severity of disease127–129.
Zmpste24−/− mice, which lack the metalloprotease required for processing prelamin A to mature lamin A, accumulate farnesylated prelamin A that remains attached to the inner nuclear membrane. These mice have a phenotype similar to that of progerin-expressing mice19, 130, so accumulation of farnesylated lamin A might mediate the disease phenotype. In support of this hypothesis, decreasing Lmna dosage in Zmpste24−/−mice using Lmna-null alleles reduces disease pathology131. Moreover, administration of a farnesyl transferase inhibitor (FTI) to Zmpste24−/− mice or progerin mice increased muscle strength and longevity and reduced rib fractures and the rate of weight loss132,133. The FTI did not fully rescue the progeria phenotype, however, and mice with a knock-in allele of non-farnesylated progerin were not completely protected from disease128. This indicates that the uncleaved C-termini in progerin and unprocessed prelamin A could also contribute to the disease phenotype, irrespective of farnesylation status. Mice that are engineered to express mature lamin A or C directly, bypassing prelamin A processing, have normal phenotypes, indicating that lamins A and C may be interchangeable134,135. Because lamin C does not require farnesylation or ZMPSTE24 proteolysis, increasing expression of lamin C, compared with lamin A, might a viable strategy for treating HGPS. A single copy of an allele that expresses only lamin C (LmnaLCO mice) eliminates the progeria disease phenotype in Zmpste24−/− mice 135, and an antisense oligonucleotide that increases lamin C at the expense of prelamin A can reduce progeria-related cardiovascular disease in progerin mice136. So, strategies to reduce farnesylated prelamin A and progerin and/or increase lamin C expression might be developed for treatment of HGPS.
Models of Muscular Dystrophy and Cardiomyopathy
The H222P mutation in LMNA causes the autosomal-dominant form of EDMD. Mice homozygous for the orthologous mutation in Lmna develop muscular dystrophy and cardiomyopathy with heart chamber dilation, conduction defects, and cardiac fibrosis and die by 9 months of age137. Mice with a mutation in Lmna that causes the amino acid substitution N195K also develop dilated cardiomyopathy with conduction defects, but not muscular dystrophy; these mice die from cardiac arrhythmias by 3 months of age138. Mice with the H222P mutation in Lmna have become the primary model for studies of cardiomyopathy associated with defects of loss of this protein, because they have late onset of disease139.
Transcriptome analysis of hearts from LmnaH222P/H222P mice indicates that dysregulation of signal transduction pathways contributes to disease pathology. Signal transduction pathways that are upregulated in LmnaH222P/H222P mice include the MAP kinase (ERK signaling to JNK and p38), Akt, and mTORC1 pathways139. MAP kinase inhibitors, of which the MEK inhibitors are the most extensively studied, have been shown to inhibit kinase activity in cardiac tissue, reduce the severity of disease, and improve heart function, indicating that constitutive activation of MAPK pathways is partly responsible for the disease phenotype139. However, it is unclear how MAPK hyperactivation occurs and how it contributes to disease progression.
mTORC1 is also hyperactivated in LmnaH222P/H222P as well as LmnaSul/Sul hearts; mTORC1 activation correlates with decreased autophagy in both these lines of mice140–143. mTORC1 inhibitors increased autophagy and improved heart function in both lines of mice140, 142, and the mTORC1 inhibitor rapamycin reversed metabolic defects in adipose tissue of LmnaSul/Sul mice124. In LmnaH222P/H222P mice, the activator of mTORC1, AKT serine/threonine kinase 1 (AKT1), is hyperactivated, so H222P LMNA could hyperactivate mTORC1 through aberrant activation of AKT1, leading to decreased autophagy and cardiomyopathy140. Like AKT1, ERK is an activator of mTORC1; these could be involved in the mechanism by which MAPK hyperactivation in LmnaH222P/H222P mice promotes cardiomyopathy140.
Based on transcriptome analysis, the Wnt pathway is also downregulated in LmnaH222P/H222P hearts, with reduced active and total β-catenin, Wnt, and Wnt10B proteins and increased frizzled-related proteins, which modulate the Wnt pathway144. Reductions in Wnt signaling were also noted in fibroblasts derived from progeroid LmnaΔ9/Δ9 mice; this reduction appears to be responsible for the decrease in extracellular matrix production by mutant fibroblasts145. These data indicate that downregulation of Wnt signaling contributes to development of cardiomyopathy in LmnaH222P/H222P mice.
Role of LMNA in the Gastrointestinal Tract—Lessons From Mice
Tissue-or cell-specific and conditional disruption of Lmna is an important strategy for studying the role of LMNA in the gastrointestinal tract, given the severity of the phenotype in Lmna-deficient mice. Lmna has been disrupted in enterocytes, hepatocytes and acinar cells. In these cells, exons 10 and 11 of Lmna were removed using the Cre-lox system. Disruption of Lmna in enterocytes of mice using a Cre transgene under the control of the villin promoter did not alter gross morphology, lifespan, intestinal histology, or cell proliferation146. However, disruption of Lmna in enterocytes of ApcMin/+ mice led to a slight increase in the total number of intestinal polyps, with as much as a 3-fold increase in the number of 2–5 mm polyps in the duodenum. These findings indicate that LMNA may act as a tumor suppressor146–148.
In contrast to the modest phenotype resulting from disruption of Lmna in mouse enterocytes146, hepatocyte-specific disruption of Lmna (using the same floxed allele as for enterocyte-specific disruption) reduces body mass but causes male-specific liver injury and steatosis that progresses to steatohepatitis and fibrosis in mice placed on a high-fat diet77. These changes correlated with upregulated transcription of genes that regulate lipogenesis, lipid transport, inflammation, type-1 interferon signaling, and fibrosis in mice on a standard diet; expression of genes that regulate fibrosis increases when mice are placed on a high-fat diet. The male specificity of the phenotype is likely due to defective growth hormone-mediated Jak2 signaling to Stat5. Activation of Stat5 regulates expression of male vs female sexually dimorphic genes in mouse livers149–151. Much like hepatocyte-specific disruption of Stat5, disruption of Lmna in hepatocytes leads to upregulation and constitutive activation of Stat1, which is necessary for inducible expression of type-1 interferon-regulated genes152–154 and might contribute to inflammation in livers of mice.
Growth hormone-mediated activation of ERK was reduced in LMNA-deficient livers of mice, although it is not clear how this finding relates to the steatohepatitis phenotype of the mice. The defect in ERK activation is opposite to what is observed in LmnaH222P/H222P cardiomyocytes, in which ERK is hyperactivated do disrupt cardiac function139, 155–157 The difference in the ERK phenotypes may be related to differences in the Lmna allele used, potential gain-of-function effects, and/or upstream pathways that are important in hepatocyte vs myocyte differentiation and function.
A major finding in the study of liver-specific LMNA-deficient mice is that disruption of Lmna leads to a cell-autonomous effect in hepatocytes to induce steatohepatitis. This result sheds a different light on the hypothesis that FPLD2-associated LMNA alleles cause lipodystrophy in humans primarily via effects on adipose tissue, with insulin resistance, metabolic syndrome, and NAFLD and NASH developing as secondary effects158, 159 This adipose tissue-centric model is supported by results from mice that overexpress the FPLD2-associated LMNA R482Q in adipose tissue. These mice develop hepatic steatosis and impaired glucose tolerance compared with control mice that overexpress full-length LMNA160. However, a more recent study in which human LMNA R482Q was expressed in mouse adipose tissue at closer to physiologic levels demonstrated fibrosis in adipose tissue similar to human FPLD2, but no overt metabolic or hepatic phenotype161. Furthermore, a recent abstract reported that deletion of TOR1AIP1 from hepatocytes of mice caused spontaneous steatosis and NASH, supporting the role of the hepatocyte nuclear envelope in steatohepatitis162. Lmna alleles might therefore act independently in adipocytes and hepatocytes to promote the FPLD2 phenotype. It should be noted, however, that human FPLD2 is caused by autosomal-dominant mutations in LMNA rather than loss of LMNA protein, and that men and women are affected, in contrast to the findings in mice after hepatocyte-specific disruption of Lmna. In addition, virtually all patients with FPLD2 and NAFLD or NASH are insulin resistant75, 108. Analysis of null and FPLD2 alleles of Lmna in adipocytes vs hepatocytes will be required to clarify the relative contributions of each cell type to the FPLD2 phenotype.
In the pancreas, inducible disruption of Lmna in acinar cells led to spontaneous pancreatitis, but only male mice developed chronic pancreatitis, based on increased Sirius-red and smooth muscle actin staining in knockout pancreata compared with controls78. Lmna-deficient pancreata from both sexes had smaller acini, smaller and fewer zymogen granules, decreased amylase expression, infiltration by inflammatory cells, and increased markers of endoplasmic reticulum stress, ductal cells, and cell proliferation78. These data indicate that acinar cell expression of LMNA is required for acinar cell homeostasis and that mutations that cause FPLD2 act in a cell-autonomous manner in acinar cells to promote FPLD2-related pancreatitis, which may also be precipitated by hypertriglyceridemia that is seen in some but not all patients75. Lmna deficiency in acinar cells also causes loss of Rb expression and concomitant activation of E2-factor F (E2F) transcription factors, based on the upregulated expression of E2F target genes78. A similar phenotype is observed in mice lacking the Lap2α, which regulates the E2F/Rb pathway as part of a complex with Lmna23, 163. Lap2α deficiency causes loss of nucleoplasmic Lmna and results in a hyperproliferative phenotype in mouse fibroblasts and epidermal and erythroid progenitor cells that has been attributed to loss of Rb-mediated E2F regulation24.
Dysregulation of the Rb/E2F pathway is likely also responsible for the increased cell proliferation seen in the Lmna-deficient pancreatitis model. This phenotype counters what is observed in myoblasts from LmnaSul/Sul mice, where Lmna deficiency leads to increased levels of Lap2α, Rb dephosphorylation and altered localization, and decreased cell proliferation164. Perhaps surprisingly, combined Lmna and Lap2α deficiency (LmnaSul/Sul; Lap2α−/−) rescues the LmnaSul/Sul phenotype, increasing lifespan, body mass, and muscle cell proliferation164. Thus, the mechanisms by which LMNA and LAP2α regulate Rb and cell proliferation may vary with cell type.
Prospects for Therapy
Our increased understanding of lamin biology and the pathophysiology of laminopathies over the last several years has led to targeted therapeutic approaches—some have shown promise in preclinical studies, and there are limited but encouraging data from small clinical trials (Fig.2). Agents tested include farnesyl transferase inhibitors, which have extensive pre-clinical data to support their utility and have been studied in clinical trials; kinase inhibitors, which have been studied in models (and some are already used for treatment of diseases unrelated to laminopathies); and small molecules designed to disrupt protein interactions or antisense oligonucleotide-based tools, which are in preclinical studies.
Figure 2.
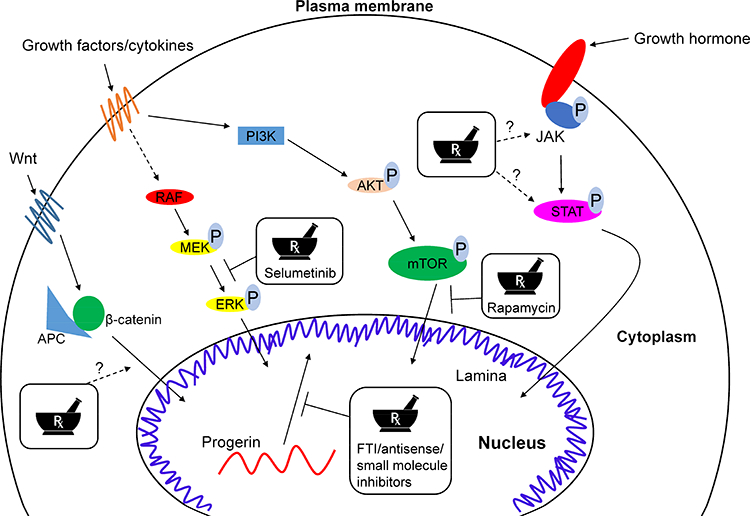
Affected pathways in laminopathies and potential avenues for therapeutic intervention. Alterations in multiple pathways contribute to the pathogenesis of laminopathies. Strategies tested in preclinical or clinical studies are indicated with solid arrows. Strategies to alter Wnt signaling to β-catenin or growth hormone signaling via Jak and Stat proteins, which are altered in mouse models of laminopathies but have not been tested in animal models or clinical trials, are indicated with dashed arrows. Agents developed to reduce or block activity of progerin, FTIs132,133,165,166, antisense oligonucleotides136, and small molecules that inhibit progerin interaction with LMNA169 have been tested.
FTIs
FTIs have been studied in experimental models and clinical trials. In mouse models of HGPS, FTIs increased muscle strength, reduced weight loss and rib fractures, and increased lifespan132, 133. These agents have been tested in uncontrolled trials of children with HGPS. In a 25 children who received the FTI lonafarnib for at least 2 years, the primary outcome (>50% weight gain) was achieved by 9 patients (36%), whereas 10 patients (40%) maintained their pre-trial rates of weight gain during the study165. A pooled analysis of all patients with HGPS given an FTI, alone or in combination with zoledronate and pravastatin, showed significant increases in survival time with the FTI compared to without166.
Kinase inhibition
Data from mouse models of laminopathies, particularly those affecting cardiac and skeletal muscle, indicate a role for aberrant MAP kinase and mTOR pathway activation (section 4). In LmnaH222P/H222P mice, administration of the MEK1/2 inhibitor selumetinib for 4 weeks inhibited ERK activation, prevented myocardial fibrosis, and prolonged survival167. Similar effects were observed when LmnaH222P/H222P mice were given other MEK1/2 inhibitors156, 168, though these compounds have not yet advanced to trials of patients with LMNA-associated cardiomyopathy. in the LmnaSul/Sul mice, rapamycin, an inhibitor of the mTOR pathway prolonged survival124. An open-label phase 1 and 2 trial of the mTOR inhibitor everolimus is underway, in combination with the FTI lonafarnib for HGPS (ClinicalTrials.gov Identifier: NCT02579044).
Other pre-clinical approaches
Based on transcriptomic data indicating alterations of Wnt signaling to β-catenin signaling in progeroid and LmnaH222P/H222P mice, these mice were given the Wnt activator 6-bromoindirubin-3’-oxime. This agent increased cardiac contractility and intraventricular conduction in these mice144. For progerin, given its binding to mature LMNA (but not LMNB1 or LMNB2) in vitro, screening a chemical library identified compounds capable of blocking the progerin-LMNA interaction169. One of these compounds improved nuclear morphology in progerin-overexpressing cells and HGPS patient-derived cells in culture, and improved weight gain, grip strength, and survival in progeroid mice.
An antisense oligonucleotide designed to alter splicing of LMNA mRNA to favor lamin C over prelamin A reduced production of progerin in fibroblasts derived from patients with HGPS. When this oligonucleotide was injected into wild-type C57BL/6J mice, it increased levels of lamin C and decreased lamin A, at the mRNA and protein levels, in liver. When injected into progeroid mice for 3-months, it decreased progerin and increased lamin C in aortic tissue and reduced aortic fibrosis136.
Overview and Future Directions
Our understanding of the structure and function of lamins and lamina-associated proteins has expanded dramatically in the last 2 decades. Fundamental studies of the mechanistic details of lamin post-translational processing have allowed FTIs to be brought into clinical use for children with HGPS, with encouraging effects on weight gain and lifespan165, 166 Similarly, based on extensive work in pre-clinical studies, kinase inhibitors hold promise for LMNA-related cardiomyopathies.
From the perspective of digestive health and disease, the effects of alterations to the nuclear lamina are becoming increasingly apparent. Gastrointestinal and hepatic manifestations of laminopathies that typically affect multiple organ systems, in addition to more recent data from mice and humans, have indicated the direct roles for lamins and their associated proteins76–78.
Targeted therapies for lamina-related gastrointestinal disease are not yet on the horizon, although the observation that hepatocyte JAK2 signaling to STAT5 is defective in the absence of LMNA provides an avenue for investigation77.
It is not clear why some LMNA mutations produce different phenotypes in different families, or even within the same family. It is likely that other genetic or environment factors and epigenetic differences determine how genotype affects phenotype—this is an important area for further study. Similarly, the relative contributions of environmental and genetic factors to common gastrointestinal conditions such as pancreatitis, and NAFLD, and NASH are unclear. We speculate that patients with such common diseases carry genetic variants that affect the nuclear lamina, without a clinically apparent laminopathy—a recent small study of twins and siblings with NAFLD supports this hypothesis76. As we move into the era of precision medicine, increasing our understanding of lamin biology and identifying disease-associated genetic variants lead to improved and targeted therapies that are even organ specific.
Supplementary Material
Acknowledgements
The authors wish to thank Drs. Roland Foisner and Howard Worman for their feedback and helpful comments pertaining to this review. We also thank all those who have provided biospecimens for the studies cited and who have participated in clinical studies to help improve our understanding and treatment of the involved diseases.
Financial Support: Our work is supported by National Institutes of Health (NIH) grants R01 DK47918 and R01 DK116548 (M.B.O.); American Liver Foundation (ALF) Liver Scholar Award (G.F.B.); and by an institutional grant (P30 DK034933) to the University of Michigan.
Abbreviations
- AMA
anti-mitochondrial antibody
- BANF1
barrier to autointegration factor
- EDMD
Emery-Dreifuss muscular dystrophy
- FTI
farnesyl transferase inhibitor
- FPLD2
type 2 (Dunnigan) familial partial lipodystrophy
- HCC
hepatocellular carcinoma
- HGPS
Hutchinson-Gilford progeria syndrome
- IF
intermediate filament proteins
- LAD
lamina-associated domain
- LAP
lamina-associated polypeptide
- LEM
LAP2, emerin, MAN1
- LGMD
limb-girdle muscular dystrophy
- LINC
Linker of the Nucleoskeleton and Cytoskeleton
- LMNA
lamin A/C
- LMNB1
lamin B1
- LMNB2
lamin B2
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PBC
primary biliary cholangitis
- Rb
retinoblastoma protein
- SREBP1
sterol regulatory element-binding protein-1
Footnotes
Disclosure(s): No potential conflicts of interest to report.
Author contributions: G.F.B., R.K., J.B.C., J.S.E., and M.B.O. jointly wrote the manuscript and generated the figures and tables.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol 2017;18:73–89. [DOI] [PubMed] [Google Scholar]
- 2.Lee YL, Burke B. LINC complexes and nuclear positioning. Semin Cell Dev Biol 2017. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Sandoval A, Gasser SM. On TADs and LADs: Spatial Control Over Gene Expression. Trends Genet 2016;32:485–95. [DOI] [PubMed] [Google Scholar]
- 4.Luperchio TR, Wong X, Reddy KL. Genome regulation at the peripheral zone: lamina associated domains in development and disease. Curr Opin Genet Dev 2014;25:50–61. [DOI] [PubMed] [Google Scholar]
- 5.Hobbs RP, Jacob JT, Coulombe PA. Keratins Are Going Nuclear. Dev Cell 2016;38:227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omary MB. “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest 2009; 119:1756–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butin-Israeli V, Adam SA, Goldman AE, et al. Nuclear lamin functions and disease. Trends Genet 2012;28:464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol 2013;14:13–24. [DOI] [PubMed] [Google Scholar]
- 9.Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem 1993;268:16321–6. [PubMed] [Google Scholar]
- 10.Lin F, Worman HJ. Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 1995;27:230–6. [DOI] [PubMed] [Google Scholar]
- 11.Biamonti G, Giacca M, Perini G, et al. The gene for a novel human lamin maps at a highly transcribed locus of chromosome 19 which replicates at the onset of S-phase. Mol Cell Biol 1992;12:3499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart C, Burke B. Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 1987;51:383–92. [DOI] [PubMed] [Google Scholar]
- 13.Rober RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 1989;105:365–78. [DOI] [PubMed] [Google Scholar]
- 14.Lin F, Blake DL, Callebaut I, et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem 2000;275:4840–7. [DOI] [PubMed] [Google Scholar]
- 15.Haraguchi T, Koujin T, Segura-Totten M, et al. BAF is required for emerin assembly into the reforming nuclear envelope. J Cell Sci 2001;114:4575–85. [DOI] [PubMed] [Google Scholar]
- 16.Burke B, Stewart CL. Functional architecture of the cell’s nucleus in development, aging, and disease. Curr Top Dev Biol 2014;109:1–52. [DOI] [PubMed] [Google Scholar]
- 17.Margalit A, Segura-Totten M, Gruenbaum Y, et al. Barrier-to-autointegration factor is required to segregate and enclose chromosomes within the nuclear envelope and assemble the nuclear lamina. Proc Natl Acad Sci U S A 2005;102:3290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stierle V, Couprie J, Ostlund C, et al. The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry 2003;42:4819–28. [DOI] [PubMed] [Google Scholar]
- 19.Bergo MO, Gavino B, Ross J, et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc Natl Acad Sci U S A 2002;99:13049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corrigan DP, Kuszczak D, Rusinol AE, et al. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J 2005;387:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck LA, Hosick TJ, Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J Cell Biol 1990;110:1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber K, Plessmann U, Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett 1989;257:411– 4. [DOI] [PubMed] [Google Scholar]
- 23.Dechat T, Korbei B, Vaughan OA, et al. Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci 2000;113 Pt 19: 3473–84. [DOI] [PubMed] [Google Scholar]
- 24.Naetar N, Korbei B, Kozlov S, et al. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol 2008;10:1341–8. [DOI] [PubMed] [Google Scholar]
- 25.Turgay Y, Eibauer M, Goldman AE, et al. The molecular architecture of lamins in somatic cells. Nature 2017;543:261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zastrow MS, Vlcek S, Wilson KL. Proteins that bind A-type lamins: integrating isolated clues. J Cell Sci 2004;117:979–87. [DOI] [PubMed] [Google Scholar]
- 27.Ozaki T, Saijo M, Murakami K, et al. Complex formation between lamin A and the retinoblastoma gene product: identification of the domain on lamin A required for its interaction. Oncogene 1994;9:2649–53. [PubMed] [Google Scholar]
- 28.Pascual-Garcia P, Debo B, Aleman JR, et al. Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol Cell 2017;66:63–76 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Capelson M, Liang Y, Schulte R, et al. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 2010;140:372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raices M, D’Angelo MA. Nuclear pore complexes and regulation of gene expression. Curr Opin Cell Biol 2017;46:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perovanovic J, Dell’Orso S, Gnochi VF, et al. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci Transl Med 2016;8:335ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finlan LE, Sproul D, Thomson I, et al. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 2008;4:e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy KL, Zullo JM, Bertolino E, et al. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature 2008;452:243–7. [DOI] [PubMed] [Google Scholar]
- 34.Zullo JM, Demarco IA, Pique-Regi R, et al. DNA sequence-dependent compartmentalization and silencing of chromatin at the nuclear lamina. Cell 2012;149:1474–87. [DOI] [PubMed] [Google Scholar]
- 35.Peric-Hupkes D, Meuleman W, Pagie L, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 2010;38:603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulsen J, Sekelja M, Oldenburg AR, et al. Chrom3D: three-dimensional genome modeling from Hi-C and nuclear lamin-genome contacts. Genome Biol 2017;18:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 1994;8:323–7. [DOI] [PubMed] [Google Scholar]
- 38.Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999;21:285–8. [DOI] [PubMed] [Google Scholar]
- 39.Mercuri E, Poppe M, Quinlivan R, et al. Extreme variability of phenotype in patients with an identical missense mutation in the lamin A/C gene: from congenital onset with severe phenotype to milder classic Emery-Dreifuss variant. Arch Neurol 2004;61:690–4. [DOI] [PubMed] [Google Scholar]
- 40.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet 2006;7:940–52. [DOI] [PubMed] [Google Scholar]
- 41.McKenna T, Baek J-H, Eriksson M Laminopathies, Genetic Disorders, Prof.Maria Puiu (Ed.), InTech. 2013. [Google Scholar]
- 42.Dhe-Paganon S, Werner ED, Chi YI, et al. Structure of the globular tail of nuclear lamin. J Biol Chem 2002;277:17381–4. [DOI] [PubMed] [Google Scholar]
- 43.Krimm I, Ostlund C, Gilquin B, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002;10:811–23. [DOI] [PubMed] [Google Scholar]
- 44.Vigouroux C, Magre J, Vantyghem MC, et al. Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy. Diabetes 2000;49:1958–62. [DOI] [PubMed] [Google Scholar]
- 45.Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol 2000;48:170–80. [PubMed] [Google Scholar]
- 46.Kayman-Kurekci G, Talim B, Korkusuz P, et al. Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: a novel gene related to nuclear envelopathies. Neuromuscul Disord 2014;24:624–33. [DOI] [PubMed] [Google Scholar]
- 47.Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000;9:1453–9. [DOI] [PubMed] [Google Scholar]
- 48.Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715–24. [DOI] [PubMed] [Google Scholar]
- 49.Merner ND, Hodgkinson KA, Haywood AF, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008;82:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Renou L, Stora S, Yaou RB, et al. Heart-hand syndrome of Slovenian type: a new kind of laminopathy. J Med Genet 2008;45:666–71. [DOI] [PubMed] [Google Scholar]
- 51.Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003;423:293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2004;101:8963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003;300:2055. [DOI] [PubMed] [Google Scholar]
- 54.Agarwal AK, Fryns JP, Auchus RJ, et al. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet 2003;12:1995–2001. [DOI] [PubMed] [Google Scholar]
- 55.Puente XS, Quesada V, Osorio FG, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet 2011;88:650–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Navarro CL, De Sandre-Giovannoli A, Bernard R, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet 2004;13:2493–503. [DOI] [PubMed] [Google Scholar]
- 57.Hellemans J, Preobrazhenska O, Willaert A, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet 2004;36:1213–8. [DOI] [PubMed] [Google Scholar]
- 58.Waterham HR, Koster J, Mooyer P, et al. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet 2003;72:1013–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet 2002;70:726–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Damiano JA, Afawi Z, Bahlo M, et al. Mutation of the nuclear lamin gene LMNB2 in progressive myoclonus epilepsy with early ataxia. Hum Mol Genet 2015;24:4483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padiath QS, Saigoh K, Schiffmann R, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 2006;38:1114–23. [DOI] [PubMed] [Google Scholar]
- 62.Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 2000;9:109–12. [DOI] [PubMed] [Google Scholar]
- 63. Shackleton S, Lloyd DJ, Jackson SN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 2000;24:153–6. [DOI] [PubMed] [Google Scholar]
- 64.Speckman RA, Garg A, Du F, et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet 2000;66:1192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hegele RA, Cao H, Liu DM, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet 2006;79:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frost B, Bardai FH, Feany MB. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr Biol 2016;26:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sheffield LG, Miskiewicz HB, Tannenbaum LB, et al. Nuclear pore complex proteins in Alzheimer disease. J Neuropathol Exp Neurol 2006;65:45–54. [DOI] [PubMed] [Google Scholar]
- 68.Belt EJ, Fijneman RJ, van den Berg EG, et al. Loss of lamin A/C expression in stage II and III colon cancer is associated with disease recurrence. Eur J Cancer 2011;47:1837–45. [DOI] [PubMed] [Google Scholar]
- 69.Willis ND, Cox TR, Rahman-Casans SF, et al. Lamin A/C is a risk biomarker in colorectal cancer. PLoS One 2008;3:e2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ali MRK, Wu Y, Ghosh D, et al. Nuclear Membrane-Targeted Gold Nanoparticles Inhibit Cancer Cell Migration and Invasion. ACS Nano 2017; 11:3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wegner L, Andersen G, Sparso T, et al. Common variation in LMNA increases susceptibility to type 2 diabetes and associates with elevated fasting glycemia and estimates of body fat and height in the general population: studies of 7,495 Danish whites. Diabetes 2007;56:694–8. [DOI] [PubMed] [Google Scholar]
- 72.Jacque JM, Stevenson M. The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature 2006;441:641–5. [DOI] [PubMed] [Google Scholar]
- 73.Moss SF, Krivosheyev V, de Souza A, et al. Decreased and aberrant nuclear lamin expression in gastrointestinal tract neoplasms. Gut 1999;45:723–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu Z, Wu L, Weng D, et al. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J Exp Clin Cancer Res 2009;28:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ajluni N, Meral R, Neidert AH, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf) 2017;86:698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brady GF, Kwan R, Ulintz PJ, et al. Nuclear lamina genetic variants, including a truncated LAP2, in twins and siblings with nonalcoholic fatty liver disease. Hepatology 2018. (in press; doi: 10.1002/hep.29522). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kwan R, Brady GF, Brzozowski M, et al. Hepatocyte-Specific Deletion of Mouse Lamin A/C Leads to Male-Selective Steatohepatitis. Cell Mol Gastroenterol Hepatol 2017;4:365–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elenbaas JS, Bragazzi Cunha J, Azuero-Dajud R, et al. Lamin A/C maintains exocrine pancreas homeostasis by regulating stability of RB and activity of E2F. Gastroenterology 2018. (in press; doi: 10.1053/j.gastro.2018.01.024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zwerger M, Ho CY, Lammerding J. Nuclear mechanics in disease. Annu Rev Biomed Eng 2011;13:397–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Denais C, Lammerding J. Nuclear mechanics in cancer. Adv Exp Med Biol 2014;773:435–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sakthivel KM, Sehgal P. A Novel Role of Lamins from Genetic Disease to Cancer Biomarkers. Oncol Rev 2016;10:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao J, Chang AC, Li C, et al. Comparative proteomics analysis of Barrett metaplasia and esophageal adenocarcinoma using two-dimensional liquid mass mapping. Mol Cell Proteomics 2007;6:987–99. [DOI] [PubMed] [Google Scholar]
- 83.Sun S, Xu MZ, Poon RT, et al. Circulating Lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J Proteome Res 2010;9:70–8. [DOI] [PubMed] [Google Scholar]
- 84.Lim SO, Park SJ, Kim W, et al. Proteome analysis of hepatocellular carcinoma. Biochem Biophys Res Commun 2002;291: 1031–7. [DOI] [PubMed] [Google Scholar]
- 85.Parise P, Finocchiaro G, Masciadri B, et al. Lap2alpha expression is controlled by E2F and deregulated in various human tumors. Cell Cycle 2006;5:1331–41. [DOI] [PubMed] [Google Scholar]
- 86.Li J, Wang T, Pei L, et al. Expression of VRK1 and the downstream gene BANF1 in esophageal cancer. Biomed Pharmacother 2017;89:1086–1091. [DOI] [PubMed] [Google Scholar]
- 87.Shen Q, Eun JW, Lee K, et al. BANF1, PLOD3, SF3B4 as Early-stage Cancer Decision Markers and Drivers of Hepatocellular Carcinoma. Hepatology 2018. (in press; doi: 10.1002/hep.29606). [DOI] [PubMed] [Google Scholar]
- 88.Sjoblom T, Jones S, Wood LD, Parsons DW, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268–74. [DOI] [PubMed] [Google Scholar]
- 89.Liggett JL, Choi CK, Donnell RL, et al. Nonsteroidal anti-inflammatory drug sulindac sulfide suppresses structural protein Nesprin-2 expression in colorectal cancer cells. Biochim Biophys Acta 2014;1840:322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Knoess M, Kurz AK, Goreva O, et al. Nucleoporin 88 expression in hepatitis B and C virus-related liver diseases. World J Gastroenterol 2006;12:5870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang ZY, Zhao ZR, Jiang L, et al. Nup88 expression in normal mucosa, adenoma, primary adenocarcinoma and lymph node metastasis in the colorectum. Tumour Biol 2007;28:93–9. [DOI] [PubMed] [Google Scholar]
- 92.Xu S, Powers MA. Nuclear pore proteins and cancer. Semin Cell Dev Biol 2009;20:620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Toh BH. Diagnostic autoantibodies for autoimmune liver diseases. Clin Transl Immunology 2017;6:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Duarte-Rey C, Bogdanos D, Yang CY, et al. Primary biliary cirrhosis and the nuclear pore complex. Autoimmun Rev 2012;11:898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Courvalin JC, Lassoued K, Worman HJ, et al. Identification and characterization of autoantibodies against the nuclear envelope lamin B receptor from patients with primary biliary cirrhosis. J Exp Med 1990;172:961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nickowitz RE, Wozniak RW, Schaffner F, et al. Autoantibodies against integral membrane proteins of the nuclear envelope in patients with primary biliary cirrhosis. Gastroenterology 1994;106:193–9. [DOI] [PubMed] [Google Scholar]
- 97.Muratori P, Muratori L, Ferrari R, et al. Characterization and clinical impact of antinuclear antibodies in primary biliary cirrhosis. Am J Gastroenterol 2003;98:431–7. [DOI] [PubMed] [Google Scholar]
- 98.Granito A, Muratori P, Muratori L, et al. Antinuclear antibodies giving the ‘multiple nuclear dots’ or the ‘rim-like/membranous’ patterns: diagnostic accuracy for primary biliary cirrhosis. Aliment Pharmacol Ther 2006;24:1575–83. [DOI] [PubMed] [Google Scholar]
- 99.Gaudy-Marqueste C, Roll P, Esteves-Vieira V, et al. LBR mutation and nuclear envelope defects in a patient affected with Reynolds syndrome. J Med Genet 2010;47:361–70. [DOI] [PubMed] [Google Scholar]
- 100.Cabane J Is Reynolds syndrome a genetic laminopathy? Gastroenterol Clin Biol 2010;34:509–10. [DOI] [PubMed] [Google Scholar]
- 101.Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. Blood 2012;120:4496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Puy H, Gouya L, Deybach JC. Porphyrias. Lancet 2010;375:924–37. [DOI] [PubMed] [Google Scholar]
- 103.Singla A, Griggs NW, Kwan R, et al. Lamin aggregation is an early sensor of porphyria-induced liver injury. J Cell Sci 2013;126:3105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maitra D, Elenbaas JS, Whitesall SE, et al. Ambient Light Promotes Selective Subcellular Proteotoxicity after Endogenous and Exogenous Porphyrinogenic Stress. J Biol Chem 2015;290:23711–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guenantin AC, Briand N, Bidault G, et al. Nuclear envelope-related lipodystrophies. Semin Cell Dev Biol 2014;29:148–57. [DOI] [PubMed] [Google Scholar]
- 106.Dutour A, Roll P, Gaborit B, et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum Mol Genet 2011;20:3779–86. [DOI] [PubMed] [Google Scholar]
- 107.Hendrikx T, Schnabl B. Lamin Deficiency in the Liver Sets the Stage for Nonalcoholic Steatohepatitis Development in Males. Cell Mol Gastroenterol Hepatol 2017;4:441–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ludtke A, Genschel J, Brabant G, et al. Hepatic steatosis in Dunnigan-type familial partial lipodystrophy. Am J Gastroenterol 2005;100:2218–24. [DOI] [PubMed] [Google Scholar]
- 109.Dechat T, Adam SA, Taimen P, et al. Nuclear lamins. Cold Spring Harb Perspect Biol 2010;2:a000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Freund A, Laberge RM, Demaria M, et al. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 2012;23:2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shimi T, Butin-Israeli V, Adam SA, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 2011;25:2579–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Coffinier C, Jung HJ, Nobumori C, et al. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell 2011;22:4683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim Y, Sharov AA, McDole K, et al. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 2011;334:1706–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vergnes L, Peterfy M, Bergo MO, et al. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 2004;101:10428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee JM, Jung HJ, Fong LG, et al. Do lamin B1 and lamin B2 have redundant functions? Nucleus 2014;5:287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang SH, Jung HJ, Coffinier C, et al. Are B-type lamins essential in all mammalian cells? Nucleus 2011;2:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang SH, Chang SY, Yin L, et al. An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum Mol Genet 2011;20:3537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jung HJ, Nobumori C, Goulbourne CN, et al. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc Natl Acad Sci U S A 2013;110:E1923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999;147:913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jahn D, Schramm S, Schnolzer M, et al. A truncated lamin A in the Lmna −/− mouse line: implications for the understanding of laminopathies. Nucleus 2012;3:463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim Y, Zheng Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem Biophys Res Commun 2013;440:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kubben N, Voncken JW, Konings G, et al. Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2011;2:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cutler DA, Sullivan T, Marcus-Samuels B, et al. Characterization of adiposity and metabolism in Lmna-deficient mice. Biochem Biophys Res Commun 2002;291:522–7. [DOI] [PubMed] [Google Scholar]
- 124.Liao CY, Anderson SS, Chicoine NH, et al. Rapamycin Reverses Metabolic Deficits in Lamin A/C-Deficient Mice. Cell Rep 2016;17:2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Solovei I, Wang AS, Thanisch K, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013;152:584–98. [DOI] [PubMed] [Google Scholar]
- 126.Stewart CL. Mouse models of the nuclear envelopathies and related diseases. Preface. Curr Top Dev Biol 2014;109:xi–xiii. [DOI] [PubMed] [Google Scholar]
- 127.Osorio FG, Navarro CL, Cadinanos J, et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med 2011;3:106ra107. [DOI] [PubMed] [Google Scholar]
- 128.Yang SH, Andres DA, Spielmann HP, et al. Progerin elicits disease phenotypes of progeria in mice whether or not it is farnesylated. J Clin Invest 2008;118:3291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mounkes LC, Kozlov S, Hernandez L, et al. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003;423:298–301. [DOI] [PubMed] [Google Scholar]
- 130.Pendas AM, Zhou Z, Cadinanos J, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002;31:94–9. [DOI] [PubMed] [Google Scholar]
- 131.Fong LG, Ng JK, Meta M, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc Natl Acad Sci U S A 2004;101:18111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fong LG, Frost D, Meta M, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006;311:1621–3. [DOI] [PubMed] [Google Scholar]
- 133.Yang SH, Meta M, Qiao X, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest 2006;116:2115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Coffinier C, Jung HJ, Li Z, et al. Direct synthesis of lamin A, bypassing prelamin a processing, causes misshapen nuclei in fibroblasts but no detectable pathology in mice. J Biol Chem 2010;285:20818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fong LG, Ng JK, Lammerding J, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J Clin Invest 2006;116:743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee JM, Nobumori C, Tu Y, et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest 2016;126:1592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Arimura T, Helbling-Leclerc A, Massart C, et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 2005;14:155–69. [DOI] [PubMed] [Google Scholar]
- 138.Mounkes LC, Kozlov SV, Rottman JN, et al. Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet 2005;14:2167–80. [DOI] [PubMed] [Google Scholar]
- 139.Muchir A, Worman HJ. Targeting Mitogen-Activated Protein Kinase Signaling in Mouse Models of Cardiomyopathy Caused by Lamin A/C Gene Mutations. Methods Enzymol 2016;568:557–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Choi JC, Muchir A, Wu W, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med 2012;4:144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Choi JC, Worman HJ. Reactivation of autophagy ameliorates LMNA cardiomyopathy. Autophagy 2013;9:110–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ramos FJ, Chen SC, Garelick MG, et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med 2012;4:144ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ramos FJ, Kaeberlein M, Kennedy BK. Elevated MTORC1 signaling and impaired autophagy. Autophagy 2013;9:108–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Le Dour C, Macquart C, Sera F, et al. Decreased WNT/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum Mol Genet 2017;26:333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hernandez L, Roux KJ, Wong ES, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell 2010;19:413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang AS, Kozlov SV, Stewart CL, et al. Tissue specific loss of A-type lamins in the gastrointestinal epithelium can enhance polyp size. Differentiation 2015;89:11–21. [DOI] [PubMed] [Google Scholar]
- 147.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990;247:322–4. [DOI] [PubMed] [Google Scholar]
- 148.Su LK, Kinzler KW, Vogelstein B, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992;256:668–70. [DOI] [PubMed] [Google Scholar]
- 149.Clodfelter KH, Holloway MG, Hodor P, et al. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol 2006;20:1333–51. [DOI] [PubMed] [Google Scholar]
- 150.Holloway MG, Cui Y, Laz EV, et al. Loss of sexually dimorphic liver gene expression upon hepatocyte-specific deletion of Stat5a-Stat5b locus. Endocrinology 2007;148:1977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Udy GB, Towers RP, Snell RG, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci U S A 1997;94:7239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cui Y, Hosui A, Sun R, et al. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 2007;46:504–13. [DOI] [PubMed] [Google Scholar]
- 153.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tassiulas I, Hu X, Ho H, et al. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat Immunol 2004;5:1181–9. [DOI] [PubMed] [Google Scholar]
- 155.Muchir A, Pavlidis P, Decostre V, et al. Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest 2007;117:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Muchir A, Shan J, Bonne G, et al. Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet 2009;18:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Muchir A, Wu W, Worman HJ. Mitogen-activated protein kinase inhibitor regulation of heart function and fibrosis in cardiomyopathy caused by lamin A/C gene mutation. Trends Cardiovasc Med 2010;20:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Huang-Doran I, Sleigh A, Rochford JJ, et al. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol 2010;207:245–55. [DOI] [PubMed] [Google Scholar]
- 159.Lee PL, Tang Y, Li H, et al. Raptor/mTORCl loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol Metab 2016;5:422–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wojtanik KM, Edgemon K, Viswanadha S, et al. The role of LMNA in adipose: a novel mouse model of lipodystrophy based on the Dunnigan-type familial partial lipodystrophy mutation. J Lipid Res 2009;50:1068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Le Dour C, Wu W, Bereziat V, et al. Extracellular matrix remodeling and transforming growth factor-beta signaling abnormalities induced by lamin A/C variants that cause lipodystrophy. J Lipid Res 2017;58:151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shin J-Y, Hernandez-Ono A, Fedotova T, et al. Novel mouse model of nonalcoholic steatohepatitis. Hepatology 2017;66:1079A. [Google Scholar]
- 163.Dorner D, Vlcek S, Foeger N, et al. Lamina-associated polypeptide 2alpha regulates cell cycle progression and differentiation via the retinoblastoma-E2F pathway. J Cell Biol 2006;173:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cohen TV, Gnocchi VF, Cohen JE, et al. Defective skeletal muscle growth in lamin A/C-deficient mice is rescued by loss of Lap2alpha. Hum Mol Genet 2013;22:2852–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 2012;109:16666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gordon LB, Massaro J, D’Agostino RB Sr. , et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 2014;130:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Muchir A, Reilly SA, Wu W, et al. Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc Res 2012;93:311–9. [DOI] [PubMed] [Google Scholar]
- 168.Wu W, Chordia MD, Hart BP, et al. Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg Med Chem 2017;25:1004–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee SJ, Jung YS, Yoon MH, et al. Interruption of progerin-lamin A/C binding ameliorates Hutchinson-Gilford progeria syndrome phenotype. J Clin Invest 2016;126:3879–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.