

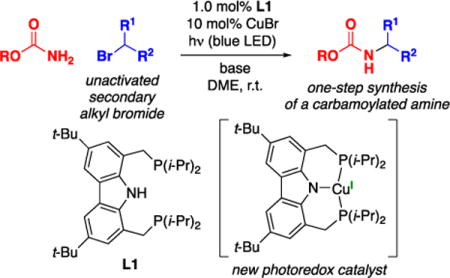
Abstract
Despite the long history of SN2 reactions between nitrogen nucleophiles and alkyl electrophiles, many such substitution reactions remain out of reach. In recent years, efforts to develop transition-metal catalysts to address this deficiency have begun to emerge. In this report, we address the challenge of coupling a carbamate nucleophile with an unactivated secondary alkyl electrophile to generate a substituted carbamate, a process that has not been achieved effectively in the absence of a catalyst; the product carbamates can serve as useful intermediates in organic synthesis as well as bioactive compounds in their own right. Through the design and synthesis of a new copper-based photoredox catalyst, bearing a tridentate carbazolide/bisphosphine ligand, that can be activated upon irradiation by blue-LED lamps, we can achieve the coupling of a range of primary carbamates with unactivated secondary alkyl bromides at room temperature. Our mechanistic observations are consistent with the new copper complex serving its intended role as a photoredox catalyst, working in conjunction with a second copper complex that mediates C–N bond formation in an out-of-cage process.
Graphical abstract
INTRODUCTION
Because primary amines play important roles in many areas of science, including biology, medicinal chemistry, and materials science, the selective synthesis of primary amines is an important challenge in organic chemistry.1 Perhaps the most obvious route, the nucleophilic substitution of an alkyl electrophile by ammonia (a commodity chemical) can be problematic due to issues such as over-alkylation with powerful electrophiles and insufficient reactivity with weak electrophiles.2 Consequently, a number of useful alternative approaches have been developed, including the Gabriel synthesis, which provides direct access to protected primary amines via an SN2 reaction between phthalimide anion and a suitable alkyl electrophile (top of Figure 1).3
Figure 1.
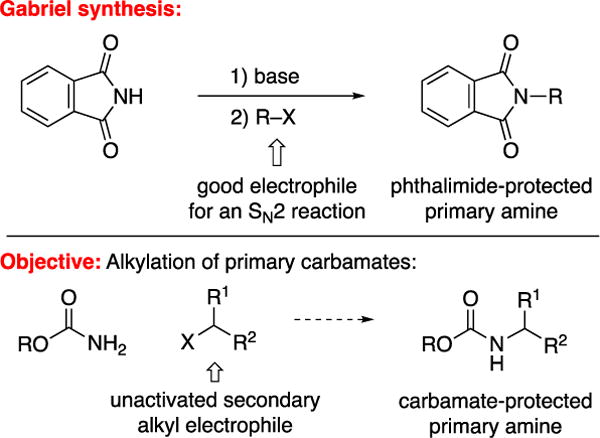
Selective mono-alkylation of nitrogen nucleophiles to generate protected primary amines.
Carbamates are among the most widely used protecting groups for amines,4 as well as being important target molecules in their own right (e.g., Ritonavir™ and carbamate insecticides).5 However, to the best of our knowledge, there are no reports of the N-alkylation of primary carbamates by unactivated secondary alkyl electrophiles (bottom of Figure 1).6 In view of our previous success in achieving the alkylation of primary carboxamides via photoinduced copper catalysis, we envisioned that it might be possible to similarly catalyze the selective mono-alkylation of a primary carbamate7,8,9 Unfortunately, the conditions that we had developed for the alkylation of carboxamides provides only a modest yield when BocNH2 is used as the nucleophile (eq 1). We therefore sought to develop a method that addresses three key shortcomings: the inefficiency of the alkylation, the need for high-energy ultraviolet irradiation, and the use of an excess of the electrophile (the more valuable coupling partner).
 |
(1) |
We have suggested that the photoinduced, copper-catalyzed alkylation of carboxamides may proceed through the pathway outlined in Figure 2.7,10 According to this hypothesis, irradiation of a copper(I)–nucleophile complex (A) generates an excited-state species (B) that engages in electron transfer with the organic electrophile (R–X) to furnish a copper(II)–nucleophile complex (C) and an organic radical (R•). These two intermediates couple to form the desired product (R–Nu) and a copper(I)–halide complex (D), which undergoes ligand substitution to regenerate the copper(I)–nucleophile complex (A).
Figure 2.
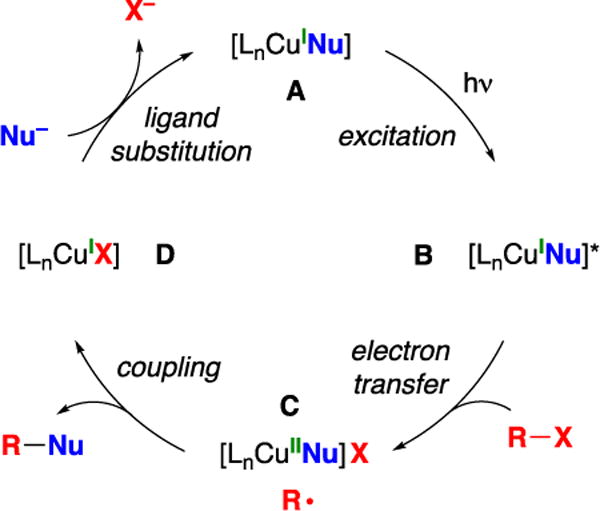
Outline of one of the possible catalytic cycles for a photoinduced, copper-catalyzed coupling reaction.
According to this mechanism, a copper–nucleophile complex is photoexcited and then engages in electron transfer to the electrophile (Figure 2; A → B → C). The photophysical properties of the copper–nucleophile complex are dependent on the structure of the nucleophile, and not all complexes will have the necessary reactivity profile to participate in this process. Therefore, to expand the scope of photoinduced, copper-catalyzed couplings, we sought to circumvent this limitation by developing a copper complex that can serve as a photoredox catalyst (Figure 3: E → F → G → E), ideally using visible light, in the presence of another copper catalyst (A) that facilitates the desired bond formation (A → C → D → A). In contrast to the pathway outlined in Figure 2, wherein R–Nu bond formation might occur through an in-cage pathway, the mechanism depicted in Figure 3 requires out-of-cage bond formation.11
Figure 3.
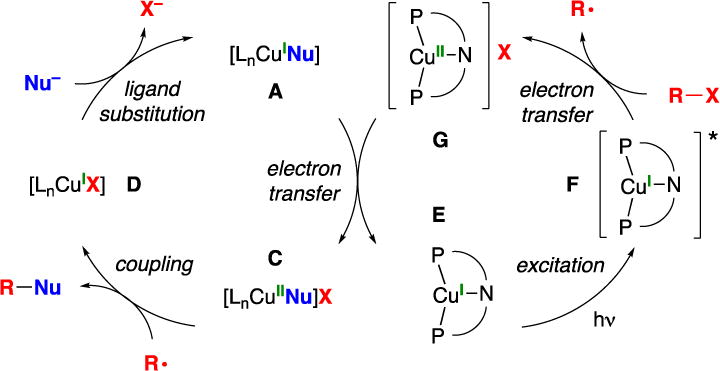
Outline of one of the possible catalytic cycles for a photoinduced, copper-catalyzed coupling reaction.
In this report, we describe the design and synthesis of a copper complex (1) that has enabled us to achieve this objective in the context of the development of a copper-catalyzed method for the selective monoalkylation of primary carbamates by unactivated secondary alkyl halides induced by irradiation with blue-LED lamps (eq 2).
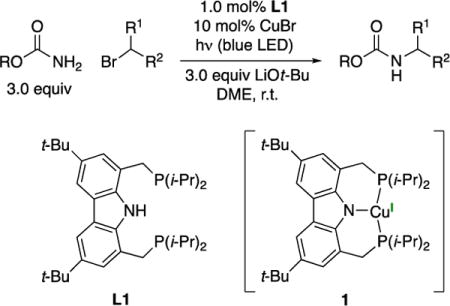 |
(2) |
RESULTS AND DISCUSSION
Design, synthesis, and properties of a new copper-based photocatalyst
We have previously reported that three-coordinate copper complexes that bear two monodentate phosphines and one carbazolide ligand (2a and 2b) can react with aryl and alkyl halides to form C–N bonds upon irradiation with CFL or blue-LED lamps.12 We hypothesized that, by linking these monodentate ligands into a tridentate ligand (L), we might be able to access a copper complex that would be stable to ligand substitution under cross-coupling conditions, while serving as an effective photocatalyst that could initiate new copper-catalyzed reactions upon exposure to visible light, independent of the nucleophile.
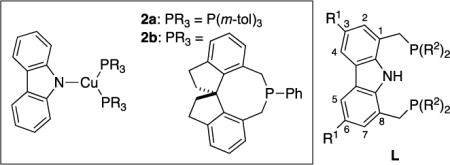 |
We anticipated that the design embodied in ligand L would minimize deleterious side reactions, i.e., the substitution at the 1 and 8 positions of the carbazole would impede functionalization at nitrogen13 and the substitution at the 3 and 6 positions would preclude the addition of radicals at these sites.14 Although several carbazole-based PNP pincer ligands were known when we began this investigation,15 at the time ligand L1 was not, although it has since been described by Gade.16
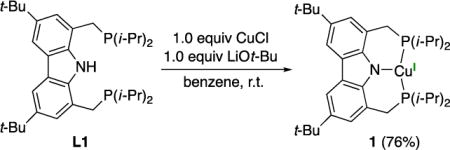 |
(3) |
Treatment of ligand L1 with CuCl and LiOt-Bu in benzene at room temperature affords complex 1 in 76% yield as a colorless solid after recrystallization (eq 3), which we have characterized by X-ray crystallography (Figure 4). The complex adopts a planar geometry, as in the case of the related complexes (2a and 2b), with a Cu–N bond distance of 1.974 Å.17
Figure 4.
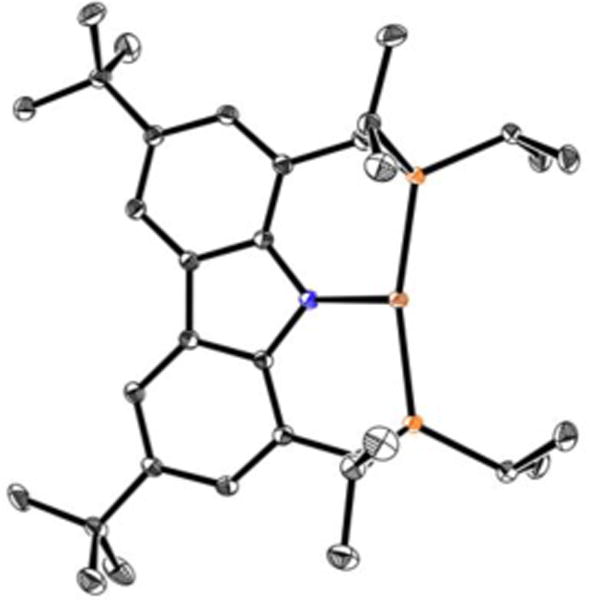
X-ray crystal structure of copper complex 1 (thermal ellipsoids are shown at 50% probability; hydrogen atoms have been omitted for clarity).
Although a solution of copper complex 1 does not absorb strongly at wavelengths greater than 400 nm, there is overlap between its absorption spectrum and the emission spectrum of a blue LED lamp (Figure 5). A cyclic voltammogram of complex 1 shows a reversible wave at ~0.6 V versus saturated calomel electrode (SCE), corresponding to the Cu(I)/Cu(II) redox couple (Figure 6). Taking into account E00 (3.1 eV), estimated from the excitation and emission spectra (see the Supporting Information), and the ground-state redox couple, the excited state of complex 1 has a predicted reduction potential of –2.5 V versus SCE, similar to that of complex 2a (–2.6 V vs. SCE).12a
Figure 5.
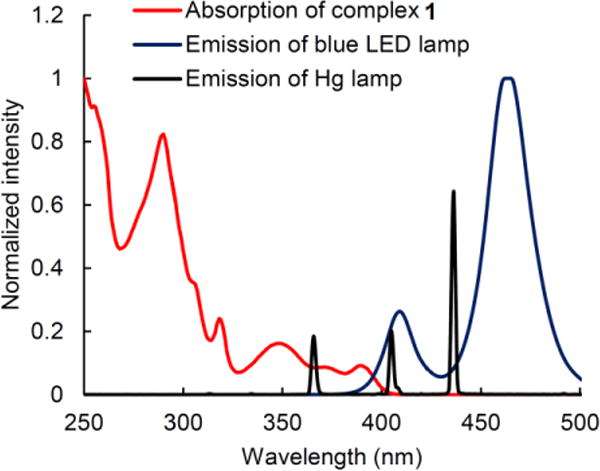
Absorption spectrum of copper complex 1 in THF; emission spectrum of a blue-LED lamp; emission spectrum of a Hg lamp.
Figure 6.
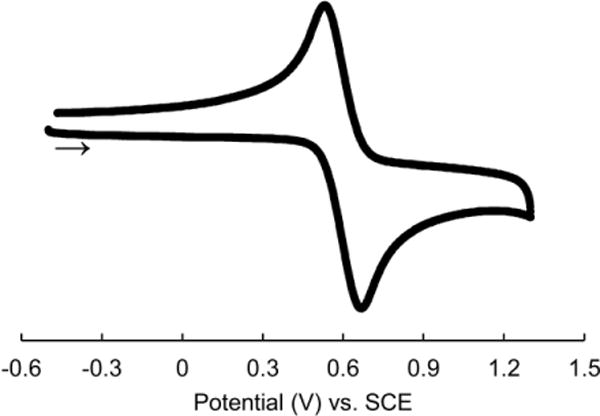
Cyclic voltammogram of copper complex 1 in THF (100 mV/s scan rate; 0.1 M in TBAPF6).
2-Bromo-4-phenylbutane quenches the luminescence of the excited state of copper complex 1; the quenching of the emission at 427 nm occurs with a Stern-Volmer constant of 14 M–1 (Figure 7). On the basis of the estimated excited-state reduction potential of complex 1 (–2.5 V vs. SCE), we postulate that this quenching is likely due to single electron transfer from the excited state of complex 1 to the alkyl bromide.
Figure 7.
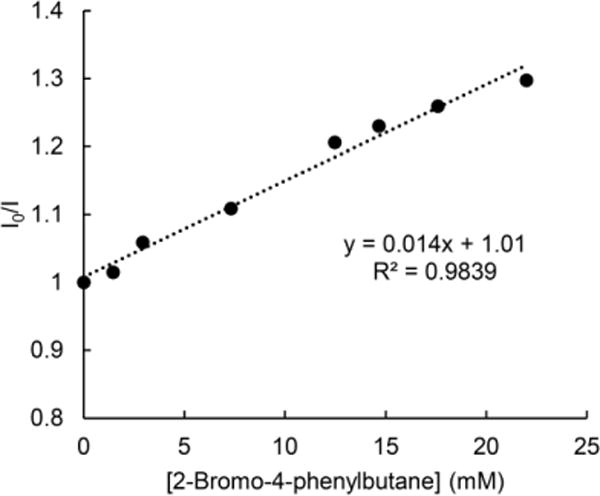
Quenching of the emission at 427 nm (irradiation at 380 nm) of a solution of copper complex 1 in THF in the presence of varying amounts of 2-bromo-4-phenylbutane.
Copper-catalyzed coupling of carbamates with unactivated secondary alkyl bromides induced by irradiation from blue-LED lamps
With data in hand on the properties of copper complex 1, we investigated its effectiveness as a catalyst for the alkylation of carbamates by unactivated secondary alkyl halides. In view of the ubiquity of N-Boc-protected amines in organic synthesis,18 we focused our initial study on t-butyl carbamate as the nucleophile. We determined that complex 1 does indeed serve as a catalyst for the coupling of t-butyl carbamate with an unactivated secondary bromide using blue-LED lamps as the light source, although the yield is modest (33%; eq 4). On the other hand, in the presence of excess CuBr, the desired alkylation proceeds in excellent yield (96%) with no detectable over-alkylation.19
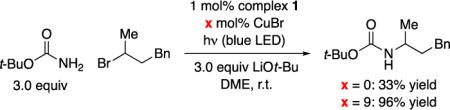 |
(4) |
We hypothesize that, under the conditions that include excess CuBr, copper complex 1 may be serving the typical role of photoredox catalysts, i.e., it is a donor and an acceptor of electrons, but it is not directly involved in the key bond-forming process (as in Figure 3);20 this stands in contrast to the scenario outlined in Figure 2, wherein a copper–nucleophile complex is engaged in both electron transfer and in bond formation. A 31P NMR study establishes that complex 1 does not dissociate ligand L1 or bind t-butyl carbamate to a significant extent in the presence of 3.0 equiv of the carbamate/LiOt-Bu.
When copper complex 1 is generated in situ from CuBr and ligand L1 (Table 1, entry 1),21 a similar yield (91%) is observed as when the discrete catalyst is used (eq 4; 96%). In the absence of ligand L1, CuBr, or light, essentially no C–N bond formation occurs (entries 2–4), and the electrophile can be recovered virtually quantitatively. Decreasing the amount of ligand L1 and CuBr, individually or simultaneously, results in a moderate loss in yield (entries 5–7), as does lowering the quantity of nucleophile (entry 8). The alkylation is air-sensitive, but not highly water-sensitive (entries 9 and 10). Although commonly used ruthenium and iridium photoredox catalysts cannot be used in place of ligand L1 (entries 11 and 12), a Hg lamp can be employed instead of blue-LED lamps (entry 13; in the absence of ligand L1: <1% yield).22 Under our standard conditions, essentially no C–N coupling is observed when an unactivated tertiary alkyl bromide is employed as the electrophile.
Table 1.
Effect of reaction parameters.

| ||
|---|---|---|
| entry | variation from the “standard” conditions | yield (%)a |
| 1 | none | 91 |
| 2 | no L1 | <1 |
| 3 | no CuBr | <1 |
| 4 | no hv | <1 |
| 5 | 0.5 mol% L1 | 75 |
| 6 | 5.0 mol% CuBr | 81 |
| 7 | 0.5 mol% L1 and 5.0 mol% CuBr | 65 |
| 8 | 2.0 equiv carbamate, 2.0 equiv LiO-t-Bu | 52 |
| 9 | under air (capped vial) | 21 |
| 10 | added H2O (0.1 equiv) | 85 |
| 11 | 1.0 mol% [Ru(bpy)3](PF6)2, instead of L1 | <1 |
| 12 | 1.0 mol% [Ir(dtbbpy)(ppy)2](PF6)2, instead of L1 | <1 |
| 13 | Hg, instead of blue-LED, lamp | 94 |
| 14 | THF, instead of DME | 90 |
Average of two runs, based on GC analysis versus an internal standard, bpy = 2,2′-bipyridine; ppy = 2-phenylpyridine; dtbbpy = 4,4′-di-f-butyl-2,2′-bipyridine.
Under the standard conditions, a variety of carbamates can be N-alkylated in good yield, providing a one-step method for the conversion of an electrophile into a carbamate-protected primary amine (Table 2). The alkyl group (R) of the carbamate can range in size from Me to t-Bu (entries 1–4).23 Although we have not detected double alkylation in these reactions, it is possible to achieve C–N bond formation in the case of an unhindered cyclic carbamate (entry 5).
Table 2.
Scope with respect to the carbamate.

| ||
|---|---|---|
| entry | carbamate | yield (%)a |
| 1 | R = Me | 90 |
| 2 | CH2CH2Ph | 67 |
| 3 | CH2CH2TMS | 76 |
| 4 | t-Bu | 87 |
|
| ||
| 5 |
|
74 |
Yield of purified product (average of two experiments).
With respect to the electrophile, we have established that a variety of acyclic and cyclic unactivated secondary alkyl bromides are suitable partners in this photoinduced, copper-catalyzed coupling process (Table 3). When 1-bromo-3-(t-butyl)cyclohexane is employed as the electrophile, moderate diastereoselectivity is observed (6.5:1 cis/trans; entry 9). The coupling is compatible with the presence of an alkene, an alkyne, a ketone, an N-alkyl indole, a secondary alcohol, a secondary amine, and an unactivated secondary alkyl chloride, whereas it proceeds with significantly lower efficiency in the presence of an N-H indole, an unactivated secondary alkyl iodide, and a pyridine (see the Supporting Information).
Table 3.
Scope with respect to the electrophile.
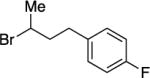
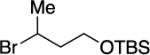
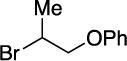
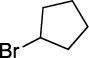
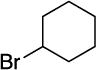
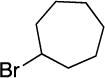
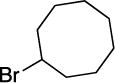
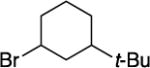
Yield of purified product (average of two experiments).
Yield of purified product when a Hg lamp is used.
Starting material: 1:5.4 cis/trans; product: 6.5:1 cis/trans.
As indicated in entry 5 of Table 3, when irradiation by blue-LED lamps leads to a modest yield of the desired alkylation product, the use instead of a Hg lamp may be beneficial (49% → 74% yield), presumably due to more efficient absorption by photocatalyst 1 (Figure 5). Additional examples of challenging N-alkylations wherein a Hg lamp has proved to be helpful are illustrated in eq 5 and eq 6. For the coupling of the pregnenolone-derived alkyl bromide (eq 6), the major diastereomer of the product is the result of retention of stereochemistry in the substitution reaction, and it can be isolated as a single isomer (>20:1) via column chromatography and then recrystallization.
 |
(5) |
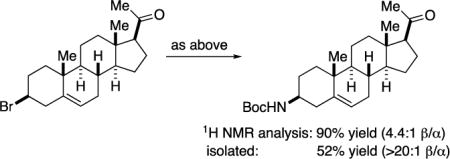 |
(6) |
Mechanistic observations
In our preliminary studies, we examined the excitation of copper complex 1 (Figure 5; E → F in Figure 3) and the quenching of this excited state by a secondary alkyl bromide (Figure 7; F → G in Figure 3). To gain further insight into the reaction of photoexcited complex 1 with an alkyl bromide (F → G in Figure 3), we have subjected 6-bromo-1-heptene to our standard coupling conditions, which generates the cyclized product with 4:1 cis/trans selectivity (eq 7). Because this is the same stereoselectivity that has been reported for the cyclization of the derived secondary alkyl radical,24 this observation supports the hypothesis that a secondary alkyl radical is an intermediate in the photoinduced, copper-catalyzed couplings reported herein.
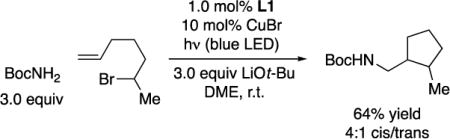 |
(7) |
This derived secondary alkyl radical cyclizes with a rate constant of 1.0 × 105 s–1 at 25 °C.24 Because this is much slower than typical rates of diffusion (generally >108 s–1),25 the radical has time to diffuse before engaging in C–N bond formation, as required by the mechanism outlined in Figure 3.
Electron transfer from the excited state of copper complex 1 to an alkyl bromide should produce a paramagnetic copper(II) intermediate (e.g., G or C in Figure 3). An EPR spectrum of the alkylation of BocNH2 with 2-bromo-4-phenylbutane reveals an S = 1/2 species with hyperfine coupling to a mononuclear Cu center (I = 3/2) (Figure 8). Because the same spectrum is observed when CuBr2 is treated with BocNH2 and LiOt-Bu (Figure 8), we suggest that this EPR-active species may be CuII(NHBoc)n (C in Figure 3).26,27 Thus, a variety of data are consistent with the mechanism outlined in Figure 3.
Figure 8.
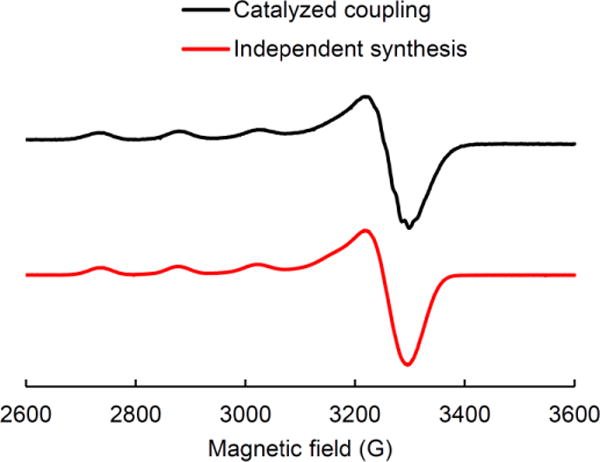
X-band EPR spectra (9.4 GHz, 77K): black: catalyzed coupling of BocNH2 with 2-bromo-4-phenylbutane; red: mixture of CuBr2, BocNH2 (10 equiv), and LiOt-Bu (10 equiv). g = [2.276, 2.063, 2.058].
Conclusions
We have developed a method for coupling an array of carbamates with unactivated secondary alkyl bromides under mild conditions (room temperature), thereby providing direct access to protected primary amines; to the best of our knowledge, the corresponding uncatalyzed nucleophilic substitution reactions have not been described. To achieve this objective, we designed a photoredox catalyst, based on copper and a tridentate carbazolide/bisphosphine ligand, that can serve as an electron donor upon irradiation by blue-LED lamps. Mechanistic studies are consistent with a reaction pathway in which the excited photoredox catalyst reduces the alkyl electrophile, producing an alkyl radical and a copper(II) intermediate; the latter oxidizes a copper(I)–nucleophile complex to a copper(II)–nucleophile intermediate, which then couples in an out-of-cage process with the alkyl radical to generate the cross-coupling product. Ongoing efforts are directed at further expanding the scope of photoinduced, copper-catalyzed coupling processes, including enantioselective reactions, and at elucidating the mechanisms of these transformations.
Supplementary Material
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, grant R01–GM109194), the Natural Sciences and Engineering Research Council of Canada (graduate research fellowship for J.M.A.), the National Science Foundation (support of the Caltech EPR Facility (NSF-1531940)), and the Arnold and Mabel Beckman Foundation (support of the Beckman Institute Laser Resource Center). We thank Kareem I. Hannoun, Lawrence M. Henling, Dr. Brian C. Sanders, Dr. Jonas Schwaben, Dr. Michael K. Takase, and Dr. Haolin Yin for assistance and helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Procedures and characterization data (PDF)
References
- 1.Ricci A, editor. Amino Group Chemistry: From Synthesis to the Life Sciences. Wiley-VCH; Weinheim, Germany: 2008. [Google Scholar]
- 2.For a discussion and leading references, see:; Marvin CC. In: Comprehensive Organic Synthesis. Knochel P, Molander GA, editors. Vol. 6. Elsevier; Amsterdam: 2015. pp. 34–99. [Google Scholar]
- 3.For leading references, see:; Ahmad NM. In: Name Reactions for Functional Group Transformations. Li JJ, Corey EJ, editors. John Wiley & Sons; Hoboken, NJ: 2007. pp. 438–450. [Google Scholar]
- 4.Wuts PGM, editor. Greene’s Protective Groups in Organic Synthesis. Wiley; Hoboken, NJ: 2014. Chapter 7. [Google Scholar]
- 5.For reviews, see:; (a) Ghosh AK, Brindisi M. J Med Chem. 2015;58:2895–2940. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuhr RJ, Dorough HW. Carbamate Insecticides: Chemistry and Biochemistry. CRC Press; Cleveland, OH: 1976. [Google Scholar]
- 6.For examples of other methods that achieve the mono-alkylation of carbamates, see:; (a) Palomo C, Oiarbide M, Halder R, Kelso M, Gomez-Bengoa E, Garcia JM. J Am Chem Soc. 2004;126:9188–9189. doi: 10.1021/ja047004e. [DOI] [PubMed] [Google Scholar]; (b) Das BG, Ghorai P. Org Biomol Chem. 2013;11:4379–4382. doi: 10.1039/c3ob40918j. [DOI] [PubMed] [Google Scholar]
- 7.Do HQ, Bachman S, Bissember AC, Peters JC, Fu GC. J Am Chem Soc. 2014;136:2162–2167. doi: 10.1021/ja4126609. [DOI] [PubMed] [Google Scholar]
- 8.N-alkylations with alkyl electrophiles are among the most commonly used reactions in medicinal chemistry. For example, see Table 2 in:; Roughley SD, Jordan AM. J Med Chem. 2011;54:3451–3479. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 9.For other reports of transition-metal-catalyzed alkylations of nitrogen nucleophiles with unactivated secondary and tertiary alkyl electrophiles, see:; (a) Bissember AC, Lundgren RJ, Creutz SE, Peters JC, Fu GC. Angew Chem Int Ed. 2013;53:5129–5133. doi: 10.1002/anie.201301202. [DOI] [PubMed] [Google Scholar]; (b) Peacock DM, Roos CB, Hartwig JF. ACS Cent Sci. 2016;2:647–652. doi: 10.1021/acscentsci.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matier CD, Schwaben J, Peters JC, Fu GC. J Am Chem Soc. doi: 10.1021/jacs.7b09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For leading references to the use of copper in photocatalysis, see:; Paria S, Reiser O. Chem Cat Chem. 2014;6:2477–2483. [Google Scholar]
- 11.We have recently described evidence for the viability of out-of-cage bond construction in a different photoinduced, copper-catalyzed process (the alkylation of carbazole):; Ahn JM, Ratani TS, Hannoun KI, Fu GC, Peters JC. J Am Chem Soc. 2017;139:12716–12723. doi: 10.1021/jacs.7b07052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647–651. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]; (b) Kainz QM, Matier CM, Bartoszewicz A, Zultanski SL, Peters JC, Fu GC. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For an example of the deleterious impact of a substituent in the 1 position on the reactivity of the carbazole nitrogen in a photoinduced, copper-catalyzed N-alkylation, see ref 9a.
- 14.For precedent for undesired reactivity in the absence of a substituent, see:; Mankad NP, Antholine WE, Szilagyi RK, Peters JC. J Am Chem Soc. 2009;131:3878–3880. doi: 10.1021/ja809834k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For one example, see:; Cheng C, Kim BG, Guironnet D, Brookhart M, Guan C, Wang DY, Krogh-Jespersen K, Goldman AS. J Am Chem Soc. 2014;136:6672–6683. doi: 10.1021/ja501572g. [DOI] [PubMed] [Google Scholar]
- 16.Plundrich GT, Wadepohl H, Gade LH. Inorg Chem. 2016;55:353–365. doi: 10.1021/acs.inorgchem.5b02498. No copper complexes were described. [DOI] [PubMed] [Google Scholar]
- 17.For the oxidized (one-electron) derivative of copper complex 1, DFT calculations are consistent with substantial spin delocalization onto ligand L1. See the Supporting Information for further details.
- 18.Two recent studies have pointed to N-Boc as the most commonly used protecting group in medicinal chemistry, accounting for about 30% of all protections/deprotections in the data sets that were analyzed: (a) See Table 3 in ref 8.; Schneider N, Lowe DM, Sayle RA, Tarselli MA, Landrum GA. J Med Chem. 2016;59:4385–4402. doi: 10.1021/acs.jmedchem.6b00153. See Figure 6 in: [DOI] [PubMed] [Google Scholar]
- 19.In contrast to CuBr, the addition of 10 mol% CoBr2, NiCl2•DME, Pd(cod)2Cl2, or Au(PPh3)Cl did not improve the yield of the N-alkylation product.
- 20.For leading references on photoredox catalysis, see:; (a) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Analysis via 31P NMR spectroscopy confirms the formation of copper complex 1 at the start of the reaction. The presence of copper complex 1 is also supported by ESI–MS analysis.
- 22.Notes: (a) When CuBr is replaced with CoBr2, NiCl2•DME, Pd(cod)2Cl2, or Au(PPh3)Cl, essentially no N-alkylation product is formed (<1%). (b) In a light-on-light-off experiment, we have determined that coupling stops when irradiation stops, and that coupling resumes when irradiation resumes. (c) The quantum yield of the reaction (irradiation at 350 nm) is 0.06, which, when considered together with the measured lifetime for copper complex 1, is not suggestive of a chain process (for a discussion of chain processes in photoredox catalysis, see:; Cismesia MA, Yoon TP. Chem Sci. 2015;6:5426–5434. doi: 10.1039/c5sc02185e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) The corresponding alkyl chloride is essentially unreactive under our standard conditions, enabling the selective substitution of the alkyl bromide in the presence of the alkyl chloride (which can be recovered in virtually quantitative yield). (e) Because some of the studies described herein (e.g., absorption and cyclic voltammetry) have been conducted in THF, entry 14 is included in order to demonstrate that the cross-coupling proceeds well in this solvent.
- 23.Under our standard conditions, benzyl carbamate and 2,2,2-trichloroethyl carbamate are not suitable nucleophilic coupling partners.
- 24.Lusztyuk J, Maillard B, Deycard S, Lindsay DA, Ingold KU. J Org Chem. 1987;52:3509–3514. [Google Scholar]
- 25.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. p. 156. [Google Scholar]
- 26.The EPR spectrum is not affected by the addition of lithiated L1.
- 27.Copper(II) complex C may be serving as the persistent radical that enables efficient cross-coupling. For discussions of persistent radicals, see:; (a) Studer A. Chem - Eur J. 2001;7:1159–1164. doi: 10.1002/1521-3765(20010316)7:6<1159::aid-chem1159>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; (b) Fischer H. Chem Rev. 2001;101:3581–3610. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.