

Abstract
Gene delivery is known to be a complicated multi-step biological process. It has been observed that subtle differences in the structure and properties of polymeric materials used for gene delivery can lead to dramatic differences in transfection efficiency. Therefore, screening of properties is pivotal to optimizing the polymer. So far, most polymeric materials are built in a "bottom-up" manner, i.e. synthesized from monomers that allow modification of polymer composition or structural factors. With this method, we previously synthesized and screened a library of biodegradable poly(amine-co-ester) (PACE) terpolymers for optimized DNA delivery. However, it can be tedious and time consuming to synthesize a polymer library for screening, particularly when small changes of a factor need to be tested, when multiple factors are involved, and when the effects of different factors are synergistic. In the present work, we evaluate the potential of PACE to deliver mRNA. After observing that mRNA transfection efficiency was highly dependent on both end group composition and molecular weight (MW) of PACE in a synergistic manner, we developed a "top-down" process we called actuation, to simultaneously vary these two factors. Some of the actuated PACE (aPACE) materials presented superior mRNA delivery properties compared to regular PACE, with up to a 106-fold-increase in mRNA transfection efficiency in vitro. Moreover, when aPACE was used to deliver mRNA coding for erythropoietin (EPO) in vivo, it produced high levels of EPO in the blood for up to 48 h without inducing systemic toxicity. This polymer constitutes a new delivery vehicle for mRNA-based treatments that provides safe yet potent protein production.
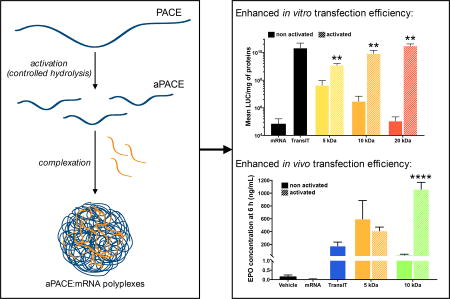
GRAPHICAL ABSTRACT
INTRODUCTION
Protein therapeutics can be used as highly effective medical treatments for a wide array of diseases [1–6]. However, the clinical use of this therapeutic class has been limited by their cost and instability after systemic administration, revealing the need for new approaches to ensure sustained, efficient, and safe delivery [7]. Gene therapies are attractive due to the promise of sustained protein secretion with low administration frequencies [8]. While DNA-based therapies utilizing viruses offer the possibility of long-term protein expression, they also raise numerous safety concerns, especially regarding risks of insertional mutagenesis, induction of severe immune responses, and difficulty in controlling protein expression levels. Recently, messenger RNA (mRNA) has emerged as a promising therapeutic modality for protein production [9, 10]. In contrast to DNA, mRNA elicits the expression of an encoded protein without entering the nucleus, thus demonstrating higher efficiency at transfecting non-dividing cells while reducing the risk of insertional mutagenesis. mRNA therapy has an additional benefit that its dosing is scalable and the treatment is transiently linked to mRNA stability in the cytosol. The in vivo use of mRNA has been previously explored [11, 12], but only limited protein expression has been observed. This is likely due to the instability of the naked mRNA [13], as well as the difficulty in its effective intracellular delivery [9].
A few studies report the use of RNA viruses to deliver mRNA [14, 15]; however, gene expression after viral transfection remains difficult to control, and carries risks of immunogenicity and inflammation. mRNA has also been delivered intradermally in a human clinical trial, using a polycationic peptide that condenses and protects mRNA from degradation by RNase [16]. Although the safety of the method was demonstrated, insufficient transfection efficiency remained as a serious challenge. Lipid nanoparticles have been used to deliver genetic materials, including mRNA, but most are limited by their toxicity [17]. Cationic polymers are promising delivery vectors because of their structural versatility. Many polymers have now been studied for mRNA transfection, including polyethylenimine (PEI) [18], DEAE-dextran [19], poly(L-lysine) (PLL) [20], poly(β-amino esters) (PBAE) [21] and dendrimers [18]. However, low transfection efficiencies and toxicity issues associated with these polycationic polymers often hamper their use.
Looking at previous reports and our own research to understand the challenges associated with mRNA delivery, we noticed that small changes in structural properties of the same material can lead to dramatically different outcomes, especially in terms of transfection efficiency. For example, a difference as small as one single methylene group in the structure of an oligoalkylamine-based cationic polymer was shown to increase mRNA transfection efficiency by 105-fold [22]. Screening of these properties thereby becomes a key step for the development of these materials as mRNA delivery vehicles [23–26]. However, the "bottom-up" construction of polymer libraries is time-consuming and labor-intensive. Moreover, depending on the maturity of the synthesis protocol, it is not always easy or feasible to obtain a series of materials with continuously evolving properties by small increments, which is sometimes necessary for optimization of the delivery system [22]. Herein, we propose a "top-down" approach for synthesizing a library of polymeric materials, as described below.
We previously reported a biodegradable material, poly(amine-co-ester) (PACE) terpolymer for gene delivery. PACE:pDNA polyplexes are among the most efficient and least toxic non-viral vectors [27]. PACE polymers were designed to possess several key features: (1) biodegradability, provided by the ester linkage in the main chain of the polymer; (2) low cationic charge density that allows for electrostatic complexation with nucleic acids while avoiding toxicity associated with highly cationic polymers; and (3) hydrophobicity provided by the lactone groups that stabilizes the polyplex. The tolerance of the lipase chemistry provided a high chemical versatility, allowing for the optimization of the polymer structure via monomer selection, to readily yield a family of different materials. Such versatility was promising to translate the PACE technology for mRNA delivery, as recent studies demonstrated that optimization of the vector is necessary when trying to deliver different nucleic acid materials because of their inherent chemical and structural differences [23–25]. Therefore, we first aimed to take advantage of the versatility of the PACE structure, and extend PACE properties to the delivery of mRNA. After an initial screening with the "bottom-up" synthesis of a library of PACE, we identified polymer MW and end group composition as two key parameters that determine the transfection efficiency of this material for mRNA. In particular, the effect of MW appeared to significantly affect the transfection efficiency within a narrow range between 5kDa and 10kDa. We then adopted a "top-down" approach to simultaneously vary PACE MW and end groups through controlled hydrolysis of high MW polymers. By doing this, we were able to expose PACE end groups with higher transfection efficiency, and fine tune MW at the same time. This method produced a new family of materials called actuated PACE (aPACE), and these biodegradable aPACE polymers yielded high transfection levels both in vitro and in vivo with negligible toxicity.
MATERIALS AND METHODS
Materials
ω-pentadecalactone (PDL), diethyl sebacate (DES), sebacic acid, N-methyldiethanolamine (MDEA), chloroform, dichloromethane, hexane, chloroform-d, chromium (III) acetylacetonate, ethylene diamine, ethanol amine, glycine, and 1,1'-carbonyldiimidazole (CDI) were purchased from Sigma Aldrich (Saint Louis, MO) and were used as received. 2-[(2-Aminoethyl)(methyl)amino]ethanol (AEMAE) was purchased from ChemBridge Corporation (San Diego, CA). Immobilized Candida antarctica lipase B (CALB) supported on acrylic resin (Novozym 435) was also obtained from Sigma Aldrich and was dried at 50 °C under 2.0 mmHg for 20 h prior to use. TransIT-mRNA transfection kit was purchased from Mirus Bio LLC (Madison, WI). Modified Fluc mRNA encoding for luciferase and murine EPO mRNA encoding for erythropoietin (EPO) were purchased from TriLink Biotechnologies (San Diego, CA). HEK293 cells, Daoy cells, and SH-SY5Y cells were purchased from ATCC (Manassas, VA).
Polymer synthesis
PACE synthesis was performed as previously described [27], including several modifications (Figure 1a) to produce polymers with different structures. Briefly, (1) to obtain either classic or acidic PACE, diethyl sebacate or sebacic acid were used for the polymerization; (2) to vary the hydrophobicity of classic PACE, different percentages of PDL (10% or 20%) were added to the reactants, and (3) to vary the molecular weight (MW) of classic PACE containing 10% PDL, the second stage reaction time was varied between 8 to 72 h, in order to obtain polymers with MW ranging from 2 kDa to 25 kDa.
Figure 1. Synthesis of a PACE library for mRNA delivery.

(a) PACE synthesis was modified to vary different parameters in the final polymer. Diethyl sebacate (R = CH2CH3), or sebacic acid (R = H) were used to make classic (ester/OH) ended or acidic (COOH/OH) ended PACE, respectively. With classic PACE, the duration of the second step of the synthesis was varied from 8 to 72 h to vary the MW. The PDL content (10% or 20%) was varied to modify the polymer hydrophobicity. (b) Screening of MW using classic PACE with 10%PDL content: transfection efficiency increased when the MW was decreased up to 5 kDa, while further decrease of the MW to 2 kDa decreased transfection efficiency. (c) Screening of PDL content using a 10 kDa classic PACE polymer: no effect of PDL content was observed on mRNA transfection efficiency. (d) Transfection efficiency of acidic PACE and Classic PACE with MW of 5kDa at 10% PDL content: transfection efficiency is increased for acidic PACE compared to classic PACE. Results are presented as mean ± SD of three independent experiments run in duplicate.
Modification of polymer end groups
To prepare PACE with different end groups, the parent polymer was synthesized with sebacic acid instead of diethyl sebacate, which yields PACE with a mixture of hydroxyl and carboxyl end groups. Both of the end groups were then activated with CDI at a molar ratio of 1:40 by stirring in dry dichloromethane overnight at room temperature. The mixture was washed three times with deionized water, followed by evaporation of DCM under vacuum to obtain the reactant, PACE-CDI (Figure 2a). PACE-CDI was reacted with amine-containing molecules to yield PACE with new end groups. Specifically, glycine and AEMAE was used to generate PACE-COOH and PACE-MAE, respectively. For conjugation, 5mM glycine or AEMAE was reacted with 0.5mM of PACE-CDI in DMSO for 40 hours at room temperature under constant stirring. After reaction, the mixture was washed with 10-fold volume of deionized water, extracted with DCM, followed by evaporation of DCM under vacuum to obtain PACE-COOH and PACE-MAE. When this protocol was adopted to synthesize 10kDa acidic PACE, the 5kDa PACE-CDI and ethylenediamine were added at an exact molar ratio of 2:1 (Figure S1).
Figure 2. End group modification to form carboxyl ended (-COOH) or hydroxyl ended (-MAE) ended PACE.

(a) To obtain PACE with -COOH or -MAE end groups, we modified acidic PACE by CDI activation (1, 2), followed by nucleophilic substitution with an amine-containing molecule (3,4; R = glycine, or AEMAE). (b) Transfection efficiency of PACE-COOH and PACE-MAE with MW of either 5 kDa or 10 kDa, synthesized by methods illustrated in (a) and Figure S1. Statistical significance was determined by Student’s t-test (indicated as follows: *, P<0.05; ***, P<0.001).
Actuation of polymers
Polymers (20 to 30 mg) with different starting MWs were spread evenly on the inner surface of glass vials, forming thin films to ensure efficient air penetration. The vials were then incubated at a controlled temperature (typically 37°C) exposing to flowing wet air for different lengths of time.
Polymer characterization
1H and 13C NMR spectra were recorded on a Bruker AVANCE 500 spectrometer. For inverse gated 13C NMR, samples were prepared at 50 mg/mL of polymer in chloroform-d, and chromium (III) acetylacetonate (Cr (acac)3) was added at a concentration of 5 mg/mL as a relaxation agent. The signal was recorded using a T1 relaxation time of 10 seconds.
The molecular weights (MW) of polymers were measured by gel permeation chromatography (GPC) using a Waters HPLC system equipped with a microSTYRAGEL column (mixed bed; pore sizes 100 Å – 106 Å). Chloroform containing 0.2 wt% triethylamine was used as the eluent at a flow rate of 1 mL/min. Sample concentrations of 2 mg/mL and injection volumes of 100 µL were used. Polymer MW was determined based on a conventional calibration curve generated by narrow polydispersity polystyrene standards from Sigma Aldrich (Saint Louis, MO). Empower II GPC software was used to run the GPC instrument and to perform MW calculations.
Polyplex preparation and characterization
Unless specified, polymer:mRNA polyplexes were prepared at a 100:1 polymer:mRNA weight ratio in 25 mM sodium acetate buffer (pH 5.8). For in vitro experiment, a solution at 10 µg mRNA/mL was prepared: 1 µL of polymer solution (100 mg/mL in DMSO) was first diluted in 50 µL sodium acetate buffer. After brief vortexing, the polymer solution was mixed with 1 µg mRNA diluted in 50 µL sodium acetate buffer, and vortexed again. The polymer:mRNA mixture was incubated at room temperature for 10 min before use. For in vivo experiments, a solution at 100 µg mRNA/mL in sodium acetate buffer was prepared by the same method.
The hydrodynamic diameter of the polyplexes was measured by Dynamic Light Scattering (DLS) using a Malvern Nano-ZS (Malvern Instruments, UK), after dilution of polyplexes in DI water at a concentration of 2 µg/mL of mRNA. To measure zeta potential, the same solution was loaded into a disposable capillary cell and analyzed on a Malvern Nano-ZS.
Encapsulation efficiency (EE) of mRNA in the polyplexes was measured using the Quant-IT RiboGreen RNA kit (Invitrogen, #R11491) according to manufacturer instructions. As the RiboGreen assay measures the amount of free mRNA in solution, this amount was substracted to the initial amount added to form the polyplexes, to obtain the amount of mRNA complexed within the polyplexes.
Cell culture
HEK293 cells and SH-SY5Y cells were cultured in 4.5 g/L glucose DMEM media (Gibco #11965) supplemented with 10% FBS and 1% pen/strep at 37°C and 5% CO2. Daoy cells were cultured in 2 mM L-glutamine, 1 mM sodium pyruvate, and 1500 mg/L sodium bicarbonate EMEM media (ATCC #30–2003) supplemented with 10% FBS and 1% pen/strep at 37°C and 5% CO2.
In vitro transfection
For in vitro transfection of Fluc mRNA, cells were seeded in 24-well plates at a density of 75,000 cells/well in 500 µL of media and incubated over-night to ensure adherence. Media was replaced by 400 µL of transfection media (culture media containing 10% FBS, but without pen/strep), and 100 µL of polyplexes (1 µg of mRNA total) was added to each well. For free mRNA control, 1 µg of mRNA was diluted in 100 µL of acetate buffer and added to the wells. The commercial mRNA transfection kit TransIT was used as a control. Briefly, 1 µg of mRNA was mixed with 0.7 µL of Boost reagent and 1.1 µL of TransIT reagent in 100 µL of OPTIMEM media (Gibco #11058021). 24 h after transfection, luciferase expression was measured. Cells were washed and lysed using 200 µL of 1× lysis buffer (Promega, #E397A) and one freeze-thaw cycle at −80°C. 20 µL of the lysate was then mixed with 100 µL of luciferase reporter reagent (Promega, #E1483), and luminescence was read on a Glomax luminometer (Promega). Lysate protein content was measured using a Pierce BCA protein assay kit (ThermoFisher, #23225). All experiments were run three independent trials in duplicate.
In vitro toxicity
To evaluate the cytotoxicity of PACE polymers and TransIT, HEK293 cells were seeded in 96-well plates at a density of 10,000 cells/well in 100 µL of media and incubated overnight to ensure adherence. The polymer:mRNA polyplexes or TransIT/mRNA complexes were formed using the same w:w ratios as for transfection experiments, and diluted in transfection media at different concentrations. 100 µL of polyplexes containing media were added to the wells to achieve final concentrations of mRNA ranging from 0.01 to 20 µg/mL. After 24 h of incubation, cell viability was measured using a MTT assay. All experiments were run three independent trials in duplicate.
Lyophilization of the polyplexes
PACE:mRNA polyplexes were prepared using sodium acetate buffer, and trehalose solutions at different concentrations (30 mg/mL or 60 mg/mL in 25 mM sodium acetate buffer, pH = 5.8) were added to the polyplex suspension at a 1:1 volume ratio to obtain final trehalose concentrations of 0, 15 or 30 mg/mL. The mixtures were then snap frozen in liquid nitrogen and lyophilized for 2 days. At the end of the lyophilization, the polyplexes were resuspended in sodium acetate buffer and transfection efficiency was evaluated in HEK293 cells as previously described. The transfection and characterization of the gene expression were performed using the methods described above.
In vivo studies
All animal work was completed at Yale University in accordance with Yale Animal Resource Center (YARC) and the Institutional Animal Care and Use Committee (IACUC) guidelines. Female BALB/c mice (20 g, Charles River, Willimantic, CT, USA) were used for the experiments. PACE:mRNA polyplexes (0.1 mg/mL in mRNA, N = 3), aPACE:mRNA polyplexes (0.1 mg/mL in mRNA, N = 3), TransIT:mRNA complexes (0.1 mg/mL in mRNA, N = 3), free mRNA diluted in sodium acetate buffer (0.1 mg/mL in mRNA, N = 3) or sodium acetate buffer (25 mM, pH 5.8, N = 3) were administered intravenously through the tail vein in a volume of 200 µL. Retro-orbital blood collections (50 µL) were performed before particle administration, and 6 h, 24 h, 48 h, 72 h and 7 days after injection. Immediately after blood collection, plasma was separated by centrifugation (3,000 g, 10 min) and frozen at −80°C until further analysis. EPO concent ration in plasma was measured using an ELISA kit (R&D Systems). 24 h and 7 days after injection, liver, kidney and spleen were collected, processed for H&E staining and scored by an external pathologist for any abnormal cellular morphology.
Statistical Analysis
GraphPad Prism 7 (GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla California USA, www.graphpad.com) was used for graphing and statistical analysis. Statistical significance was tested using a two-tailed unpaired student’s t-test with a level of confidence of 95%.
RESULTS AND DISCUSSION
PACE polymers for mRNA delivery
PACE is a family of terpolymers formed through enzymatic copolymerization of diesters/diacids with amino-substituted diols and lactones. We have previously shown that PACE can efficiently deliver pDNA [27], microRNA [28], and siRNA [29]. However, given the inherent structural differences between mRNA and these other nucleic acids, we hypothesized that improved mRNA delivery will likely require either optimization of existing PACE structures, or development of new PACE variants [23]. As previously described, the high tolerance of the lipase catalyst offers a versatility of structures, especially in terms of hydrophobicity and MWs. Starting from one of the most efficient PACE compositions for delivery and transfection of DNA (classic PACE), we generated different polymers with varied MWs and lactone contents in order to specifically improve mRNA delivery and transfection (Figure 1a).
Gel Permeation Chromatography (GPC) was used to determine polymer MW (Table S1). All the PACE polymers were able to complex mRNA, and formed nanosized polyplexes with neutral or negative surface charges (Table S2). When evaluated by the RiboGreen assay, all the polymers were able to encapsulate mRNA with efficiencies ranging from 55–76% (Table S2). The ability of the different PACE polymers to transfect luciferase (LUC) expressing mRNA in HEK-293 cells was used to screen in vitro performance. None of these PACE polymers, representing different structural modifications, provided transfection levels comparable to the commercial agent TransIT, which was used as a positive control (Figure 1). However, we observed a clear trend in transfection levels with the MW of the polymers (Figure 1b). As polymer MW decreased from 20 kDa to 5 kDa an increase in transfection efficiency of 2 orders of magnitude (1.1 × 105 RLU/mg and 4.4 × 107 RLU/mg, respectively) were observed. However, this trend appears to plateau at 5 kDa since the level of transfection dramatically decreased using PACE MW of 2 kDa, comparable to the level obtained with free mRNA. Previous studies have reported that the strength and the stability of electrostatic complexation between polycations and polyanions increase exponentially with the length of the polycation [21, 30–32]. These results support our observation that very short polymers (2 kDa) are inefficient for transfection, likely due to poor complexation of the mRNA. On the other hand, above the 2 kDa threshold, shorter polymers (5 kDa) are more efficient at delivering mRNA than higher MW polymers (10 kDa or 20 kDa), likely due to the inefficient release of mRNA from high MW PACE polyplexes inside the cells. High MW PACE chains contain a large number of positive charges and hydrophobic domains, which lead to thermodynamically stable binding with mRNA and prevent its release from the polyplexes. Our results suggest that efficient mRNA delivery comes from a fine balance of the MW of the polymer: polymer chain length needs to be long enough to ensure complexation of the mRNA and stabilization of the polyplex, but short enough for mRNA release inside the cells. On the other hand, increasing the PDL content from 10% to 20% of a 10 kDa PACE polymer did not significantly affect transfection efficiency (Figure. 1c), which is contrary to what has been observed with pDNA, and confirming that the polymer structure requires optimization to each genetic material.
PACE transfection efficiency is highly dependent on its end group composition
Classic PACE, synthesized by terpolymerization of diethyl sebacate, PDL, and MDEA, contains a mixture of methyl (from diethyl sebacate) and hydroxyl (from MDEA) end groups. We notice that when the monomer diethyl sebacate was replaced by sebacic acid for PACE synthesis to form acidic PACE, its mRNA transfection efficiency doubled (Figure 1d). It has been previously reported that different end groups on the same polymer can dramatically affect transfection efficiency of pDNA [25, 26]. Therefore, we hypothesized that end group compositions can also affect mRNA transfection, and modified the PACE end groups to further optimize PACE for mRNA delivery. To this end, we started with acidic PACE, which contains a mixture of carboxyl and hydroxyl end groups with a molecular weight around 5 kDa. Both of these two end groups can be activated by 1,1'-Carbonyldiimidazole (CDI), which was further substituted by amine-containing molecules including glycine or AEMAE, to form a carboxyl (-COOH) end group (PACE-COOH) or a (methylamino)ethanol (-MAE) end group (PACE-MAE), respectively (Figure 2). These two monomers were chosen to mimic the naturally occurring mix of end groups found in acidic PACE, in order to identify the end group with higher transfection efficiency. The reaction mechanism ensured more than 90% conversion rates for both end groups, as confirmed by NMR spectroscopy (data not shown). We then evaluated the abilities of these polymers to transfect mRNA in vitro. Significant differences in transfection efficiency were observed between PACE with different end groups, as the 5 kDa PACE-MAE polymer demonstrated two orders of magnitude higher transfection efficiency than PACE-COOH of the same MW (Figure 2b). This effect can be explained by the difference in EE of these two polymers (Table S2), as PACEMAE encapsulated 98% of total mRNA, PACE-COOH were only able to encapsulate 18%.
Although acidic PACE at molecular weights higher than 5 kDa were not directly synthesized due to technical challenges (difficulty in removing the water byproduct as the polymer chain grows longer), 10 kDa acidic PACE can be obtained by CDI activation of the 5 kDa polymers followed by crosslinking two polymer chains with an ethylene diamine molecule (Figure S1). Remarkably, when 10 kDa acidic PACE was modified with either carboxyl or hydroxyl end groups, its transfection efficiency was significantly improved compared with its 5 kDa counterparts (Figure 2b), meaning that for PACE-COOH and PACE-MAE polymers, transfection efficiency increases when the MW increases. This behavior is dramatically different from classic PACE whose transfection efficiency decreases as MW increases, further demonstrating the importance of end groups in transfection efficiency. Although the EE of PACE-COOH increased from 18% to 45% when its MW increased from 5 kDa and 10 kDa, the EE of PACE-MAE actually did not change much (98% at 5 kDa vs. 95% at 10 kDa, Table S2), suggesting that an increase in EE is not the only explanation for the transfection efficiency improvement. Considering the complicated biological steps involved in intracellular mRNA delivery [10], these data suggest that the contribution of PACE end groups and MW are likely synergistic: while the nature of PACE end groups appears to contribute to its capability of complexation with mRNA, the MW also plays a role in complexation, and can be involved in down-stream biological steps, such as endosomal escape and/or mRNA release in the cytosol. Given the synthetic challenges associated with the simultaneous control of both MW and end groups, a new method was needed to obtain polymers with a broader range of properties. Hence, we developed a "top-down" actuation process that exposes the hydroxyl and carboxyl groups in classic PACE, while easily controlling MW.
"Top-down" actuation of PACE for mRNA delivery
Actuated PACE polymers (aPACE) were produced by controlled hydrolysis of the ester backbone. Exposure to air under moderate temperature is expected to provide mild conditions for hydrolysis of PACE, thus decreasing its MW and exposing hydroxyl and carboxyl end groups. To confirm this, we characterized aPACE by NMR spectroscopy and GPC (Figure 3). As expected, NMR analysis of aPACE demonstrated that the actuation process resulted in the exposure of hydroxyl (Figure 3a) and carboxyl (Figure 3b) end groups, as an increase in the area under the hydroxyl group peak (Figure 3a, 58.2 and 58.9 for aPACE and classic PACE, respectively) and the appearance of a clear carboxyl peak at 178 ppm (Figure 3b) in aPACE compared to classic non-actuated PACE were observed. GPC showed that the actuation process decreased the MW of all PACE polymers (Figure 3c). Hydrolysis of polyesters has been well studied [33] and these reactions can usually proceed at high temperatures, high pressures, and/or in the presence of a catalyst. Accordingly, we observed that the actuation could be accelerated when performed at 100°C compared to 37°C (Figure S2). However, using milder temperatures provided for a more reproducible process, that resulted in the desired molecular weights of the aPACE we were looking to achieve.
Figure 3. Characterization of actuated polymers using NMR and GPC.

(a–b) NMR spectra of non-actuated 20 kDa polymer (red line) and the same polymer actuated for 30 days (blue line), zoomed on the hydroxyl group region around 58 ppm (a) and the carboxyl group region around 178 ppm (b). (c) Evolution of weight-average MW of aPACE polymers during the actuation process.
To explore the effect of the actuation protocol on PACE transfection efficiency for mRNA, we tested aPACE produced from different starting MW with different periods of actuation. Self-assembled polyplexes produced from aPACE and mRNA were highly effective at transfecting HEK293 cells, leading to levels of luciferase expression comparable to the positive control, TransIT (Figure 4a–b). Transfection efficiency appeared to be dependent on the actuation time, and the initial MW of the actuated polymer, with an optimal actuation time for each initial MW (Figure 4a, 5 days for the 5 kDa polymer, 10 days for the 10 kDa polymer, and 30 days for the 20 kDa polymer). For these optimized actuation times, all aPACE polymers provided comparable levels of transfection to TransIT, and significantly higher transfection levels compared to their non-actuated counterparts (Figure 2b, p < 0.005). When tested for cytotoxicity at different concentrations, TransIT induced considerable cell death, while for similar amounts of mRNA delivered, all aPACE formulations were non-cytotoxic (Figure 4c). Finally, the accelerated actuation process where 20 kDa polymer was actuated for 6 h at 100°C resulted in similar transfection levels as th e 20 kDa polymer actuated for 30 days at 37°C (Figure 4d). Despite an accelerated actuation process, a similar kinetic profile was observed when performing the actuation at 100°C compared to 37°C: that is, both processes exhibited an increase in transfection efficiency up to an optimum, followed by a drop of activity. This observation suggests that a similar time-dependent process is occurring at both temperatures (Figure S2).
Figure 4. In vitro characterization of aPACE polymers (HEK293 cells).

(a) Luciferase mRNA transfection efficiency depending on the actuation time demonstrating an optimal actuation time for each MW. (b) Transfection efficiency using non-actuated PACE and actuated PACE of different initial MW at their optimal actuation time (5 days for the 5 kDa polymer, 10 days for the 10 kDa polymer, and 30 days for the 20 kDa polymer, **p < 0.005). (c) Cytotoxicity profiles of mRNA:aPACE polyplexes compared to the mRNA:TransIT complexes. (d) Transfection efficiency of aPACE using different temperature of actuation (**p < 0.005). All results are presented as mean ± SD of three independent experiments run in duplicate.
Notably, the MW of these optimized aPACEs were close to each other, ranging from 6 kDa to 8 kDa (Figure 4c). Considering the differences in their transfection efficiency, our data indicates rapid changes in transfection efficiency within a narrow range of aPACE MW, highlighting the value of using a "top-down" actuation approach for screening. As previously mentioned, delivery vehicles should be optimized for carriage of different genetic materials [23]. Hence, a controlled "top-down" actuation process constitutes a straightforward but powerful way to optimize the combination of MW and endgroup for each genetic material. The advantage of this process involves the accuracy in fine-tuning key factors like MW, as well as the simplicity of synthesizing a discrete library of polymers at actuation stages from the same starting material. Overall, our results confirmed that our approach provided an optimal combination of MW and end-group tailored for mRNA delivery and transfection.
To ensure broad efficacy of aPACE, these polymers were also tested for transfection in Daoy cells, a human medulloblastoma cell line, and in SH-SY5Y cells, a human neuroblastoma cell line. As observed in the HEK293 cells, the actuation process significantly increased the transfection efficiency, compared to non-actuated PACE in both cell lines (Figure S3). Finally, as a development benchmark, we evaluated the effect of lyophilization on aPACE:mRNA polyplexes. Trehalose was used as a cryoprotectant, and transfection efficiency was assessed in HEK293 cells after reconstitution of the polyplexes. Lyophilized polyplexes prepared with aPACE were as efficient as fresh polyplexes, even in the absence of cryoprotectant (Figure S4), while the addition of high concentration of trehalose (6%) slightly increased the transfection efficiency.
aPACE for mRNA delivery in vivo
Numerous delivery strategies ranging from commercial transfection reagents to other nanoparticulate systems have recently been used to deliver mRNA in vivo [12, 23, 34, 35]. To evaluate aPACE polymers for delivery of therapeutically relevant mRNA, we investigated their ability to deliver EPO-expressing mRNA in mice. Two aPACE polymers (PACE 5 kDa actuated for 5 days and PACE 10 kDa actuated for 10 days) were tested and compared to the positive control TransIT, which was selected because of its proven effectiveness for mRNA delivery in vivo [12]. PACE 20 kDa actuated for 30 days was not tested, as the formed polyplexes were not stable enough in terms of size, at high concentrations required for in vivo administration. EPO mRNA:aPACE polyplexes were intravenously administered to wild-type mice to deliver a total dose of 20 µg of mRNA, and blood was collected at different time-points after injection to measure EPO levels by ELISA. mRNA polyplexes using optimized aPACE polymers demonstrated high efficacy in the delivery of EPO mRNA, as reflected in subsequent EPO production. Six hours after injection, the non-actuated PACE 5 kDa, which was the best of non-actuated polymers, produced a high level of EPO (530 ng/mL), higher than the positive control TransIT (170 ng/mL). The use of the 10 kDa non-actuated PACE produced a lower EPO level (14 ng/mL) at 6 h, confirming the trend observed in vitro: for the non-actuated polymers, polyplexes formed from polymers with MW higher than 5 kDa provide lower transfection efficiency. The actuated 5 kDa PACE did not significantly increase the EPO production compared to its non-actuated form, but the actuation of the 10 kDa for 10 days at 37°C, in creased the EPO level significantly, to 1,100 ng/mL (Figure 5a, p < 0.0001). In vivo EPO production has been previously studied to evaluate the potency of mRNA delivery systems. The commercial transfection agent, TransIT, has been prior used to deliver mRNA with modified structures [12]: administering a total dose of 0.1 µg mRNA per animal intraperitoneally, the authors reported an increase in EPO levels up to around 10 ng/mL, when measured 6 h after administration. More recently, EPO production has been used as a surrogate to optimize a lipid-based nanoparticle formulation via fractional factorial and definitive screening design [23]. In this study, the optimized formulation provided an EPO production of 7 500 ng/mL, 6 h after IV administration of a total dose of 15 µg of mRNA, which is around seven times higher than what we obtained with our best actuated polymer. However, this formulation was evaluated under different conditions (such as the use of a different strain of mice), and was not evaluated for expression of the mRNA at times longer than 6 h. Here, we observed that, for both TransIT and the aPACE polyplexes, the EPO production peaked at 6 h post-injection (Figure 5b), as previously described [12]. The injection of acetate buffer (vehicle) followed by repeated bleeding induced a slight increase in EPO blood levels over time, but the levels of EPO after administration of 10 kDa actuated PACE polyplexes were significantly higher compared to the levels obtained after administration of TransIT for up to 48 h (Figure 5b, p < 0.001 at 6 h and p < 0.05 at 24 h and 48 h). It has been suggested that the duration of EPO expression might be more important than the maximal EPO expression level to induce a physiological response, such as red blood cell production [36]. EPO delivery using DNA has been explored and provided long-term EPO production, however lethal polycythemia due to uncontrolled production of the protein has been reported [37, 38]. Using our best aPACE polymer (10 kDa actuated for 10 days), we obtained significant EPO production for up to 48 h, which is significantly longer than the blood half-lifetime of free EPO (around 2 h). Finally, blood chemistry (Figure 5c) and histology analysis (Figure 5d, Table S3) demonstrated that aPACE did not induce systemic toxicity, 24 h or 7 days after administration.
Figure 5. In vivo transfection of EPO mRNA using aPACE.

(a) EPO blood concentration 6 h after IV administration of mRNA (20 µg total) using TransIT, 5 kDa non-actuated PACE, 5 kDa aPACE actuated for 5 days, 10 kDa non-actuated PACE, or 10 kDa aPACE actuated for 10 days. Results are presented as mean ± SD of N = 3 animals (****p < 0.0001). (b) Time course of EPO production following IV administration of mRNA (20 µg total) using TransIT, 5 kDa non-actuated PACE, 5 kDa aPACE actuated for 5 days, 10 kDa non-actuated PACE, or 10 kDa aPACE actuated for 10 days. Results are presented as mean ±SD of N = 3 animals (***p < 0.001 and *p < 0.05). (c) Blood chemistry analysis 24 h and 7 days after IV administration of acetate buffer, free mRNA or mRNA:aPACE polyplexes. Results are presented as mean ± SEM of N = 3 animals. (d) Histology analysis of liver and spleen 24 h after IV administration of acetate buffer or mRNA:aPACE polyplexes. Images are representative of N = 3 animals.
CONCLUSION
Gene therapy represents an exciting opportunity for the treatment of numerous chronic diseases. DNA-based treatments are associated with long-term safety risks and lack of ability to control protein expression and mRNA-based therapies address these concerns, but require an efficient delivery method which is safe enough. In this study, we observed that the end group and MW of PACE affects its mRNA transfection efficiency synergistically. Based on this observation, we produced a unique polymeric structure obtained by "top-down" actuation through the controlled hydrolysis of classic PACE terpolymers. The combination of MW and end-groups in the actuated polymers enabled efficient mRNA complexation and transfection in vitro and in vivo, while the low cation density of PACE ensured a low toxicity profile. Actuation of the PACE terpolymer opens the way for new mRNA-based treatments using a biodegradable delivery system that has been engineered to safely augment protein production.
Supplementary Material
Acknowledgments
This work was supported by Alexion Pharmaceuticals Inc. ACK and GTT were supported by fellowships from the US National Insititutes of Health (T32 DK101019 and F32 HL131270, respectively). YW and WMS were partially supported by US National Institutes of Health grant R01 CA149128.
C.J. Cheng was an employee of Alexion Pharmaceuticals Inc. at the time this work was performed.
Footnotes
DATA AVAILABILITY
All the data needed to reproduce the work performed and evaluate the conclusions made are presented in the paper and/or the Supplemental Materials. Additional raw/processed data forms part of an ongoing study and may be requested from the authors.
References
- 1.Harris NM, et al. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacology Biochemistry and Behavior. 2016;150–151:48–56. doi: 10.1016/j.pbb.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang Y, et al. SOD1 nanozyme salvages ischemic brain by locally protecting cerebral vasculature. Journal of Controlled Release. 2015;213:36–44. doi: 10.1016/j.jconrel.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Natarajan G, et al. Nanoformulated copper/zinc superoxide dismutase exerts differential effects on glucose vs lipid homeostasis depending on the diet composition possibly via altered AMPK signaling. Translational Research. 2017;188:10–26. doi: 10.1016/j.trsl.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Efremenko EN, et al. A simple and highly effective catalytic nanozyme scavenger for organophosphorus neurotoxins. Journal of Controlled Release. 2017;247:175–181. doi: 10.1016/j.jconrel.2016.12.037. [DOI] [PubMed] [Google Scholar]
- 5.Jiang Y, et al. Nanoformulation of Brain-Derived Neurotrophic Factor with Target Receptor-Triggered-Release in the Central Nervous System. Advanced Functional Materials. 2018;28(6):1703982. doi: 10.1002/adfm.201703982. n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, et al. SOD1 nanozyme with reduced toxicity and MPS accumulation. Journal of Controlled Release. 2016;231:38–49. doi: 10.1016/j.jconrel.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 7.Carter PJ. Introduction to current and future protein therapeutics: a protein engineering perspective. Experimental cell research. 2011;317(9):1261–1269. doi: 10.1016/j.yexcr.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Weissman D. mRNA transcript therapy. Expert Rev Vaccines. 2015;14(2):265–81. doi: 10.1586/14760584.2015.973859. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto A, et al. Current prospects for mRNA gene delivery. Eur J Pharm Biopharm. 2009;71(3):484–9. doi: 10.1016/j.ejpb.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 10.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov. 2014;13(10):759–80. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 11.Kormann MS, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29(2):154–7. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 12.Kariko K, et al. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther. 2012;20(5):948–53. doi: 10.1038/mt.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoerr I, et al. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30(1):1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Agapov EV, et al. Noncytopathic Sindbis virus RNA vectors for heterologous gene expression. Proc Natl Acad Sci U S A. 1998;95(22):12989–94. doi: 10.1073/pnas.95.22.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitzer M, et al. Sendai virus vectors as an emerging negative-strand RNA viral vector system. J Gene Med. 2003;5(7):543–53. doi: 10.1002/jgm.426. [DOI] [PubMed] [Google Scholar]
- 16.Kreiter S, et al. Tumor vaccination using messenger RNA: prospects of a future therapy. Curr Opin Immunol. 2011;23(3):399–406. doi: 10.1016/j.coi.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Li B, et al. Effects of local structural transformation of lipid-like compounds on delivery of messenger RNA. Sci Rep. 2016;6:22137. doi: 10.1038/srep22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettinger T, et al. Peptide-mediated RNA delivery: a novel approach for enhanced transfection of primary and post-mitotic cells. Nucleic Acids Res. 2001;29(18):3882–91. doi: 10.1093/nar/29.18.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A. 1989;86(16):6077–81. doi: 10.1073/pnas.86.16.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher KJ, Wilson JM. The transmembrane domain of diphtheria toxin improves molecular conjugate gene transfer. Biochem J. 1997;321(Pt 1):49–58. doi: 10.1042/bj3210049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akinc A, et al. Synthesis of Poly(β-amino ester)s Optimized for Highly Effective Gene Delivery. Bioconjugate Chemistry. 2003;14(5):979–988. doi: 10.1021/bc034067y. [DOI] [PubMed] [Google Scholar]
- 22.Jarzebinska A, et al. A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced mRNA Delivery. Angew Chem Int Ed Engl. 2016;55(33):9591–5. doi: 10.1002/anie.201603648. [DOI] [PubMed] [Google Scholar]
- 23.Kauffman KJ, et al. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015;15(11):7300–6. doi: 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- 24.Dong Y, et al. Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo. Nano Lett. 2016;16(2):842–8. doi: 10.1021/acs.nanolett.5b02428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunshine JC, et al. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules. 2011;12(10):3592–600. doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm. 2012;9(11):3375–83. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, et al. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nat Mater. 2012;11(1):82–90. doi: 10.1038/nmat3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams BD, et al. miR-34a Silences c-SRC to Attenuate Tumor Growth in Triple- Negative Breast Cancer. Cancer Res. 2016;76(4):927–39. doi: 10.1158/0008-5472.CAN-15-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui J, et al. Ex vivo pretreatment of human vessels with siRNA nanoparticles provides protein silencing in endothelial cells. Nature Communications. 2017;8(1):191. doi: 10.1038/s41467-017-00297-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuchida E, Osada Y. The role of the chain length in the stability of polyion complexes. Die Makromolekulare Chemie. 1974;175(2):593–601. [Google Scholar]
- 31.Choosakoonkriang S, et al. Biophysical characterization of PEI/DNA complexes. Journal of pharmaceutical sciences. 2003;92(8):1710–1722. doi: 10.1002/jps.10437. [DOI] [PubMed] [Google Scholar]
- 32.Schaffer DV, et al. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnology and bioengineering. 2000;67(5):598–606. doi: 10.1002/(sici)1097-0290(20000305)67:5<598::aid-bit10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 33.Göpferich A. Mechanisms of polymer degradation and erosion. Biomaterials. 1996;17(2):103–114. doi: 10.1016/0142-9612(96)85755-3. [DOI] [PubMed] [Google Scholar]
- 34.Thess A, et al. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol Ther. 2015;23(9):1456–64. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finn JD, et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22(9):2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 36.Elliott S, Pham E, Macdougall IC. Erythropoietins: a common mechanism of action. Exp Hematol. 2008;36(12):1573–84. doi: 10.1016/j.exphem.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Villeval JL, et al. Retrovirus-mediated transfer of the erythropoietin gene in hematopoietic cells improves the erythrocyte phenotype in murine beta-thalassemia. Blood. 1994;84(3):928–33. [PubMed] [Google Scholar]
- 38.Johnston J, et al. Regulated expression of erythropoietin from an AAV vector safely improves the anemia of beta-thalassemia in a mouse model. Mol Ther. 2003;7(4):493–7. doi: 10.1016/s1525-0016(03)00043-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.