

Abstract
Objective
We evaluated the safety, tolerability, and effectiveness of prednisone in patients with ocular myasthenia gravis (OMG) concurrently treated with pyridostigmine.
Methods
Randomized, double-blind, placebo-controlled trial. Participants whose symptoms failed to remit on pyridostigmine were randomized to receive placebo or prednisone, initiated at 10mg every other day and titrated to a maximum of 40mg/day over 16-weeks. Primary outcome measure was treatment failure.
Results
Fewer subjects were randomized than the 88 planned. Of the 11 randomized, 9 completed 16-weeks of double-blind therapy. Treatment failure incidence was 100% (95% CI 48–100%) in the placebo group (n=5) vs. 17% (95% CI 0–64%) in the prednisone group, P = 0.02 (n=6). Median time to sustained minimal manifestation status (MMS) was 14-weeks, requiring an average prednisone dosage of 15mg/day. Adverse events were infrequent and generally mild in both groups.
Conclusions
A strategy of low-dose prednisone with gradual escalation appears to be safe, well tolerated, and effective in treating OMG.
Keywords: ocular myasthenia, prednisone, steroids, clinical trial, neuromuscular
Introduction
Myasthenia gravis (MG) is a generalized disorder that often manifests initially as focal weakness. The most common focal presentation involves weakness of the extraocular muscles, eyelid levators and orbicularis oculi, with symptoms of ptosis and diplopia. The estimated prevalence of MG is approximately 10 per 100,000, and approximately 60% of patients initially present with isolated ocular symptoms1–3. Estimates of the frequency with which these patients progress to develop generalized myasthenia gravis vary widely from 50% to 80%2, 4–11,12.
The goals of treatment for ocular myasthenia gravis (OMG) are to return the individual to a state of clear vision and to prevent the development or limit the severity of generalized myasthenia gravis (GMG). Treatments proposed for OMG include drugs with a purely symptomatic effect such as cholinesterase inhibitors, as well as drugs that suppress the immune system, such as corticosteroids. Proponents of steroids point to the limited efficacy of pyridostigmine, the possibility that chronic cholinesterase-inhibitor therapy may exacerbate the cholinergic deficit in myasthenia13, the potentially greater beneficial effects of prednisone, and the potential for steroids to reduce the risk of progression from ocular to generalized disease. Opponents of steroids emphasize the potential risk of serious side effects and question whether these risks are justified in the setting of purely ocular symptoms.
There has been 1 prior randomized controlled trial (RCT) relevant to the use of steroids in OMG5, 9, 14. This trial, however, did not permit any conclusion regarding the efficacy of steroid therapy, as patients were only treated for 8 days and outcomes were reported solely in terms of the degree of ophthalmoplegia. There have also been 7 non-randomized observational studies15–21, 5 of which suggested a possible benefit of steroids in reducing the risk of progression to GMG15–17, 19, 21 and 2 of which suggested a favorable symptomatic effect20, 21. However, in view of the paucity and limited methodological quality of the available data, controversy persists regarding the optimal approach to the treatment of patients with OMG22–24. The importance of the clinical question and the absence of convincing evidence of efficacy and safety, combined with the equipoise among neuromuscular specialists, provide justification for an RCT to evaluate the safety and efficacy of prednisone in the treatment of OMG14.
Materials and Methods
Study design25
The Efficacy of Prednisone in the Treatment of Ocular Myasthenia (EPITOME) trial was a randomized, double blind, parallel group, placebo-controlled trial of prednisone for treatment of OMG. Following enrollment, all subjects were treated with pyridostigmine for a period of 4–6 weeks, with dosage escalated until efficacy [i.e., minimal manifestation status (MMS)26] or toxicity that could not be mitigated with a selective muscarinic antagonist; the maximum pyridostigmine dosage permitted was 480mg/day. Subjects whose symptoms failed to remit continued to take pyridostigmine and were randomized to either prednisone or placebo for 16 weeks (Stage 1). Following this 16-week period of double-blind therapy, subjects whose symptoms failed to remit were crossed over to treatment with high dosagee prednisone (60mg/day) for a period of 16 weeks (Stage 2). For subjects whose symptoms did remit during Stage 1, study drug was tapered in a double-blind manner during Stage 2 (Figure 1). The institutional review board (IRB) of each participating center approved the study protocol, and all subjects provided written informed consent. The study was registered with clinicaltrials.gov (NCT NCT00995722).
Figure 1. Study Schema.
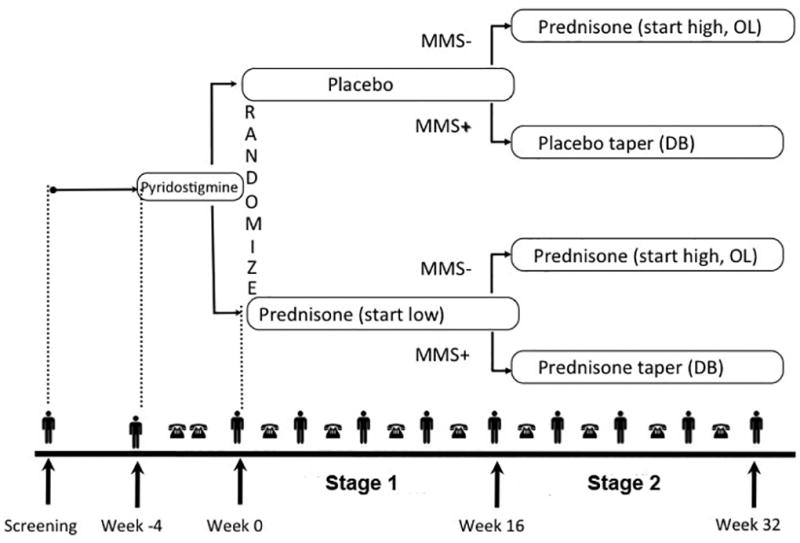
Following enrollment (Week -4), study participants were treated with escalating doses of pyridostigmine for a period of 4 weeks. Those who did not achieve MMS were randomized to receive prednisone or placebo in a double-blind manner (Stage 1). Outcomes were assessed at Week 16, following which those participants who had not yet achieved MMS were crossed over to receive open-label high-dose prednisone (Stage 2). MMS – minimal manifestation status; OL – open label; DB – double-blind.
 indicates in-person visit;
indicates in-person visit;
 indicates telephone visit.
indicates telephone visit.
Study participants
Trial eligibility criteria (Supplementary table S1, available online) aimed to enroll a sample of patients with new or recent onset of purely ocular myasthenia who had not previously received immunosuppressive or modulating therapy and who had not already received prednisone. Patients were enrolled at 6 academic centers across the United States and Canada including the University of Miami (Miami, FL), Duke University (Durham, NC), Kansas University Medical Center (Kansas City, MS), Yale University (New Haven, CT), University of Vermont (Vermont, NH), and University of Toronto (Toronto, Ontario).
Investigational Product
Prednisone and placebo tablets were over-encapsulated by the University of Iowa Research Pharmacy to produce matching capsules. The Clinical Materials Services Unit (CMSU) at the University of Rochester was responsible for investigational product packaging, labeling, and distribution to study sites.
Randomization and Blinding
Participants were randomized 1:1 to receive either prednisone or matching placebo. The randomization schedule was developed by the Muscle Study Group (MSG) Biostatistics Center at the University of Rochester. Randomization was stratified by center and included blocking to ensure approximate balance of treatment group assignments within each center at any point during the trial. Randomization was implemented using a secure web-based module that was accessible by each site; the module returned the appropriate drug pack number following confirmation of subject eligibility. All study staff, other than the programmer in the MSG Biostatistics Center who generated the randomization plan and the CMSU research pharmacist, remained blinded to treatment assignments throughout the study. To promote maintenance of blinding, an evaluator blinded to drug assignment and the occurrence of adverse events performed all efficacy outcome assessments. The treating neurologist was responsible for reviewing safety data.
Interventions
During Stage 1, the trial evaluated a prednisone dosing strategy of starting low and titrating upward as needed based on efficacy and safety/tolerability, rather than a fixed dose of prednisone. Prednisone was started at a dosage of 10mg every other day; it was then initially increased to 10mg/day, then 20mg alternating with 10mg, and so forth, with adjustments in dose made no more frequently than every 2 weeks. The maximum dosage allowed during Stage 1 was 40mg/day. The dose was titrated according to whether MMS had been attained and the presence and nature of adverse events. Dosage escalation was constrained by toxicity that did not respond to appropriate medical intervention. The strategy of starting low (and utilizing an alternate day approach) was motivated by the empiric belief that higher doses of prednisone are more likely to be associated with steroid-induced side effects, especially in patients with relevant comorbidities (e.g. diabetes). During Stage 2, participants who had not achieved MMS during Stage 1 were assigned to receive high dosage prednisone (60mg/day), and the dose was tapered thereafter based on efficacy and safety/tolerability. The dose was initially reduced to 50mg/day, then 40mg/day, and so forth down to 10mg/day, with adjustments made every 2 weeks. After 2 weeks at 10mg/day, the dose could be reduced to 10mg every other day, the lowest dosage permitted during open-label treatment.
Outcomes
The schedule of in-person visits and telephone contacts is outlined in Figure 1. The primary outcome measure was treatment failure, defined as failure to achieve sustained MMS26, progression to GMG, or toxicity leading to discontinuation of study drug by Week 16. Sustained MMS was defined as the appearance of MMS that was maintained across 2 consecutive in-person evaluations 4 weeks apart. Progression to GMG was based on a clinical assessment by the treating neurologist that included MG-specific manual muscle testing27 and careful inquiry about the presence of swallowing or breathing symptoms that could be attributed to MG. Secondary outcome measures included the time to sustained MMS, change in ocular Quantitative Myasthenia Gravis (QMG) score25, changes in quality of life as measured by the National Eye Institute Visual Function Questionnaire (NEI-VFQ-25)28, the 10-item neuro-ophthalmological supplement to the NEI-VFQ-2529, and the MG-Qol-1530, each administered at baseline and Week 16, as well as the occurrence of individual adverse events. To enhance detection of steroid-related side effects (even if not reported subjectively), all study participants underwent glucose tolerance tests (or hemoglobin A1c measurement for known diabetics) as well as bone dual energy x-ray absorptiometry (DEXA) scans and ophthalmological examinations (for cataracts and glaucoma) prior to randomization and again at the time of study completion. All outcome assessments were performed at least 12 hours after the last dose of pyridostigmine.
Sample size
Preliminary studies indicated that 75% of OMG patients would achieve remission on prednisone plus pyridostigmine (after failing to remit on pyridostigmine alone), compared to 31% on pyridostigmine alone20. Sample size estimates were based on the assumptions that (a) fewer than 75% of the prednisone treated participants would achieve MMS, i.e. that at least 25% would experience treatment failure; (b) that < 31% of the placebo treated group would remit, i.e. ≥ 69% would experience treatment failure; and (c) the rate of adverse events requiring drug discontinuation would be ~10% and, as a result, ≥ 35% (≥ 25% + 10%) of subjects in the prednisone group would experience treatment failure. A total sample size of 80 subjects (40 per group) was determined to provide at least 80% power to detect a group difference of 30%–35% in the incidence of treatment failure using a chi-square test and a 5% significance level, as long as the proportion of treatment failures in the placebo group was at least 75%. We planned to enroll 88 subjects to account for a projected loss-to-follow-up rate of ≤ 10%.
Statistical methods
The Fisher exact test was used to compare the incidence of treatment failure in the treatment groups. All randomized subjects were included in the analysis in accordance with the intention-to-treat principle. Analysis of covariance was used to compare mean values of secondary outcome variables (changes in ocular QMG score and quality of life scores) between treatment groups at Week 16, adjusting for the baseline value of the outcome variable. A significance level of 5% (two-tailed) was used for hypothesis testing.
Safety monitoring
An independent data and safety monitoring board (DSMB), comprising 2 neurologists and a statistician, approved the study protocol and periodically reviewed study data for participant safety, study conduct, and progress. An independent and blinded medical monitor reviewed all serious adverse events (SAEs).
Results
Participants
The trial was open for enrollment for 34 months between January 2011 and October 2013 at 9 clinical centers in the United States and Canada. The trial was closed early due to slow participant accrual. This decision was made by the Trial Steering Committee (with approval of the DSMB) while blinded to treatment group and study results. One hundred forty-five patients were approached for participation; 23 declined to participate, and 107 failed pre-screening. Among the remaining 15 patients assessed for eligibility, 11 were randomized, 6 (55%) to prednisone and 5 (45%) to placebo. One of the participants randomized to placebo withdrew consent prior to receipt of study drug, but was included in the analysis in accordance with the intention-to-treat principle. One participant stopped treatment at Week 12 due to early trial closure. The remaining 9 participants completed 16 weeks of double-blind treatment (Figure 2). There were no substantial differences between treatment groups with respect to baseline characteristics (Table 1).
Figure 2. Participant Flow for Double-Blind Phase.
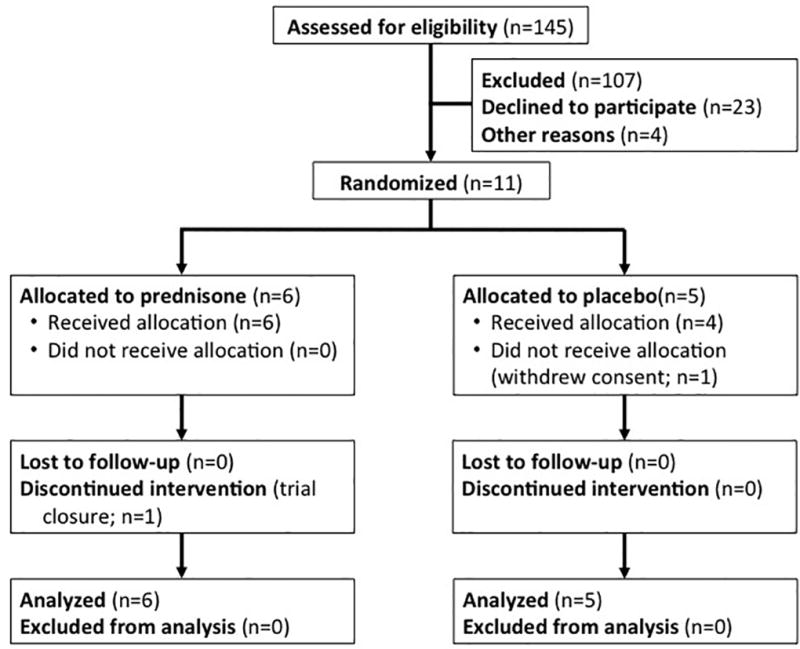
Of the 145 patients assessed for eligibility, 11 were randomized, and 10 received study drug. No participants were lost to follow-up, and all 11 participants were included in the primary analysis.
Table 1.
Pre-Randomization Characteristics
| Placebo (n = 5) |
Prednisone (n = 6) |
|
|---|---|---|
|
| ||
| Men, n (%) | 2 (40%) | 4 (67%) |
|
| ||
| Age (years), mean (SD) | 62 (9) | 64 (18) |
|
| ||
| White, n (%) | 4 (80%) | 4 (67%) |
|
| ||
| Disease duration (months), median (range) | 5 (4–17) | 7.5 (1–18) |
|
| ||
| Diagnostic tests, n (%) | ||
| AChR Antibodies | 3 (60%) | 2 (33%) |
| Single fiber EMG | 2 (40%) | 2 (33%) |
| Ice test | 1 (20%) | 3 (50%) |
|
| ||
| Pyridostigmine dosage (mg/day), median (range) | 360 (240–480) | 330 (0–480) |
|
| ||
| Medical history, n (%) | ||
| Impaired glucose tolerance | 2 (40%) | 2 (40%) |
| Diabetes | 0 | 0 |
| Glaucoma | 0 | 0 |
| Cataracts | 3 (60%) | 2 (33%) |
| Osteopenia or osteoporosis | 3 (60%) | 2 (33%) |
|
| ||
| MG-QoL-15 score, median (range) | 3 (0–13) | 6 (2–31) |
|
| ||
| NEI-VFQ-25 score, median (range) | 87 (82–100) | 85 (72–91) |
|
| ||
| NEI-VFQ-25 10-item neuro-ophthalmological supplement score, median (range) | 61 (45–80) | 68 (60–91) |
|
| ||
| Ocular QMG score, median (range) | 6 (3–11) | 6.5 (3–10) |
Efficacy
No subjects progressed to GMG or discontinued study drug due to toxicity. All 5 placebo-treated subjects (100%) experienced treatment failure by failing to achieve sustained MMS (95% CI 48–100%), compared to 1 of 6 subjects (17%) in the prednisone-treated group (95% CI 0–64%) (P = 0.02, Fisher exact test). The 1 prednisone-treated patient who did not achieve MMS was withdrawn at week 16 due to early trial closure. The prednisone-treated subjects achieved MMS at Weeks 4 (n=1), 8 (n=2), 12 (n=1), and 16 (n=1). The median time to sustained MMS was 14 weeks. The median (range) prednisone dosage at the time of sustained MMS was 15mg/day (15–25mg/day). Per protocol, study subjects who achieved sustained MMS successfully tapered their prednisone dosage to a median (range) dose of 10mg/day (10–15mg/day) without relapse of symptoms. Observed mean responses on the secondary outcome measures were better in the prednisone group than in the placebo group, but the group differences were not statistically significant (Table 2). Individual subject responses are displayed in Figure 3.
Table 2.
Secondary Outcome Measures at End of Double-Blind Treatment
| Outcome Measure | Prednisone | Placebo | Rx Effect |
95% CI | p-value |
|---|---|---|---|---|---|
| MG-QoL-15 score 1 | −6.30 | −2.50 | −3.81 | −9.37 to 1.75 | 0.15 |
| NEI-VFQ-25 score 2 | 10.70 | 4.14 | +6.56 | −2.59 to 15.71 | 0.13 |
| 10-item supplement 2 | 15.28 | −1.70 | + 16.98 | −9.22 to 43.17 | 0.16 |
| Ocular QMG score 3 | −2.25 | −0.05 | −2.20 | −7.20 to 2.80 | 0.37 |
Values presented are mean changes from baseline to Week 20, treatment effects (prednisone – placebo), 95% confidence intervals (CI), and P-values, all obtained from an analysis of covariance model that adjusts for the baseline value of the outcome variable.
MG-Qol-15 score ranges from 0–60, where 0 is normal, and a reduction in score represents an improvement
NEI-VFQ-25 and the 10-item neuro-ophthalmological supplement scores range from 0–100, where 100 is normal, and an increase in score represents an improvement.
Ocular QMG score ranges from 0–15, where 0 is normal, and a reduction in score represents an improvement.
Figure 3. Secondary Outcome Measures.
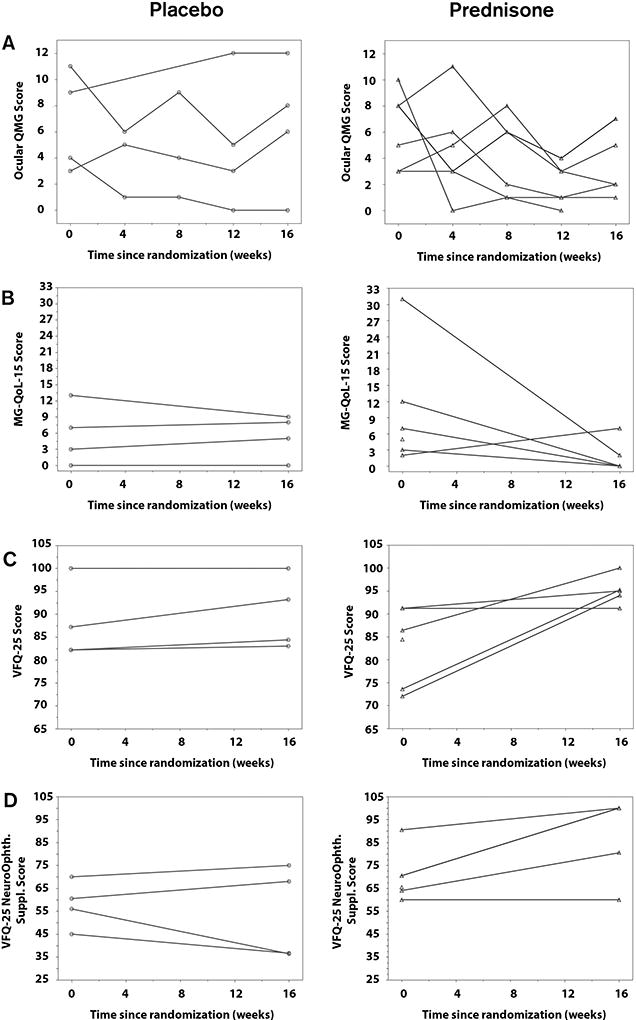
A. Ocular QMG scores at each time point between randomization (Week 0) and the end of the double-blind phase of the trial (Week 16); scores range from 0 (normal) to a maximum of 15. B. MG-QoL-15 scores at randomization and Week 16; scores range from 0 (normal) to a maximum of 60. C. NEI-VFQ-25 scores at randomization and Week 16; scores range from 0 to 100 (normal). D. NEI-VFQ-25 10-item neuro-ophthalmological supplement scores at randomization and Week 16; scores range from 0 to 100 (normal).
Open-label high-dosage prednisone
Three of 5 placebo-treated participants switched to high-dosage prednisone (60mg/day) with rapid taper. Sustained MMS was attained in 2 subjects within 4 and 8 weeks, respectively. Of the other 2 participants randomized to receive placebo, 1 never received study drug and the other was withdrawn early due to study closure.
Safety
Thirty adverse events (AEs) were reported during the double-blind phase of the study, 8 of which led to a reduction in dose of study medication (Table 3). Twenty-two of these AEs occurred in the prednisone group, and 8 occurred in the placebo group. Four serious adverse events were reported during the course of the trial, 3 in the placebo group and 1 in the prednisone group, but none were judged to be related to study medication. Based on two-hour glucose tolerance tests and DEXA bone scans, there were no new diagnoses of impaired glucose tolerance, diabetes, or osteopenia/osteoporosis during the course of the trial. Weight gain in 2 prednisone-treated participants during the double-blind phase of the trial prompted a reduction in the dose of study drug, but both participants still achieved sustained MMS, 1 by Week 12 and 1 by Week 20.
Table 3.
Adverse Events Following Randomization
| Placebo | Prednisone | |
|---|---|---|
| Gastrointestinal | ||
| Constipation | 0 | 1 |
| Diarrhea | 1 | 2 * |
| Flatulence | 0 | 1 |
| Nausea | 0 | 2 |
| Dysphagia | 0 | 1 |
| Neurological | ||
| Agitation | 0 | 1 * |
| Anxiety | 1 | 0 |
| Blepharospasm | 0 | 1 |
| Blurred vision | 0 | 1 |
| Headaches | 0 | 1 |
| Insomnia | 1 * | 0 |
| Eye fasciculations | 0 | 1 |
| Face fasciculations | 0 | 1 |
| Cardiovascular | ||
| Arrhythmia | 0 | 1 |
| Conduction disorder | 1 | 0 |
| Palpitations | 1 * | 0 |
| Sinus tachycardia | 1 * | 0 |
| Respiratory | ||
| Bronchitis | 0 | 1 |
| Endocrine/Metabolic | ||
| Impaired glucose tolerance | 0 | 1 * |
| Weight gain | 0 | 2 * |
| Miscellaneous | ||
| Dizziness | 1 | 0 |
| Edema | 0 | 2 |
| Hematoma | 0 | 1 |
| Myalgia/Cramping | 0 | 1 |
| Eye pain (poked in eye) | 1 | 0 |
| TOTAL | 8 | 22 |
Indicates that AE led to a reduction in dose of study medication.
Dose was reduced because of diarrhea in only 1 of the 2 patients in the prednisone group.
Discussion
The major limitation of this study is the small sample size, which was a function of slow patient enrollment (approximately 1 eligible patient identified every 2 months, with approximately 1 patient enrolled every 3 months across the 9 study centers). Although initially planned as a 5 center clinical trial, a decision was made soon after enrollment opened to expand the number of enrolling centers. The reasons for slow patient accrual include strong patient preferences (either to be on steroids or not to take steroids), the availability of prednisone outside of the trial, prednisone initiation prior to referral to study centers, and logistics [patient travel, number of visits, and the complexity of coordinating availability of 2 physicians (treating neurologist and blinded evaluator) for each study visit]. Slow recruitment appears to be a common problem in MG RCTs, as many studies have terminated prematurely for this reason31.
Notwithstanding the small number of participants enrolled in this trial, the primary efficacy analysis demonstrated a clinically and statistically significant benefit of prednisone compared to placebo. Five of 6 participants (83%) in the prednisone group achieved the primary end-point of sustained MMS at a median of 14 weeks on a median prednisone dosage of 15mg/day, compared to none of the 5 participants in the placebo group. The observation that none of the patients in the placebo group achieved MMS may, at least in part, be a function of an important element of study design – that all patients were initially treated with pyridostigmine and that those who responded were not randomized to receive study drug (i.e. those ”patients who responded to pyridostigmine alone were not randomized). Secondary outcome measures also favored the prednisone group (although not reaching statistical significance). The estimated effect of prednisone of 3.8 points in mean MG-QoL score would be considered clinically meaningful, but the effect is imprecisely estimated in this small trial
It is of interest that the 2 of the 3 placebo-treated patients who switched over to high-dose prednisone achieved MMS within 4 and 8 weeks, respectively. While EPITOME was designed to shed light on the utility of high-dose prednisone, the small number of subjects enrolled, the even smaller number switched over to high dose prednisone, and the unblinded nature of the data make it difficult to draw conclusions about the relative utility of this treatment approach.
Adverse events were typically mild and did not generally require drug discontinuation. However, it should be noted that the duration of steroid treatment was relatively short (maximum of 36 weeks), and so the long-term safety of low-dose steroids in this population remains unclear. Nevertheless, at the dosage needed to achieve clinical improvement, prednisone appears to be safe and well tolerated. These data support the strategy of treating patients with OMG whose symptoms have failed to remit on pyridostigmine therapy alone with an initial low-dosage of prednisone followed by a gradual titration until efficacy is achieved.
Supplementary Material
Acknowledgments
The study was sponsored by the FDA Orphan Products Development Program (1R01FD003710). This publication was supported, in part, by an Institutional Clinical and Translational Science Award, NIH/NCATS Grant Number UL1TR000001 to Richard J. Barohn MD. We are grateful to members of the DSMB (John Kissel MD, Henry Kaminski MD, and Gary Cutter, PhD) as well as the independent medical monitor (Gregory Martin, MD).
MSG EPITOME Study Group:
University of Miami: Michael Benatar MD, PhD (PI), Alexandre Waltz (project manager), Sara-Claude Michon PhD (coordinator)
University of Rochester: Rabi Tawil (co-PI), Alexis Smirnow (project manager), Farheen Hussain (project manager), Patricia C Smith (project manager), Nuran Dilek (data manager), Michael P McDermott PhD (biostatistician), Joanne Janciuras (biostatistical programmer)
Clinical Material Services Unit (CMSU): Cornelia Kamp, MBA (Executive Director, Strategic Initiatives), Tim Hackett (Director, Regulatory and Technical Affairs), Pat Bolger, R.Ph., MBA (Director, Clinical and Business Affairs), Joan Woodcook, MS (Associate Director, Quality Affairs)
Kansas University Medical Center: Richard J Barohn MD (site-PI), April L McVey MD (blinded evaluator), Mazen M Dimachkie (sub-I), Mamatha Pasnoor MD (blinded evaluator), Thomas J Whittaker JD MD (sub-I), Laura Herbelin (sub-I), Gabrielle Rico (coordinator)
University of Vermont: Michael Hehir MD (site-PI), Waqar Waheed MD (blinded evaluator), Shannon Lucy (coordinator)
Yale University: Richard J Nowak MD, MS (site-PI), Daniel B Dicapua MD (blinded evaluator), Jonathan M Goldstein MD (blinded evaluator), Benison Keung MD (blinded evaluator), Joan Nye BS (coordinator)
University of Toronto: Hans Katzberg (site-PI), Vera Bril (co-I), Carolina Barnett Tapia MD (blinded evaluator), Ari Breiner MD (blinded evaluator), Seint Kokokyi BSc MSc (coordinator)
University of Virginia: Ted Burns MD (site-PI), Guillermo Solorzano MD (co-I), Amruta Joshi (coordinator)
University at Buffalo: Gil I Wolfe, MD (site-PI), Nicholas J Silvestri MD (blinded evaluator), Kara Patrick (coordinator)
University of Texas Southwestern Medical Center: Srikanth Muppidi MD (site-PI), Sharon Nations MD (blinded evaluator), Steve Hopkins (coordinator)
Duke University Medical Center: Vern C Juel MD (site-PI), Jeffrey T Guptill MD (blinded evaluator), Lisa D Hobson MD (co-I), Janice M Massey MD (co-I), Katherine Beck RN (coordinator)
ABBREVIATIONS
- AE
Adverse Event
- CI
Confidence Interval
- CMSU
Clinical Material Services Unit
- DEXA
Dual Energy X-ray Absorptiometry
- DSMB
Data Safety Monitoring Board
- EPITOME
Efficacy of Prednisone for Treatment of Ocular Myasthenia
- GMG
Generalized Myasthenia Gravis
- IRB
Institutional Review Board
- MG-QoL-15
Myasthenia Gravis Quality of Life – 15 item Scale
- MMS
Minimal Manifestation Status
- MSG
Muscle Study Group
- QMG
Quantitative Myasthenia Gravis Score
- NEI-VFQ-25
National Eye Institute Visual Function Questionnaire 25
- NCT
Clinicaltrials.gov Identifier
- OMG
Ocular Myasthenia Gravis
- RCT
Randomized Controlled Trial
- SAE
Serious Adverse Event
Footnotes
Statistical Analysis. Performed by Michael P McDermott, PhD (University of Rochester)
Conflicts of Interest
None
References
- 1.Ferguson FR, Hutchinson EC, Liversedge LA. Myasthenia gravis; results of medical management. Lancet. 1955;269:636–639. doi: 10.1016/s0140-6736(55)92480-6. [DOI] [PubMed] [Google Scholar]
- 2.Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle & nerve. 2008;37:141–149. doi: 10.1002/mus.20950. [DOI] [PubMed] [Google Scholar]
- 3.Sanders D. Approach to Diseases of the Neuromuscular Junction. In: Tawil R, Venance S, editors. Neuromuscular Disorders. First. Vol. 2011. John Wiley & Sons Ltd; pp. 113–117. [Google Scholar]
- 4.Aiello I, Pastorino M, Sotgiu S, et al. Epidemiology of myasthenia gravis in northwestern Sardinia. Neuroepidemiology. 1997;16:199–206. doi: 10.1159/000109688. [DOI] [PubMed] [Google Scholar]
- 5.Benatar M, Kaminski H. Medical and surgical treatments for ocular myasthenia. Cochrane Database Syst Rev. 2006 doi: 10.1002/14651858.CD005081.pub2. [DOI] [PubMed] [Google Scholar]
- 6.Bever CT, Aquino AV, Penn AS, Lovelace RE, Rowland LP. Prognosis of ocular myasthenia. Annals of neurology. 1983;14:516–519. doi: 10.1002/ana.410140504. [DOI] [PubMed] [Google Scholar]
- 7.Casetta I, Fallica E, Govoni V, Azzini C, Tola M, Granieri E. Incidence of myasthenia gravis in the province of Ferrara: a community-based study. Neuroepidemiology. 2004;23:281–284. doi: 10.1159/000080093. [DOI] [PubMed] [Google Scholar]
- 8.Lavrnic D, Jarebinski M, Rakocevic-Stojanovic V, et al. Epidemiological and clinical characteristics of myasthenia gravis in Belgrade, Yugoslavia (1983–1992) Acta Neurologica Scandinavica. 1999;100:168–174. doi: 10.1111/j.1600-0404.1999.tb00733.x. [DOI] [PubMed] [Google Scholar]
- 9.Mount F. Corticotropin in Treatment of Ocular Myasthenia - A Controlled Clinical Trial. Archives of neurology. 1964;11:114–124. doi: 10.1001/archneur.1964.00460200010002. [DOI] [PubMed] [Google Scholar]
- 10.Phillips LH, Torner JC, Anderson MS, Cox GM. The epidemiology of myasthenia gravis in central and western Virginia. Neurology. 1992;42:1888–1893. doi: 10.1212/wnl.42.10.1888. [DOI] [PubMed] [Google Scholar]
- 11.Robertson N, Deans J, Compston D. Myasthenia gravis: a population based epidemiological study in Cambridgeshire, England. Journal of Neurology, Neurosurgery & Psychiatry. 1998;65:492–496. doi: 10.1136/jnnp.65.4.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Incidence of myasthenia gravis in the Emilia-Romagna region: a prospective multicenter study. Emilia-Romagna Study Group on Clinical and Epidemiological Problems in Neurology. Neurology. 1998;51:255–258. doi: 10.1212/wnl.51.1.255. [DOI] [PubMed] [Google Scholar]
- 13.Brenner T, Hamra-Amitay Y, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:214–222. doi: 10.1096/fj.02-0609com. [DOI] [PubMed] [Google Scholar]
- 14.Benatar M, Kaminski HJ. Evidence report: the medical treatment of ocular myasthenia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2007;68:2144–2149. doi: 10.1212/01.wnl.0000263481.14289.90. [DOI] [PubMed] [Google Scholar]
- 15.Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Archives of neurology. 2003;60:243–248. doi: 10.1001/archneur.60.2.243. [DOI] [PubMed] [Google Scholar]
- 16.Mee J, Paine M, Byrne E, King J, Reardon K, O'Day J. Immunotherapy of ocular myasthenia gravis reduces conversion to generalized myasthenia gravis. J Neuroophthalmol. 2003;23:251–255. doi: 10.1097/00041327-200312000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. Journal of the neurological sciences. 2004;217:131–133. doi: 10.1016/j.jns.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Papapetropoulos TH, Ellul J, Tsibri E. Development of generalized myasthenia gravis in patients with ocular myasthenia gravis. Archives of neurology. 2003;60:1491–1492. doi: 10.1001/archneur.60.10.1491-b. [DOI] [PubMed] [Google Scholar]
- 19.Sommer N, Sigg B, Melms A, et al. Ocular myasthenia gravis: response to long-term immunosuppressant treatment. Journal of Neurology, Neurosurgery and Psychiatry. 1997;62:156–162. doi: 10.1136/jnnp.62.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhanushali MJ, Wuu J, Benatar M. Treatment of ocular symptoms in myasthenia gravis. Neurology. 2008;71:1335–1341. doi: 10.1212/01.wnl.0000327669.75695.38. [DOI] [PubMed] [Google Scholar]
- 21.Mittal MK, Barohn RJ, Pasnoor M, et al. Ocular myasthenia gravis in an academic neuro-ophthalmology clinic: clinical features and therapeutic response. Journal of clinical neuromuscular disease. 2011;13:46–52. doi: 10.1097/CND.0b013e31821c5634. [DOI] [PubMed] [Google Scholar]
- 22.Agius M. Treatment of ocular myasthenia with corticosteroids: yes. Archives of neurology. 2000;57:750–751. doi: 10.1001/archneur.57.5.750. [DOI] [PubMed] [Google Scholar]
- 23.Hachinski V. Treatment of ocular myasthenia. Archives of neurology. 2000;57:753. doi: 10.1001/archneur.57.5.753. [DOI] [PubMed] [Google Scholar]
- 24.Kaminski H, Daroff R. Treatment of ocular myasthenia: steroids only when compelled. Archives of neurology. 2000;57:752–753. doi: 10.1001/archneur.57.5.752. [DOI] [PubMed] [Google Scholar]
- 25.Benatar M, Sanders D, Wolfe G, McDermott M, Tawil R. Design of the Efficacy of Prednisone in the Treatment of Ocular Myasthenia (EPTIOME) Trial. Annals of the New York Academy of Science. 2012;1275:17–22. doi: 10.1111/j.1749-6632.2012.06780.x. [DOI] [PubMed] [Google Scholar]
- 26.Jaretzki A, Barohn R, Ernstoff R, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55:16–23. doi: 10.1212/wnl.55.1.16. [DOI] [PubMed] [Google Scholar]
- 27.Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann N Y Acad Sci. 2003;998:440–444. doi: 10.1196/annals.1254.057. [DOI] [PubMed] [Google Scholar]
- 28.Mangione CM. [Accessed 25 December 2008];Version 2000. The National Eye Institute 25-Item Visual Function Questionnaire (VFQ-25) [online] Available at: http://www.nei.nih.gov/resources/visionfunction/manual_cm2000.pdf.
- 29.Raphael BA, Galetta KM, Jacobs DA, et al. Validation and test characteristics of a 10-item neuro-ophthalmic supplement to the NEI-VFQ-25. Am J Ophthalmol. 2006;142:1026–1035. doi: 10.1016/j.ajo.2006.06.060. [DOI] [PubMed] [Google Scholar]
- 30.Burns TM, Grouse CK, Wolfe GI, Conaway MR, Sanders DB. The MG-QOL15 for following the health-related quality of life of patients with myasthenia gravis. Muscle & nerve. 2011;43:14–18. doi: 10.1002/mus.21883. [DOI] [PubMed] [Google Scholar]
- 31.Benatar M, Sanders DB, Burns TM, et al. Recommendations for myasthenia gravis clinical trials. Muscle & nerve. 2012;45:909–917. doi: 10.1002/mus.23330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.