

Abstract
An acetyl-protected aminoethyl amine (APAA) chiral ligand based on an ethylenediamine backbone was developed to achieve Pd-catalyzed enantioselective C(sp3)–H arylation of cyclopropanecarboxylic and 2-aminoisobutyric acids without using exogenous directing groups. This new chiral catalyst affords new disconnection for preparing diverse chiral carboxylic acids from simple starting materials that are complementary to the various ring forming approaches.
Graphical abstract
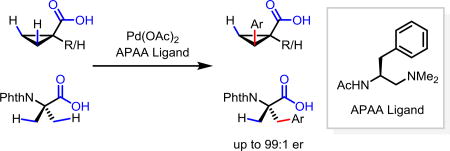
Desymmetrization through C–H activation holds the potential to become a broadly useful chiral technology due to the widespread presence of symmetric prochiral C(sp3)–H bonds in the majority of organic molecules.1 Pd(II)-catalyzed enantioselective intermolecular C(sp3)–H activation was recently made possible by a combination of weakly coordinating directing group and chiral bidentate ligand.2–5 This strategy was firstly demonstrated by the development of N-perfluoroaryl amide-directed enantioselective C–H cross-coupling of α-quaternary cyclopropanecarboxamides using mono-N-protected amino acids (MPAA) as the chiral ligands.2a Recently, chiral bidentate quinoline ligands were developed to realize enantioselective functionalization of methylene C(sp3)–H bond of acyclic N-perfluoroaryl carboxamides to construct β-chiral centers,2c while bidentate oxazoline ligands enabled enantioselective C(sp3)–H functionalization of gem-dimethyl of N-perfluoroaryl or methoxy carboxamides for the construction of α-chiral centers.2d However, substrates in these reactions require pre-installed directing groups which need to be removed after C–H functionalization. Following the same notion of achieving protecting group free synthesis,6 we embarked on the development of enantioselective C–H activation of carboxylic acids without using exogenous directing groups.
Directed functionalization of C(sp3)–H bonds of carboxylic acids without installing external directing group remains a significant challenge despite recent advances using pyridine/quinoline and MPAA ligands.7 These difficulties escalate in the development of enantioselective C–H activation reactions. First, C(sp3)–H activation reactions of free carboxylic acids suffer from low reactivity due to the weak directing ability of the carboxyl groups. Second, the conformation of the metal-carboxylate complex is more flexible than that of the metal-amide directing group complex, which could cause problems for stereocontrol. Indeed, our previously developed bidentate acetyl-protected aminoethyl quinoline ligand only had limited success with a single special substrate, phthalyl-protected 1-aminocyclopropanecarboxylic acid.2c Therefore, we set out to develop a new type of ligands that could achieve more effective enantioselective control with free carboxylic acids. Herein we report the development of ethylenediamine derived chiral ligand that enables enantioselective C–H arylation of a broad range of cyclopropanecarboxylic acid, as well as the 2-aminoisobutyric acid.
Development of asymmetric syntheses of chiral cyclopropane8 continues to attract attention because of their prevalence in biologically active natural products and pharmaceuticals.9 We, therefore selected cyclopropanecarboxylic acid as a model substrate for our ligand development. Notably, our previous enantioselective C–H coupling of cyclopropanecarboxamides with Ar-Bpin requires the presence of α-quaternary carbon centers (Scheme 1).2a,10
Scheme 1.

Bidentate Ligands Developed for Enantioselective C(sp3)–H Functionalization of Carboxylic Acids
Based on our previous chiral bidentate MPAA, quinoline, and oxazoline ligands, it is known that the acetyl-protected amino group (NHAc) is a privileged moiety of chiral ligands for promoting C–H cleavage.2b,2c,2d We, therefore, decided to keep this motif intact while replacing the carboxyl, quinoline, or oxazoline with other σ-donor for chelation; Tertiary amine was chosen as the other σ-donor, because of its distinct stereochemical implication compared with quinoline and oxazoline, as well as the more diffuse lone pair of sp3 nitrogen than sp2 nitrogen. Consequently, we synthesized a series of acetyl-protected aminoethyl amine (APAA) ligands to achieve enantioselective C(sp3)–H functionalization of free carboxylic acids (Table 1). First, various N-alkyl tertiary amine ligands were tested. Despite moderate background reaction in the absence of ligands (Table 1), effective binding of the ligands and possible ligand acceleration afforded significant enantioselectivity. Comparison of the results from L1 to L4 indicates that steric hindrance on the tertiary amine reduces the reactivity. For example, diisopropylamine ligand only provided 8% yield of the product with almost no enantioselectivity. Cyclic amine ligand L5 and L6 are inferior in both reactivity and enantioselectivity. Notably, replacing acetyl with other protecting groups led to a complete loss of reactivity (L7–L10). Ligands with different side chains were also examined. Among different substituents, benzyl group (L1) gave the best yield of 82% and highest er of 97:3, while, isopropyl (L11), sec-butyl (L12) tert-butyl group (L13) and isobutyl (L14) gave slightly lower yield and enantioselectivity. Surprisingly, phenyl group substitution (L15) provided only 20% yield and low enantioselectivity. Less hindered homobenzyl group (L16) also reduced the reactivity and selectivity. Hence, we focused on the modification of the benzyl group. Introducing substituent to the para and ortho position on the phenyl group, as well as replacing phenyl with the naphthalenyl group lowered the yield (L17–L21). Finally, our previous three classes of chiral ligands all gave modest/poor yields or enantioselectivity (L22–L23).
Table 1.

|
Conditions: 1 (0.2 mmol), 2a (2.0 equiv), Pd(OAc)2 (10 mol%), ligand (20 mol%), Ag2CO3 (1.5 equiv), Na2CO3 (1.5 equiv), HFIP (0.25 mL), 80 °C, air, 16 h.
1H NMR yields, using CH2Br2 as an internal standard.
With the high-yielding and highly selective conditions in hand, we examined the scope of aryl iodides (Table 2). Majority of the aryl iodides containing electron-withdrawing and electron-donating group afforded desired products in good yields and high enantioselectivities (up to 98:2 er). Aryl iodides bearing electron-withdrawing groups such as para-methoxycarbonyl (3a), para-acetyl (3b), para-trifluoromethyl (3d), meta-trifluoromethyl (3k) and ortho-methoxycarbonyl (3t) gave slightly higher yields than other aryl iodides. However, aryl iodide with a nitro group (3c) enantioselectivity. Note that aryl iodides containing bromo (3h), phosphonate (3j), and aldehyde (3m) afforded the desired products in high yields and good enantioselectivity. In addition to substituted phenyl iodides, heteroaryl iodides such as 2-acetyl-5-iodothiophene (3u) and 5-iodo-2-furaldehyde (3v) could also be tolerated in this reaction, providing moderate yields and high er. The absolute configuration of the arylated compound was confirmed by optical rotation (see supporting information). The reaction using methyl iodobenzoate as the limiting reagent and a lower loading of silver salt also afforded a higher yield and enantioselectivity (3a).
Table 2.

|
Conditions 1 (0.2 mmol), 2 (2.0 equiv), Pd(OAc)2 (10 mol%), L1 (20 mol%), Ag2CO3 (1.5 equiv), Na2CO3 (1.5 equiv), HFIP (0.25 mL), 80 °C, air, 16 h.
Isolated yields.
Conditions 2a (0.2 mmol), 1 (2.0 equiv), Pd(OAc)2 (10 mol%), L1 (20 mol%), Ag2CO3 (1.0 equiv), Na2CO3 (1.5 equiv), HFIP (0.25 mL), 80 °C, air, 16 h.
Using AgOAc (3.0 equiv) instead of Ag2CO3 (1.5 equiv), NaHCO3 (1.5 equiv) instead of Na2CO3 (1.5 equiv).
Using 2 (1.5 equiv).
A wide range of α-substituted cyclopropanecarboxylic acids are also tested using methyl iodobenzoate as the coupling partner (Table 3). 1-Aryl-1-cyclopropanecarboxylic acids (5a–d), which are an important motif in pharmaceutical chemistry,11 were arylated to give the desired products in excellent yield and enantioselectivity. Interestingly, C(sp2)–H arylation of the α-phenyl groups did not occur. Chloro (5b), bromo (5c) and trifluoromethyl (5d) substituents on the phenyl group of substrates were all well tolerated in this reaction. α-Alkyl cyclopropanecarboxylic acids are also suitable substrates for this reaction. Arylation of α-ethyl (5e), butyl (5f), and chloropentyl (5h) cyclopropanecarboxylic acids under 60 °C afforded the mono-arylated products in good yields and er. Surprisingly, α-phenylpropyl substitution reduced the yield to 58% (5g). Although α-Benzyl containing substrates (5i and 5j) decomposed under these conditions, replacement of Ag2CO3 with AgOAc provided desired products in moderate yield and high enantioselectivity. Benzyl-protected 1-hydroxymethyl (5k) and phthalyl-protected 1-aminomethyl cyclopropanecarboxylic acids (5l) provided good yield and excellent enantioselectivity. These β-hydroxyl and β-amino-cyclopropanecarboxylic acid motifs are recurrent structures in bioactive molecules.12
Table 3.

|
Conditions 4 (0.2 mmol), 2a (2.0 equiv), Pd(OAc)2 (10 mol%), L1 (20 mol%), Ag2CO3 (1.5 equiv), Na2CO3 (1.5 equiv), HFIP (0.25 mL), 80 °C, air, 16 h.
Isolated yields.
60 °C.
Using AgOAc (3.0 equiv) instead of Ag2CO3 (1.5 equiv), NaHCO3 (1.5 equiv) instead of Na2CO3 (1.5 equiv), 60 °C.
The performance of this new chiral ligand was further evaluated in the desymmetrization of geminal dimethyl groups for the enantioselective arylation of phthalyl-protected 2-aminoisobutyric acids (Table 4). Such reaction could provide a simple avenue for the synthesis of diverse chiral α-amino acids. While aryl iodides bearing different substituents gave similar yields of products, the enantioselectivity varied. Electron-neutral group-substituted aryl iodides gave good enantioselectivity, aryl iodides containing electron-withdrawing groups (7a, 7b, and 7g) provided lower er. Since aryl iodides are not involved in the enantio-determining C–H activation step, it is possible that one of the chiral palladacycle intermediates from this particular substrate is less reactive in the oxidation addition step with aryl iodide, thereby contributing to the enantioselectivity partially. A further extensive mechanistic study will be conducted to rationalize this observation. The absolute configuration of the product was confirmed by X-ray crystallographic analysis (see Supporting Information)
Table 4.

|
Conditions: 1 (0.1 mmol), 7 (2.5 equiv), Pd(OAc)2 (10 mol%), L1 (20 mol%), AgOAc (3.0 equiv), NaHCO3 (1.5 equiv), HFIP, 80 °C, air, 24 h.
Isolated yields.
Isolated as the corresponding methyl ester.
To further demonstrate the utility of this new methodology, a late-stage C–H functionalization of a promising drug candidate on neurological disorders, Itanapraced,13 was performed. The reaction proceeded smoothly, and the modified molecule was obtained in high yield and with excellent enantioselectivity (eq 1).
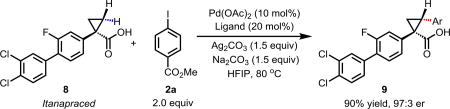 |
(1) |
In summary, we have developed a new class of chiral acetyl-protected aminoethyl amine ligands which enable the enantioselective C–H activation of free carboxylic acids without using exogenous directing groups. Enantioselective C–H arylation of simple cyclopropanecarboxylic acid and phthalyl-protected 2-aminoisobutyric acid provides a new synthetic disconnection for asymmetric synthesis of diverse chiral carboxylic acids. The successful design of this new ligand to match the weakly coordinating carboxylic acid for stereocontrol offers a framework for understanding the chiral induction in C(sp3)–H activation.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS 2R01 GM084019) and Shanghai RAAS Blood Products Co. Ltd. for financial support. We thank Dr. Jason Chen from Automated Synthesis Facility, The Scripps Research Institute for his assistance with 2D HPLC/SFC analysis.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For selected reviews of enantioselective C–H Functionalization: Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Chem. Soc. Rev. 2009;38:3242. doi: 10.1039/b816707a.Newton CG, Wang S-G, Oliveira CC, Cramer N. Chem. Rev. 2017;117:8908. doi: 10.1021/acs.chemrev.6b00692.Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F, Yu J-Q. Science. 2018;359:759. doi: 10.1126/science.aao4798.
- 2.(a) Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q. J. Am. Chem. Soc. 2011;133:19598. doi: 10.1021/ja207607s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiao K-J, Lin DW, Miura M, Zhu R-Y, Gong W, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2014;136:8138. doi: 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen G, Gong W, Zhuang Z, Andra MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q. Science. 2016;353:1023. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q. Science. 2017;355:499. doi: 10.1126/science.aal5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For the preliminary result of enantioselective C(sp3)–H functionalization using mono-N-protected amino acid ligands (MPAA) and pyridine directing group: Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem. Int. Ed. 2008;47:4882. doi: 10.1002/anie.200801030.
- 4.For selected Pd(0)-catalyzed intramolecular enantioselective C–H functionalization see: Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew. Chem. Int. Ed. 2011;50:7438. doi: 10.1002/anie.201102639.Anas S, Cordi A, Kagan HB. Chem. Commun. 2011;47:11483. doi: 10.1039/c1cc14292e.Saget T, Cramer N. Angew. Chem. Int. Ed. 2012;51:12842. doi: 10.1002/anie.201207959.Martin N, Pierre C, Davi M, Jazzar R, Baudoin O. Chem. Eur. J. 2012;18:4480. doi: 10.1002/chem.201200018.Ladd CL, Charette AB. Org. Lett. 2016;18:6046. doi: 10.1021/acs.orglett.6b02982.Pedroni J, Cramer N. J. Am. Chem. Soc. 2017;139:12398. doi: 10.1021/jacs.7b07024.Zhu C, Wang D, Zhao Y, Sun W-Y, Shi Z. J. Am. Chem. Soc. 2017;139:16486. doi: 10.1021/jacs.7b10365.
- 5.For Pd (II)-catalyzed enantioselective C–H functionalization using phosphoric acids/amides ligands Yan S-B, Zhang S, Duan W-L. Org. Lett. 2015;17:2458. doi: 10.1021/acs.orglett.5b00968.Wang H, Tong H-R, He G, Chen G. Angew. Chem. Int. Ed. 2016;55:15387. doi: 10.1002/anie.201609337.Jain P, Verma P, Xia G, Yu J-Q. Nat. Chem. 2016;353:1023.
- 6.Young IS, Baran PS. Nat. Chem. 2009;1:193. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 7.(a) Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunder LB, Yu J-Q. J. Am. Chem. Soc. 2007;129:3510. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]; (b) Chen G, Zhuang Z, Li G-C, Saint-Denis TG, Xiao Y, Joe CL, Yu J-Q. Angew. Chem. Int. Ed. 2017;56:1506. doi: 10.1002/anie.201610580. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu Y, Chen X, Yuan C, Li G, Zhang J, Zhao Y. Nat. Commun. 2017;8:14904. doi: 10.1038/ncomms14904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ghosh KK, van Gemmeren Manual. Chem. Eur. J. 2017;23:17697. doi: 10.1002/chem.201705449. [DOI] [PubMed] [Google Scholar]
- 8.For selected reviews and reactions of asymmetric cyclopropanation, see: Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; New York: 1998. Doyle MP, Protopopova MN. Tetrahedron. 1998;54:7919.Denmark SE, Beutner G. In: In Cycloaddition Reactions in Organic Synthesis. Kobayashi SJ, ørgensen KA, editors. Wiley-VCH; Weinheim, Germany: 2002. pp 85ff.Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e.Lou Y, Horikawa M, Kloster RA, Hawryluk NA, Corey EJ. J. Am. Chem. Soc. 2004;126:8916. doi: 10.1021/ja047064k.Goudreau SR, Charette AB. J. Am. Chem. Soc. 2009;131:15633. doi: 10.1021/ja9074776.Zhu S, Cui X, Zhang XP. Eur. J. Inorg. Chem. 2012:430.Wang Y, Wen X, Cui X, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2017;139:1049. doi: 10.1021/jacs.6b11336.
- 9.Talele TT. J. Med. Chem. 2016;59:8712. doi: 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- 10.For other intermolecular enantioselective C–H functionalizations of cyclopropanes, see Chan KSL, Fu H-Y, Yu J-Q. J. Am. Chem. Soc. 2015;137:2042. doi: 10.1021/ja512529e.Lee T, Hartwig JF. Angew. Chem. Int. Ed. 2016;55:8723. doi: 10.1002/anie.201603153.
- 11.Peretto I, Radaelli S, Parini C, Zandi M, Raveglia LF, Dondio G, Fontanella L, Misiano P, Bigogno C, Rizz A, Riccardi B, Biscaioli M, Marchetti S, Puccini P, Catinella S, Rondelli I, Cenacchi V, Bolzoni PT, Caruso P, Villetti G, Facchinetti F, Del Giudice E, Moretto N, Imbimbo BP. J. Med. Chem. 2005;48:5705. doi: 10.1021/jm0502541. [DOI] [PubMed] [Google Scholar]
- 12.(a) MITSUBISHI TANABE PHARMA CORPORATION. Sakurai O, Saruta K, Hayashi N, Goi T, Morokuma K, Tsujishima H, Sawatomo H, Shitama H, Imashiro R. WO2012/81736. 2012 [Google Scholar]; (b) Edgar DM, Hangauer DG, Shiosaki K, Solomon M, White JF. US2006/63755. 2006 [Google Scholar]
- 13.CHIESI FARMACEUTICI S.P.A. WO2004/74232. 2004 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.