

Abstract
Background & Aims
Hepatic stellate cells (HSCs) contribute to desmoplasia and stiffness of liver metastases by differentiating into matrix-producing myofibroblasts. We investigated whether stiffness due to the presence of tumors increases activation of HSCs into myofibroblasts and their tumor-promoting effects, as well as the role of E1A binding protein p300 (EP300 or p300), a histone acetyltransferase that regulates transcription, in these processes.
Methods
HSCs were isolated from liver tissues of patients, mice in which the p300 gene was flanked by 2 loxP sites (p300F/F mice), and p300+/+ mice (controls). The HSCs were placed on polyacrylamide gels with precisely defined stiffness, and their activation (differentiation into myofibroblasts) was assessed by immunofluorescence and immunoblot analyses for alpha-smooth muscle actin. In HSCs from mice, the p300 gene was disrupted by cre recombinase. In human HSCs, levels of p300 were knocked down with small hairpin RNAs or a mutant form of p300 that is not phosphorylated by AKT (p300S1834A) was overexpressed. Human HSCs were also cultured with inhibitors of p300 (C646), PI3K signaling to AKT (LY294002), or RHOA (C3 transferase) and effects on stiffness-induced activation were measured. RNA sequencing and chromatin immunoprecipitation quantitative PCR were used to identified HSC genes that changed expression levels in response to stiffness. We measured effects of HSC-conditioned media on proliferation of HT29 colon cancer cells and growth of tumors following subcutaneous injection of these cells into mice. MC38 colon cancer cells were injected into portal veins of p300F/Fcre and control mice and liver metastases were measured. p300F/Fcre and control mice were given intraperitoneal injections of CCl4 to induce liver fibrosis. Liver tissues were collected and analyzed by immunofluorescence, immunoblot, and histology.
Result
Substrate stiffness was sufficient to activate HSCs, leading to nuclear accumulation of p300. Disrupting p300 level or activity blocked stiffness-induced activation of HSC. In HSCs, substrate stiffness activated AKT signaling via RHOA to induce phosphorylation of p300 at serine 1834; this caused p300 to translocate to the nucleus, where it upregulated transcription of genes that increase activation of HSCs and metastasis, including CXCL12. MC38 cells, injected into portal veins, formed fewer metastases in livers of p300F/Fcre mice than control mice. Expression of p300 was increased in livers of mice following injection of CCl4; HSC activation and collagen deposition were reduced in livers of p300F/Fcre mice compared with control mice.
Conclusions
In studies of mice, we found liver stiffness to activate HSC differentiation into myofibroblasts, which required nuclear accumulation of p300. P300 increases HSC expression of genes that promote metastasis.
Keywords: tumor progression, epigenetic modification, chromatin remodeling, mechanotransduction
Graphical abstract

Introduction
Liver is a common site for metastatic gastrointestinal cancer cells to colonize and grow. Current treatment options for liver metastasis are very limited, contributing to high mortality of patients. Hepatic stellate cells (HSCs) are a component of the prometastatic liver microenvironment for their transdifferentiation into tumor-promoting myofibroblasts (MFs) via a TGF-β-dependent mechanism1-3. Activated-HSC/MFs regulate liver metastatic growth by paracrine mechanisms, including releasing growth factors, cytokines, extracellular matrix (ECM) and matrix metalloproteinases4, 5. Understanding key molecules and pathways governing HSC activation, in addition to TGF-β signaling, may lead to novel strategies to target the hepatic tumor microenvironment for liver metastasis.
Matrix stiffening is a prominent hallmark of the tumor microenvironment and the role of stiffness on cancer has received intensive investigations. Stiffness modifies malignant phenotypes of cancer cells by directly regulating their growth and motility6-8, angiogenesis9 and tumor-associated inflammation10, 11. Additionally, stiffness activates YAP1 so as to maintain MF phenotypes of breast cancer12. Although activated-HSC/MFs are a major contributor to desmoplasia of liver metastases, it is unknown if tumor stiffness can in return regulate the biology of the activated-HSC/MFs and their tumor-promoting effects. This study therefore focused on whether and how tumor stiffness influenced MF activation of HSCs aiming to identify a novel mechanosignaling and positive feedback loop for liver metastatic growth.
Transcription coactivator p300 promotes gene transcription in different cell types, critical for cellular functions such as proliferation, apoptosis, and differentiation13. P300 interacts with a large number of transcription factors (TFs) and modifies their functions by adding an acetyl group to their lysine residues14, 15. P300 promotes gene transcription by epigenetic mechanisms; it: (a) acetylates histones to loosen chromatin into an active transcription state, (b) acetylates TFs, activators, and coactivators to increase their activities, and (c) provides a bridge connecting TFs to chromatin and the basal transcriptional machinery. P300 acetyltransferase activity is regulated by its post-translational modifications such as phosphorylation, its binding proteins and its subcellular localization16-20. In fibroblasts of scleroderma mice and patients, p300 protein levels are elevated and p300 promotes TGF-β/SMAD-dependent collagen synthesis21, 22. Since the role of p300 in HSCs has received scant attention, this study explored a novel p300-dependent epigenetic mechanism for stiffness-mediated HSC activation.
Using polyacrylamide gels with incremental stiffness as culture substrates for HSCs, including primary murine and human HSCs, we showed that substrate stiffness alone induced MF activation of HSCs by activating a RHOA-AKT-p300 signaling pathway. This mechanosignaling induced p300 phosphorylation and its nuclear targeting, leading to transcription of α-SMA and a panel of tumor-promoting factors, critical for liver metastatic growth. In agreement with this, conditioned medium of HSCs on a stiff substrate promoted colorectal cancer growth in vitro and in mice as compared to that of HSCs on a soft substrate. Cre-loxP-mediated p300 gene deletion in activated-HSC/MFs suppressed colorectal liver metastatic growth in mice and CCl4-induced HSC activation and liver fibrosis. Thus, tumor-associated stiffness, built by activated-HSC/MFs, in return, further enhances HSC activation and tumor growth so as to form “an amplification loop” for metastatic growth in the liver.
Materials and Methods
Cell culture
Human primary HSCs were purchased from ScienCell (#5300). LX2 cells were obtained from Dr. Scott Friedman. HT29 human colorectal cancer cells were from ATCC (HTB-38). L3.6 human pancreatic cancer cells were obtained from Dr. Raul Urrutia (Mayo Clinic), and MC38 murine colorectal cancer cells were from Dr. Steven A. Rosenberg (National Cancer Institute)3. Cells were authenticated by short tandem repeat (STR) DNA profiling by Genetica, and routinely tested for mycoplasma contamination with a MycoAlert detection kit (Lonza).
Polyacrylamide hydrogels with incremental stiffness
Polyacrylamide hydrogels with stiffness ranging from 0.4 kilopascal (kPa) to 25.6 kPa were prepared according to formula listed in Suppl. Fig. 1A23. A detailed procedure for making them is described in Supplemental Materials and Methods.
RNA Sequencing (RNA-seq) and ChIP-qPCR
HSCs cells on 0.4 kPa or 25.6 kPa were subjected to RNA isolation. Experimental details for RNA-seq and ChIP-qPCR are described in Supplemental Materials and Methods. The dataset is in Gene Expression Omnibus (GSE101343).
Study Approval
Experiments with mice were approved by the Institutional Animal Care and Use Committee of University of Minnesota and Mayo Clinic. Experiments with patient's biopsies were approved by the Institutional Review Board at Mayo Clinic.
Coinjection of HT29 and HSC-conditioned medium (CM) into mice
HT29 cells serum-starved for 24 hours were lifted up and divided into 4 parts, each containing 5 × 106 cells. After centrifugation, each cell pellet was re-suspended in 1 ml each of following CMs: CM of HSCs on 0.4 kPa, CM of HSC-NT shRNA on 25.6 kPa, CM of HSC-p300 shRNA on 25.6 kPa, and CM of HSCs on a plastic dish. HT29 cells were then incubated with CM at 37°C allowing cells to uptake paracrine factors from CM prior to injection. 2 hours later, HT29 were re-suspended with the CM and HT29/CM were coinjected into SCID mice subcutaneously (each injection contained 100 μl HT29/CM mixture, 0.5 × 106 HT29 cells). This protocol was adapted from recent publications24, 25. Mice were killed at day 7 and tumor volume was calculated by equation: volume = (length × width2)/2.
Intrasplenic and portal vein tumor injection mouse models
P300F/F mouse line was generated by Kasper LH et al.26 and bred with a collagen1A1-cre transgenic line (kindly provided by Dr. Tatiana Kisseleva at UCSD). PCR was used to genotype the mice and aged-matched p300+/+cre and p300F/Fcre mice were used for portal vein injection.
Conditional p300 knockout mice or SCIDs older than 8-week-old were used as tumor injection recipients. Under a general anesthesia by isoflurane (2-5%), a 1-2 cm surgical incision was made on the left side of mouse abdomen to expose the spleen or portal vein. To generate MC38 or HT29 liver metastases, 2 × 106 cancer cells suspended in 100 μl 1× PBS were injected into portal vein of mice3, 27. To generate L3.6 liver metastases, 1 × 106 L3.6 cells suspended in 33 μl 1× PBS were injected to the spleen28. The incision was closed with suture after bleeding stopped.
Statistical analysis
Data are presented as Mean ± S.E.M. The difference between groups was evaluated by Student's t-test or ANOVA followed by a post hoc test using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA). P<0.05 was regarded as statistically different.
Results
Substrate stiffness induces MF activation of HSCs and p300 nuclear accumulation in murine and human HSCs
To test if tumor stiffness in return regulates HSC activation, we used polyacrylamide hydrogels of variable but precisely defined stiffness as supports for in vitro culture of HSCs23, 29 (Suppl. Fig. 1A). As shown in Fig. 1A, primary murine HSCs seeded on a 0.4 kPa or 25.6 kPa hydrogel showed dramatically different morphology, consistent with data of rat HSCs29. Cells on 0.4 kPa failed to spread and expressed a low α-SMA level as revealed by immunofluorescence (IF). In contrast, cells on 25.6 kPa were well-spread and developed multiple cellular protrusions positive for α-SMA, characteristics of activated-HSCs (Fig. 1A and B). Since stiffness activated fibroblasts independent of TGF-β signaling23, 29, we attempted to find a novel epigenetic mechanism for stiffness-mediated HSC activation. To this end, we performed Western blot analysis (WB) and IF to test if stiffness influenced p300 acetyltransferase, a transcription coactivator and epigenetic regulator of cells. We found that 25.6 kPa stiffness enhanced p300 protein level and concurrently p300 nuclear accumulation in HSCs as compared 0.4 kPa (P<0.05 by t-test, Fig. 1B and C), suggesting p300 as a mechanosensitive molecule that may participate in mechanotransduction of HSCs.
Fig. 1. Substrate stiffness promotes p300 nuclear accumulation and MF activation of HSCs.
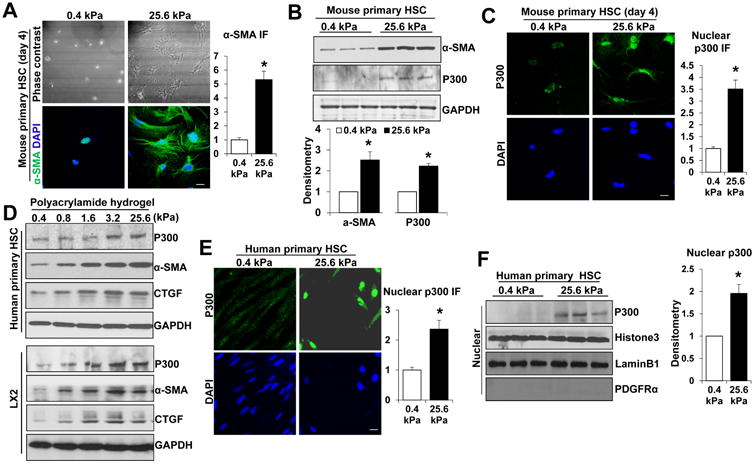
A and B. HSCs isolated from mice were plated on a 0.4 or 25.6 kPa hydrogel and subjected to phase contrast microscopy, IF and WB with quantitative data shown. 25.6 kPa stiffness promoted HSC activation and p300 protein level. *, P<0.05 by t-test, n>20 cells per group for IF and n=3 repeats for WB. Bar, 20 μm. C. P300 IF showed that 25.6 kPa stiffness induced p300 nuclear accumulation as compared to 0.4 kPa. *, P<0.05 by t-test, n>20 cells per group. Bar, 20 μm. D. Primary human HSCs and LX2 cells plated on polyacrylamide hydrogels were collected for WB. E. and F. IF and subcellular fractionation assay were used to analyze stiffness-induced p300 nuclear accumulation. Cell nuclei were counterstained with DAPI. *, P<0.05 by t-test, n>40 cells per group for IF and n=3 for WB. Bar, 20 μm.
To further confirm the finding, we seeded primary human HSCs (ScienCell) and LX2 cells, immortalized HSCs, on polyacrylamide gels with incremental stiffness, 0.4, 0.8, 1.6, 3.2 and 25.6 kPa for WB and found that protein levels of α-SMA and CTGF, markers of HSC activation, increased proportionally to stiffness (Fig. 1D). P300 protein levels increased proportionally to stiffness as well (Fig. 1D). IF and subcellular fractionation assay demonstrated that 25.6 kPa stiffness indeed induced p300 nuclear accumulation as compared to 0.4 kPa (P<0.05 by t-test, Fig. 1E and 1F, Suppl. Fig. 1B and 1C). These data further support p300 as a mechanotransducer connecting mechanical stimulation to gene transcription of HSCs. This hypothesis was tested by experiments below.
Stiffness-induced HSC activation is suppressed by cre-loxP-mediated p300 gene deletion, shRNA-mediated p300 knockdown, or C646-mediated p300 inhibition
We first used murine HSCs to study the role of p300 in stiffness-mediated HSC activation. HSCs isolated from p300F/F mice26 were seeded on 0.4 kPa or 25.6 kPa gel and transduced with Adcre-GFP to disrupt p300 gene. AdlacZ viruses were used as controls. Live cell imaging and WB confirmed that Adcre-GFP transduction indeed resulted in a 55 kd cre-GFP fusion protein dominantly nuclear in distribution (Suppl. Fig. 1D). As revealed by α-SMA/p300 double IF (Fig. 2A), 25.6 kPa induced p300 nuclear accumulation (arrows) and α-SMA-positive stress fibers (arrowheads) in lacZ-expressing cells, and these stiffness-mediated phenotypes were inhibited in cre-GFP-expressing cells. This was further supported by a confocal fluorescence image containing both cre-GFP-negative and -positive cells (Suppl. Fig. 1E), demonstrating that p300 accumulated in the nucleus and α-SMA-positive stress fibers formed in a control HSC (arrowhead) but not in two adjacent p300-null HSCs (arrows, cre-GFP positive). Additionally, WB confirmed that stiffness-mediated HSC activation was indeed suppressed in p300-null cells (p<0.05 by ANOVA Fig. 2B). Thus, p300 is required for stiffness-mediated murine HSC activation in vitro.
Fig. 2. Stiffness induces HSC activation by a p300-dependent mechanism.
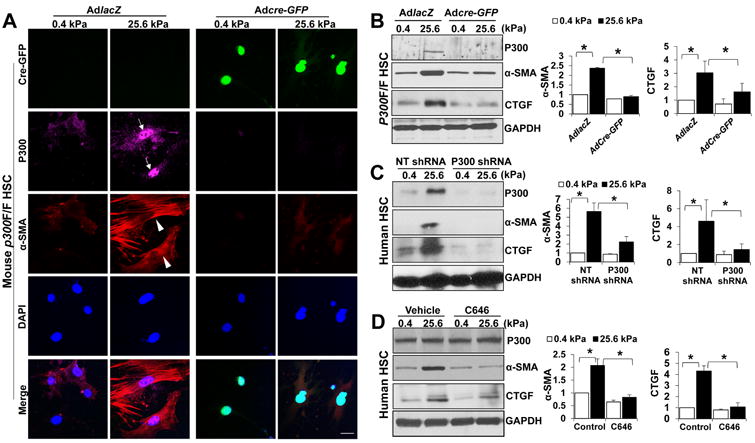
A P300F/F HSCs seeded on a 0.4 or 25.6 kPa gel were transduced with adenoviruses encoding lacZ (AdLacZ) or cre-GFP (Adcre-GFP) for IF. Stiffness-mediated upregulation of α-SMA and p300 was inhibited by p300 gene deletion (rows 2 and 3). Cell nuclei were counterstained by DAPI. Bar, 20 μm. B. WB revealed that stiffness-mediated HSC activation was inhibited by p300 gene deletion. *, P<0.05 by ANOVA, n=3. C. Primary human HSCs on 0.4 or 25.6 kPa were transduced with lentiviruses encoding NT shRNA or p300 shRNA. Stiffness-mediated HSC activation was inhibited by p300 knockdown. *, P<0.05 by ANOVA, n=3. D. Stiffness-mediated HSC activation was suppressed by p300 inhibitor C646. *, P<0.05 by ANOVA, n=3.
We next used p300 shRNA or p300 inhibitor C646 to test the role of p300 in mechanotransduction of primary human HSCs. HSCs transduced with lentiviruses encoding non-targeting shRNA (NT shRNA; control) or p300 shRNA were seeded on 0.4 kPa or 25.6 kPa gel and harvested for WB, which revealed that p300 knockdown suppressed stiffness-mediated upregulation of α-SMA and CTGF in human HSC (p<0.05 by ANOVA, Fig. 2C). Consistently, pharmacological inhibition of p300 acetyltransferase by C646 also suppressed stiffness-induced HSC activation (p<0.05 by ANOVA, Fig. 2D). Thus, our data, generated with both primary murine and human HSCs, support that stiffness-mediated HSC activation requires p300.
P300 nuclear accumulation by stiffness is mediated by RHOA
RHOA is a known mechanotransducer of the cell12, 30, 31. We performed WB and found that RHOA protein increased proportionally to stiffness in primary human HSCs and LX2 cells (Fig. 3A) and that RHOA activity was enhanced by stiffness (p<0.05 by t-test, Suppl. Fig. 2A). Next two approaches were utilized to test the role of RHOA in stiffness-mediated p300 nuclear accumulation: (1) inhibiting RHOA activity by C3 transferase and (2) overexpressing RHOAQ63L, a constitutively active RHOA mutant. Indeed, stiffness-mediated upregulation of α-SMA and p300 protein levels and p300 nuclear accumulation was significantly suppressed by C3 transferase in both primary human HSCs and LX2 cells (p<0.05 by ANOVA, Fig. 3B and 3C, Suppl. Fig. 2C and 2D). Furthermore, cells expressing lacZ (control) or RHOAQ63L by retroviral transduction were plated on a soft gel and RHOAQ63L overexpression in cells on 0.4 kPa led to upregulation of α-SMA and p300 proteins and p300 nuclear accumulation, similar to the effects of stiffness (p<0.05 by ANOVA, Fig. 3D and 3E, Suppl. Fig. 2E and 2F). Thus, RHOA is critical for stiffness-induced p300 nuclear accumulation and HSC activation.
Fig. 3. P300 nuclear accumulation by stiffness is mediated by RHOA.
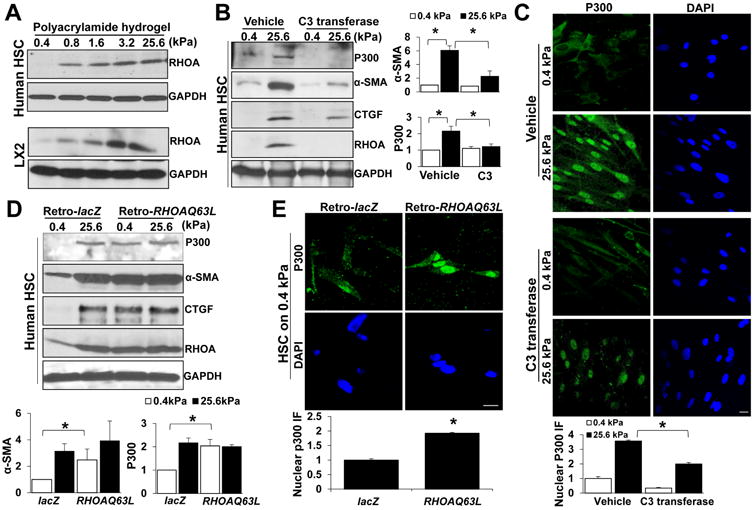
A Primary human HSCs and LX2 cells on hydrogels were collected for WB. RHOA protein was increased by stiffness. B and C. Primary human HSCs on 0.4 or 25.6 kPa were incubated with vehicle or RHOA inhibitor C3 transferase. Stiffness-mediated p300 upregulation and nuclear accumulation and HSC activation was inhibited by C3 transferase. *, P<0.05 by ANOVA, n=3 for WB and n>40 cells per group for IF. Bar, 20 μm. D. and E. Primary human HSCs transduced with retroviruses encoding lacZ (Retro-lacZ) or RHOAQ63L (Retro-RHOAQ63L) were seeded on 0.4 or 25.6 kPa for WB and IF. RHOAQ63L overexpression led to HSC activation, p300 upregulation and nuclear accumulation in HSCs on 0.4 kPa. *, P<0.05 by ANOVA, n=3 repeats for WB and n>40 cells per group for IF. Bar, 20 μm.
Stiffness induces p300 phosphorylation at serine 1834 and its nuclear targeting by activating a RHOA-AKT pathway
P300 mRNA levels were not enhanced by stiffness, as revealed by reverse transcription followed by quantitative real-time PCR (qPCR) (p>0.05 by t-test, Suppl. Fig. 3A), suggesting that stiffness may regulate p300 post-transcriptional modifications to promote its nuclear accumulation. Indeed, stiffness increased p300 phosphorylation at serine 1834, the ratio of p-p300(S1834) to total p300, and p-p300 (S1847) nuclear accumulation in HSCs (P<0.05 by t-test, Fig. 4A). Stiffness also induced AKT phosphorylation at serine 473 (P<0.05 by t-test, Fig. 4A). To test if stiffness-induced p300 phosphorylation is mediated by AKT, we treated cells with AKT phosphorylation inhibitor LY294002 and found that LY294002 suppressed stiffness-mediated p300 phosphorylation (Fig. 4B). To investigate if p-p300(S1834) mediates p300 nuclear targeting, we generated a p300S1834A mutant by replacing serine 1834 by alanine20. Both wild-type p300 and p300S1834A cDNA were tagged by FLAG and their cellular localization was determined by IF for FLAG. As shown in Fig. 4C, stiffness induced nuclear accumulation of FLAG-p300WT but not FLAG-p300S1834A fusion protein, supporting that p-p300(S1834) by AKT indeed mediated stiffness-induced p300 nuclear targeting.
Fig. 4. Stiffness induces p300 phosphorylation at S1834 by activating RHOA-AKT pathway.
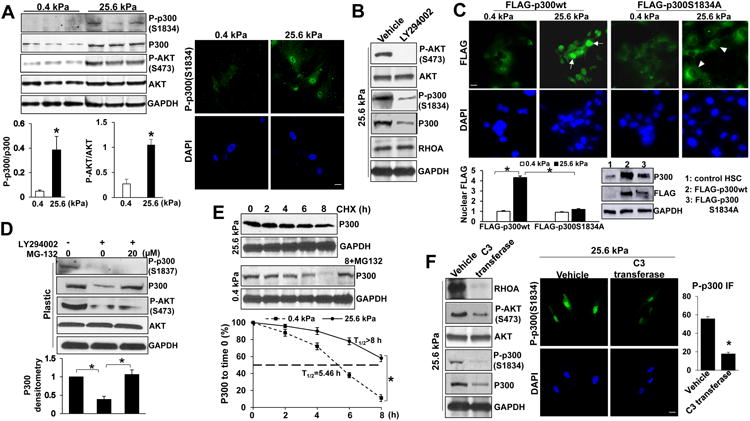
A WB revealed that stiffness induced p300 phosphorylation at S1834 and AKT phosphorylation at S473. The ratio of p-p300 to total p300, the ratio of p-AKT to total AKT, and IF for p-p300(S1834) are shown. *, P<0.05 by t-test, n=3. B. AKT inhibitor LY294002 reduced p-AKT(S473), p-p300(S1834) and total p300 level of HSCs. Data represent multiple repeats with similar results. C. Stiffness induced nuclear accumulation of FLAG-p300wt but not FLAG-p300S1834A mutant. FLAG-tagged p300 fusion proteins were detected by IF and WB for FLAG. *, P<0.05 by ANOVA, n=25 cells per group. Bar, 20 μm. D. LY294002 reduced p300 protein level with its effect reversed by proteasome inhibitor MG-132. *, P<0.05 by ANOVA, n=3 repeats. E. P300 protein stability in HSCs was analyzed by WB in the presence of cycloheximide (CHX). The half-life (T1/2) of p300 in HSCs on 0.4 kPa was 5.46 hours and >8.0 hours in HSCs on 25.6 kPa. *, P<0.05 by ANOVA, n=3. F. RHOA inhibitor C3 transferase reduced p-AKT, p-p300 as well as total p300 level of HSCs. *, P<0.05, by t-test, n>20 cells per group. Bar, 20 μm.
Since LY294002 downregulated total p300 level (Fig. 4B), we next treated HSCs with a proteasome inhibitor MG-132 and found that LY294002-mediated downregulation of p300 protein and p300 nuclear accumulation was reversed by MG-132 (p<0.05 by ANOVA, Fig. 4D, Suppl. Fig. 3B). Moreover, in the presence of a protein synthesis inhibitor cycloheximide, p300 protein of HSCs on 0.4 kPa degraded faster than it did on 25.6 kPa (p<0.05 by ANOVA, Fig. 4E). Thus, p300 of HSCs on 0.4 kPa underwent proteasomal degradation whereas p300 of HSCs on 25.6 kPa was targeted to the nucleus and stabilized by AKT-induced p300 phosphorylation at serine 1834.
We next treated cells with C3 transferase and found that C3 transferase reduced stiffness-mediated phosphorylation of AKT and p300 and p-p300 (S1834) nuclear accumulation (Fig. 4F), suggesting that RHOA is required for stiffness-mediated phosphorylation of AKT and p300. As expected, stiffness-mediated p300 nuclear targeting and HSC activation was suppressed by AKT inhibitor LY294002 (p<0.05 by ANOVA, Suppl. Fig. 3C and D). Additionally, stiffness upregulated β1-integrin protein and β1-integrin-FAK signaling (Suppl. Fig. 3E). Thus, stiffness promotes p300 nuclear accumulation and HSC activation by activating a RHOA-AKT-p300 mechanosignaling of HSCs.
Stiffness-p300 axis epigenetically turns on transcription of tumor-promoting factors of HSCs
We next performed RNA sequencing (RNA-seq) to study if stiffness-p300 axis modulated transcription of HSCs (Suppl. Fig. 4A and GSE101343 in Gene Expression Omnibus). As analyzed with software packages edgeR and cuffdiff2 (fold change ≥ 2 & FDR < 0.05 and fold change ≥ 1.5 & FDR < 0.01), 1196 genes were identified to be transcriptionally targeted by stiffness (Fig. 5A, Suppl. Fig. 4B). Ingenuity Pathway Analysis identified that genes related to HSC activation were among those affected by stiffness (Fig. 5B). Moreover, stiffness promoted transcription of more than 20 tumor-promoting factors, including CXCL12, IL11, IL6, VEGFA, PDGFA and B, FGF, and CTGF (Fig. 5C), suggesting that stiffness-p300 axis may promote tumor-promoting effects of HSCs by epigenetic regulation of gene transcription. Since CXCR4/CXCL12 axis regulates organ-specific metastasis, including liver metastasis of breast or prostate cancers32, 33, we used CXCL12 as a prototype to test our larger hypothesis and model. Indeed, WB detected that stiffness increased CXCL12 protein level through HSC p300 and qPCR confirmed that CXCL12 mRNA level was enhanced by stiffness (p<0.05 by t-test, Fig. 5D and Fig. 5E). ELISA demonstrated that CXCL12 concentration in culture medium was promoted by stiffness-p300 axis as well (Suppl. Fig, 5A). Thus, stiffenss-p300 axis promoted HSCs to produce and release tumor-promoting factors, such as CXCL12.
Fig. 5. Stiffness epigenetically promotes gene transcription of HSCs.
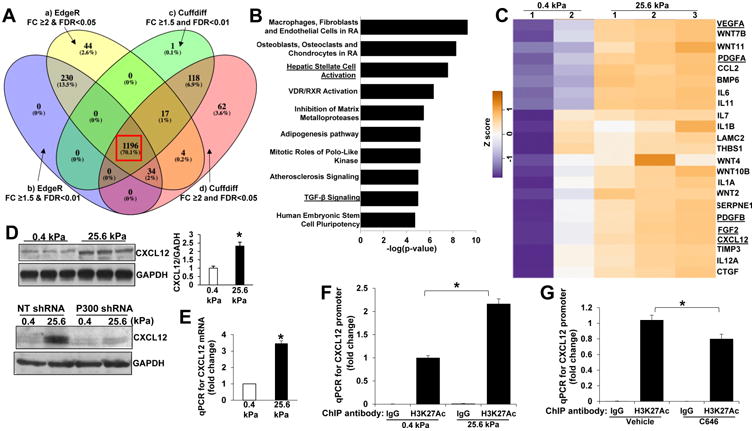
A HSCs on 0.4 or 25.6 kPa were subjected to RNA-seq. 1196 genes were identified as stiffness targets by 4 bioinformatic analyzing approaches (4 colors). B. Ingenuity pathway analysis of the 1196 genes. C. A panel of tumor-promoting factors transcriptionally turned on by stiffness is shown by a heatmap. D. Stiffness promoted HSCs to produce CXCL12 protein through HSC p300. *, P<0.05 by t-test, n=3. E. Stiffness increased CXCL12 mRNA level as revealed by qPCR after reverse transcription. *, P<0.05 by t-test, n=6. F. Histone 3/DNA complexes were pulled down by control IgG or anti-histone H3-acetyl K27 for ChIP-qPCR and qPCR was performed for CXCL12 promoter with primer pair 1. Stiffness increased H3K27AC on CXCL12 promoter. *, P<0.05 by ANOVA, n=3. G. C646 reduced H3K27AC on CXCL12 promoter of HSCs. *, P<0.05 by ANOVA, n=3.
We next used chromatin immunoprecipitation (ChIP)-qPCR to analyze histone acetylation on CXCL12 gene promoter. To this end, an antibody recognizing H3K27AC (histone 3 acetylation at lysine 27), a histone mark for active promoters, was used for ChIP-qPCR, which revealed that stiffness increased H3K27 acetylation on CXCL12 promoter (P<0.05 by ANOVA, Fig. 5F. qPCR data using 2nd pair of primers are in Suppl. Fig. 4C). Additionally, C646 reduced CXCL12 promoter acetylation (P<0.05 by ANOVA, Fig. 5G. Data using 2nd pair of primers are in Suppl. Fig. 4C. Because of a minimal dose (15 μM) of C646 used to avoid cytotoxicity, only 20-30% of inhibition was detected). Thus, stiffness-p300 axis promoted histone acetylation so as to induce an active chromatin structure for gene transcription of HSCs.
Stiffness-p300 axis potentiates paracrine tumor-promoting effects of HSCs in vitro
We have shown that conditioned medium (CM) of HSCs promoted tumor growth3, 34. CM of HSCs on 0.4 kPa or 25.6 kPa were next collected for MTS and Boyden chamber assay to test their role on HT29 cell proliferation and migration. CM of HSCs on 0.4 kPa promoted HT29 proliferation and migration as compared to basal medium (control), and importantly, these effects of HSC CM were potentiated by stiffness (P<0.05 by ANOVA, Fig. 6A and Suppl. Fig. 5B). Since HT29 cells expressed CXCL12 receptor CXCR4 (Suppl. Fig. 5C), we next tested the role of CXCL12 for HT29 cells and found that recombinant CXCL12 dose-dependently promoted HT29 proliferation (P<0.05 by ANOVA, Fig. 6B) and that a neutralizing CXCL12 antibody dose-dependently suppressed the effect of 25.6 kPa CM on HT29 proliferation (P<0.05 by ANOVA, Suppl. Fig. 5D). Furthermore, the effects of 25.6 kPa CM on HT29 proliferation and migration were reduced by p300 knockdown in HSCs (P<0.05 by ANOVA, Fig. 6C, Suppl. Fig. 5E). Thus, stiffness-p300 axis stimulates HSCs to produce paracrine factors, such as CXCL12, to potentiate the tumor-promoting effects of HSCs.
Fig. 6. Stiffness potentiates tumor-promoting effects of HSCs through HSC p300.
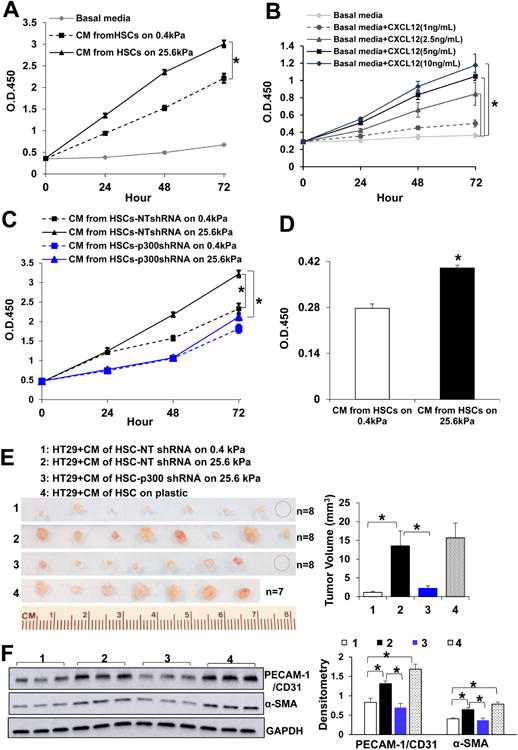
A HSC-conditioned media (CMs) were used as stimulants for HT29 proliferation assay. HSC 25.6 kPa CM promoted HT29 proliferation as compared to 0.4 kPa CM. *, p<0.05 by ANOVA, n=5. B. Recombinant CXCL12 dose-dependently promoted HT29 proliferation. *, p<0.05 by ANOVA, n=5. C. The effect of HSC-CM on HT29 proliferation was suppressed by p300 knockdown in HSCs. *, p<0.05 by ANOVA, n=5. D. Serum-starved HT29 cells stimulated with HSC-CM at 37°C for 2 hours were plated in a 96-well plate (30,000 cells/well) and cultured in serum-free DMEM for additional 24 hours. MTS assay revealed that in vitro stimulation with HSC-CM accelerated cell proliferation. *, p<0.05 by t-test, n=6. E. Serum-starved HT29 were pretreated with CM at 37°C for 2 hours and resuspended in CM, and HT29/CM were coinjected into SCID mice subcutaneously. Tumor nodules were isolated and quantitated at day 7. *, P<0.05 by ANOVA, n=7 or 8 per group. F. Tumor lysates were subjected to WB for α-SMA and PECAM-1/CD31. *, P<0.05 by ANOVA, n=3.
Stiffness-p300 axis potentiates tumor-promoting effects of HSCs in mice
We adapted a tumor cell/CM coimplantation mouse model24, 25 to validate the role of stiffness-p300 axis for in vivo tumor growth. CMs tested included CM of control HSCs on 0.4 kPa or 25.6 kPa, CM of p300 knockdown HSCs on 25.6 kPa, and CM of HSCs on a plastic culture dish as an additional control. Their CXCL12 concentrations were quantitated by ELISA and a highest concentration of CXCL12 was detected in plastic-derived culture medium (Suppl. Fig. 5A). To allow HT29 cells to internalize paracrine factors of CM, we added a step by incubating serum-starved HT29 cells with CM at 37°C for 2 hours before HT29 and CM were re-mixed and coinjected into SCID mice subcutaneously. Indeed, this preincubation step promoted HT29 proliferation as revealed by a MTS assay in a separate study (p<0.05 by t-test, Fig. 6D). Mice were killed 7 days later after HT29/CM coimplantation. As revealed by Fig. 6E, 25.6 kPa CM promoted HT29 growth in mice as compared to 0.4 kPa CM (Group 2 vs. Group 1, P<0.05 by ANOVA), and this effect of 25.6 kPa CM on HT29 growth was suppressed by p300 knockdown in HSCs (Group 3 vs. Group 2, P<0.05 by ANOVA). As expected, CM of HSCs on plastic (>giga Pa) exhibited a strongest effect on promoting tumor growth (Fig. 6E). Since RNA-seq data showed that stiffness increased transcripts of VEGFA, FGF2, IL6, CCL2, and PDGFA, potent angiogenic factors, we next compared the effects of CM on recruiting stromal cells by performing WB for PECAM-1/CD31 (endothelial marker) and α-SMA. Indeed, tumors arising from HT29/25.6 kPa CM and HT29/ plastic CM contained higher levels of PECAM-1/CD31 and α-SMA as compared to other groups, suggesting that stiffness-p300 axis potentiated the effects of HSCs on promoting tumor angiogenesis and desmoplasia in mice (P<0.05 by ANOVA, Fig. 6F).
P300 is upregulated in the MFs of murine and patient liver metastases
We next generated murine liver metastases to investigate p300 expression in activated-HSC/MFs of liver metastases. Two liver metastasis mouse models were used to generate liver metastases: (1) portal vein injection of MC38 murine colorectal or HT29 cells into SCID mice3, 27 and (2) intrasplenic injection of L3.6 human pancreatic cancer cells into SCID mice28. Although both α-SMA and desmin labeled activated-HSC/MFs of MC38 liver metastases, desmin but not α-SMA labeled quiescent HSCs of mouse liver (Suppl. Fig. 6A, top). Both α-SMA/p300 and desmin/p300 double IF revealed strong p300 IF in the activated-HSC/MFs of MC38 liver metastases (Suppl. Fig. 6A bottom and Fig. 7A), which was more than 3 times higher than that of quiescent HSCs (p<0.05 by t-test, Fig. 7A). Similarly, elevated p300 IF was seen in the MFs of HT29 or L3.6 liver metastases as compared quiescent HSCs (Suppl. Fig. 7A). Interestingly, all MC38, HT29 and L3.6 cancer cells expressed high levels of p300 as detected by IF and WB (Suppl. Fig. 6A, 7A and 7B). Additionally, atomic force microscopy (AFM), which measured Young' modulus (stiffness) of tissue cryosections, demonstrated that the stroma of murine liver metastases was significantly stiffer than that of liver (p<0.05 by ANOVA, Suppl. Fig. 7C).
Fig. 7. P300 inactivation in activated-HSC/MFs suppresses liver metastasis in mice.
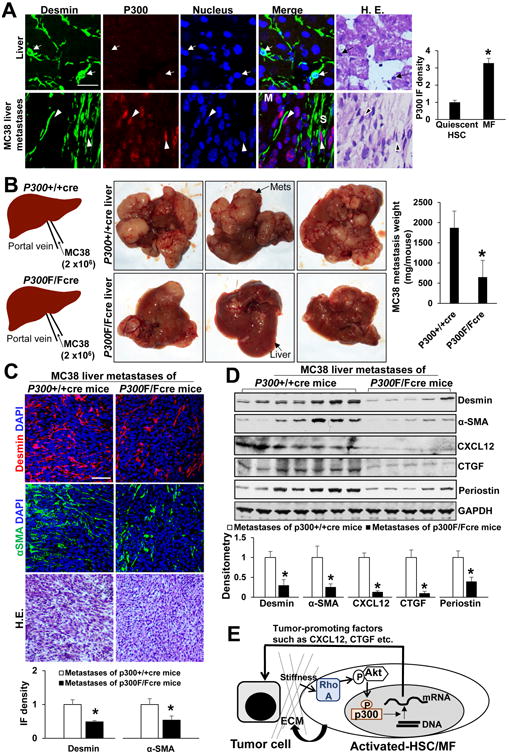
A MC38 colorectal cancer cells were implanted into SCID mice by portal vein injection. Desmin IF labeled both quiescent HSCs and activated-HSC/MFs of liver metastases. P300 IF in activated-HSC/MFs of liver metastases was more than 3 times stronger than it in quiescent HSCs (red channel). *, p<0.05 by t-test, n=12, 10. M: metastasis; S: stroma. Bar, 50 μm. B. P300F/F mice were bred with collgen1A1-cre transgenic mice and age-matched male p300F/Fcre and p300+/+cre mice received MC38 portal vein injection. P300F/Fcre mice developed fewer MC38 liver metastases as compared to matched p300+/+cre mice. *, p<0.05 by t-test, n=7, 7. Mets: metastases. C. and D. IF and WB revealed that MC38 liver metastases of p300F/Fcre mice contained reduced levels of desmin, α-SMA, CXCL12, CTGF, and periostin, as compared to those of p300+/+cre mice. *, p<0.05 by t-test, n=7, 7 for IF and n=7, 5 for WB. Bar: 100 μm. E. A schematic illustration of “an amplification loop” for liver metastatic growth.
Next AFM was performed with colorectal liver metastases of patients and their control liver tissues, which revealed that the stroma of colorectal liver metastases was stiffer than that of liver (p<0.05 by t-test, Suppl. Fig. 8A). Consistent with data of murine liver metastases, p300 IF in the stroma of patient liver metastases was increased as compared with that in the control liver (p<0.05 by t-test, Suppl. Fig. 8B and 8C). Together, these data support the clinical relevance of stiffness-p300 mechanosignaling for colorectal liver metastatic growth.
Liver metastatic growth and CCl4-induced fibrosis were reduced in conditional p300 knockout mice
To study the role of HSC p300 for liver metastatic growth, we generated p300F/Fcre mice by breeding p300F/F mice with cre transgenic mice (collgen1A1-cre). The collagen1A1-cre mice were chosen for crossing because the collagen promoter drove GFP expression in the activated-HSC/MFs after CCl4 treatment35, and additionally, α-SMA and collagen1A1 proteins colocalized to activated-HSC/MFs of liver metastases (Suppl. Fig. 6B). Age-matched male p300F/Fcre and p300+/+cre mice were subjected to portal vein injection of MC38 cells and p300F/Fcre mice developed significantly fewer MC38 liver metastases as compared to p300+/+cre mice (p<0.05 by t-test, Fig. 7B). IF revealed that p300 in the MFs of p300F/Fcre liver metastases was indeed knocked down by cre-loxP system (Suppl. Fig. 9A). IF and WB also revealed that α-SMA and desmin, markers of activated-HSC/MFs, and additionally, CXCL12, CTGF and periostin were reduced in p300F/Fcre metastases as compared to p300+/+cre metastases (p<0.05 by t-test, Fig. 7C and 7D). We also performed WB for RHOA and p-AKT and found that RHOA and p-AKT level of p300F/Fcre metastases were reduced as compared to those of p300+/+cre metastases (p<0.05 by t-test, Suppl. Fig. 9B). Moreover, we gave CCl4 to mice by intraperitoneal injection and found that p300 was upregulated in murine livers after CCl4 injection and that CCl4-mediated HSC activation and collagen deposition was reduced in p300F/Fcre livers as compared to p300+/+cre livers (p<0.05 by t-test, Suppl. Fig. 10). Thus, p300 of HSCs is a key factor for activation of HSCs into MFs in both liver injury and cancer metastasis mouse models.
Discussion
Cancer cells recruit HSCs to build a favorable stromal microenvironment to support their implantation, growth and invasion in the liver 2, 3, 34. However, the role of tumor stroma on the biology of the activate HSC/MFs remains poorly understood. In this regard, we demonstrate for the first time that tumor stiffness in return further enhances HSC activation through HSC p300. Stiffness induces p300 nuclear targeting and p300-dependent transcription of α-SMA and CTGF, markers of HSC activation, and a panel of tumor-promoting factors, including CXCL12, IL11, IL6, PDGFA and B, and VEGFA, so as to enhance HSC activation and their tumor-promoting effects. Our data illustrate “an amplification loop” for metastatic growth in the liver (Fig. 7E). Since histone modifications and chromatin dynamics governing HSC activation remain poorly investigated, representing a gap of current knowledge in the field. Our data also reveal a novel mechanotransduction that connects extracellular mechanical cues to p300-dependent epigenetics in HSCs.
TGF-β is a most potent cytokine inducing MF activation of HSCs and we have reported that p300 knockdown suppressed TGF-β-mediated activation of HSCs into tumor-promoting MFs in vitro, as assessed by IF and WB for α-SMA36. Because TGF-β enhanced, but was not required for stiffness-mediated HSC activation and lung fibroblast activation23, 29, our results support that p300 is required for both TGF-β-induced HSC activation and stiffness-induced HSC activation. Additionally, we show here that p300 disruption in activated-HSC/MFs suppressed CCl4-mediated HSC activation and liver fibrosis in mice (Suppl. Fig. 10). Cell senescence is implicated in the pathogenesis of numerous human diseases and understanding mechanisms governing HSC senescence may lead to novel approaches to reverse liver fibrosis and suppress the prometastatic liver microenvironment37, 38. So we performed additional experiments and found that p300 knockdown or C646 inhibition indeed increased the number of senescent HSCs and cellular p16 protein levels (Suppl. Fig. 11), indicating that p300 acetyltransferase represses HSC senescence in vitro. Taken together, our data support p300 as an important target for suppressing HSC activation, liver fibrosis and the prometastatic liver microenvironment.
We have seen that C3 transferase suppressed RHOA protein levels (Fig. 3B and Fig. 4F), without significantly reducing its mRNA levels (p>0.05 by t-test, Suppl. Fig. 2B). Reduction of RHOA protein by C3 transferase has also been seen in numerous cell lines39. Since C3 transferase induces ADP-ribosylation at asparagine 41 of RHOA, it is possible that this post-translational modification reduces the stability of RHOA protein. In fact, another form of post-translational modification, such as methylation of RHOA, has been shown to increase the half-life of RHOA40. It is interesting to investigate if the stability of RHOA protein is indeed regulated by C3 transferase-mediated ADP-ribosylation or by another mechanism.
Up to 90% of cases of HCC arise in the setting of advanced fibrosis or cirrhosis41, supporting the concept that a stiff microenvironment indeed promotes cancer development in human. However, whether colorectal liver metastasis occurs more on cirrhotic liver of patients still remains as a matter of debate. It has been shown that colorectal liver metastases were enhanced by alcohol-induced or steatohepatitis--induced fibrosis in colorectal cancer patients42, 43. On contrary, lower incidence of colorectal liver metastasis was found in patients with chronic liver diseases44. The exact reasons for these conflicting clinical data remain unknown. However, given the global rise in incidence of chronic liver diseases related previously to hepatitis C and now to steatohepatitis related cirrhosis, it is important to understand the association between liver fibrosis and liver cancer, both primary and metastasis. Regardless, the “amplification loop” identified by this study is related to how tumor-associated stiffness regulates tumor growth in the liver. In our model, cancer cells, disseminated into the liver, interact and activate HSCs to induce a desmoplastic tumor stroma, and the tumor-associated stiffness, in return, enhances metastatic growth and further activates HSCs. Interestingly, an intense expression of p300 was detected in metastatic cells (Suppl. Fig. 7A and B), suggesting that p300 of cancer cells may play a role in cancer cell survival and proliferation in the liver, critical for the development of liver metastasis. Indeed, curcumin, a natural bioactive compound targeting against p300 activity, has been proved in clinical trials as a promising therapeutic agent for breast cancer, pancreatic cancer, and colorectal cancer45. Thus, p300 may represent as a novel target for suppressing both the prometastatic liver microenvironment and metastatic cells.
Supplementary Material
Acknowledgments
The authors wish to acknowledge Mrs. Juan Abrahante and Aaron Becker at the University of Minnesota Genomic Center for their outstanding work on RNA sequencing and data analysis, Dr. Makiko Fujii at Aichi Cancer Center Research Institute, Japan, for providing pcDNA3-FLAG-p300wt plasmid, and Dr. Tatiana Kisseleva at UCSD for the collagen1A1-cre transgenic mouse line. The authors also wish to thank the Clinic Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology for providing patient samples.
Funding: NIH grants R01 CA160069 to N. Kang, R01 HL133320 to V. H. Shah and D. Tschumperlin, and Cell Biology Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567). C. Dou, Z. Liu and C. Chen are funded by China Scholarship Council.
Abbreviations used in this paper
- HSC
hepatic stellate cell
- E1A
adenovirus early region 1A gene
- CCl4
carbon tetrachloride
- MF
myofibroblast
- CM
conditioned medium
- IF
immunofluorescence
- WB
Western blot analysis
- GFP
green fluorescent protein
- CHX
cycloheximide
- WT
wild-type
- ChIP-qPCR
chromatin immunoprecipitation-quantitative polymerase chain reaction
- AFM
atomic force microscopy
- SCID
severe combined immunodeficiency
- cre
cre recombinase
- ELISA
enzyme-linked immunosorbent assay
Footnotes
Conflict of Interest: All authors have declared that no conflict of interest exists
Author Contribution: Changwei Dou, Zhikui Liu, Kangsheng Tu, Hongbin Zhang, Usman Yaqoob, Delphine Sicard, Chen Chen, Yuanguo Wang, Jialing Wen, and Hongzhi Zou performed in vitro experiments and tumor implantation studies in mice. Jan van Deursen and Wei-Chien Huang generated experiment reagents for this study. Daniel Tschumperlin and Raul Urrutia provided reagents and directions to this project. Ningling Kang and Vijay Shah directed this project, analyzed data and wrote this manuscript together with Changwei Dou.
RNA sequencing data: Gene Expression Omnibus (GSE101343).
Additional procedures are in Supplemental Materials and Methods.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vidal-Vanaclocha F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008;1:113–29. doi: 10.1007/s12307-008-0011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology. 2011;54:707–13. doi: 10.1002/hep.24384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu C, Billadeau DD, Abdelhakim H, et al. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest. 2013;123:1138–56. doi: 10.1172/JCI63836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang N, Shah VH, Urrutia R. Membrane-to-Nucleus Signals and Epigenetic Mechanisms for Myofibroblastic Activation and Desmoplastic Stroma: Potential Therapeutic Targets for Liver Metastasis? Mol Cancer Res. 2015;13:604–612. doi: 10.1158/1541-7786.MCR-14-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen SR, Quaranta V, Linford A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549–60. doi: 10.1038/ncb3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrader J, Gordon Walker TT, Aucott RL, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paszek MJ, Zahir N, Johnson KR, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ingber DE. Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circulation research. 2002;91:877–887. doi: 10.1161/01.res.0000039537.73816.e5. [DOI] [PubMed] [Google Scholar]
- 10.Hur EM, Youssef S, Haws ME, et al. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nature immunology. 2007;8:74–83. doi: 10.1038/ni1415. [DOI] [PubMed] [Google Scholar]
- 11.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature cell biology. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. Journal of Biological Chemistry. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 14.Bedford DC, Brindle PK. Is histone acetylation the most important physiological function for CBP and p300? Aging (Albany NY) 2012;4:247–55. doi: 10.18632/aging.100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–77. [PubMed] [Google Scholar]
- 16.Bricambert J, Miranda J, Benhamed F, et al. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. The Journal of clinical investigation. 2010;120:4316–4331. doi: 10.1172/JCI41624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lv L, Xu YP, Zhao D, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Molecular cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji M, Zhang Q, Ye J, et al. Myostatin induces p300 degradation to silence cyclin D1 expression through the PI3K/PTEN/Akt pathway. Cellular signalling. 2008;20:1452–1458. doi: 10.1016/j.cellsig.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Girdwood D, Bumpass D, Vaughan OA, et al. P300 transcriptional repression is mediated by SUMO modification. Molecular cell. 2003;11:1043–1054. doi: 10.1016/s1097-2765(03)00141-2. [DOI] [PubMed] [Google Scholar]
- 20.Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol Cell Biol. 2005;25:6592–602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharyya S, Ghosh AK, Pannu J, et al. Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor beta. Arthritis Rheum. 2005;52:1248–58. doi: 10.1002/art.20996. [DOI] [PubMed] [Google Scholar]
- 22.Yuan H, Reddy MA, Sun G, et al. Involvement of p300/CBP and epigenetic histone acetylation in TGF-beta1-mediated gene transcription in mesangial cells. Am J Physiol Renal Physiol. 2013;304:F601–13. doi: 10.1152/ajprenal.00523.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–57. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu J, Ye X, Fan F, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer cell. 2013;23:171–185. doi: 10.1016/j.ccr.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau EY, Lo J, Cheng BY, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016;15:1175–89. doi: 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 26.Kasper LH, Fukuyama T, Biesen MA, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol. 2006;26:789–809. doi: 10.1128/MCB.26.3.789-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker NK, Abdelmoneim SS, Yaqoob U, et al. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol. 2008;173:1002–12. doi: 10.2353/ajpath.2008.080158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruns CJ, Harbison MT, Kuniyasu H, et al. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen AL, Bloomer SA, Chan EP, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–8. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang X, Yang N, Fiore VF, et al. Matrix stiffness–induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. American journal of respiratory cell and molecular biology. 2012;47:340–348. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Görtzen J, Schierwagen R, Bierwolf J, et al. Interplay of matrix stiffness and c-SRC in hepatic fibrosis. Frontiers in physiology. 2015;6:359. doi: 10.3389/fphys.2015.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 33.Sakai N, Yoshidome H, Shida T, et al. CXCR4/CXCL12 expression profile is associated with tumor microenvironment and clinical outcome of liver metastases of colorectal cancer. Clin Exp Metastasis. 2012;29:101–10. doi: 10.1007/s10585-011-9433-5. [DOI] [PubMed] [Google Scholar]
- 34.Tu K, Li J, Verma VK, et al. VASP promotes TGF-β activation of hepatic stellate cells by regulating Rab11 dependent plasma membrane targeting of TGF-β receptors. Hepatology (Baltimore, Md) 2015;61:361. doi: 10.1002/hep.27251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo L, Xiang X, Shah HV, Kang N. P300 promotes TGF-β stimulated nuclear translocation of SMADs and activation of hepatic stellate cells into liver metastasis promoting myofibroblasts. Hepatology (Baltimore, Md) 2015;62:1157A. Abstract. [Google Scholar]
- 37.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–9. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohrbeck A, von Elsner L, Hagemann S, et al. Uptake of clostridium botulinum C3 exoenzyme into intact HT22 and J774A.1 cells. Toxins (Basel) 2015;7:380–95. doi: 10.3390/toxins7020380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Backlund PS., Jr Post-translational processing of RhoA. Carboxyl methylation of the carboxyl-terminal prenylcysteine increases the half-life of Rhoa. J Biol Chem. 1997;272:33175–80. doi: 10.1074/jbc.272.52.33175. [DOI] [PubMed] [Google Scholar]
- 41.Wallace MC, Friedman SL. Hepatic fibrosis and the microenvironment: fertile soil for hepatocellular carcinoma development. Gene Expr. 2014;16:77–84. doi: 10.3727/105221614X13919976902057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maeda M, Nagawa H, Maeda T, et al. Alcohol consumption enhances liver metastasis in colorectal carcinoma patients. Cancer. 1998;83:1483–8. doi: 10.1002/(sici)1097-0142(19981015)83:8<1483::aid-cncr2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 43.Kondo T, Okabayashi K, Hasegawa H, et al. The impact of hepatic fibrosis on the incidence of liver metastasis from colorectal cancer. Br J Cancer. 2016;115:34–9. doi: 10.1038/bjc.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Augustin G, Bruketa T, Korolija D, et al. Lower incidence of hepatic metastases of colorectal cancer in patients with chronic liver diseases: meta-analysis. Hepatogastroenterology. 2013;60:1164–8. doi: 10.5754/hge11561. [DOI] [PubMed] [Google Scholar]
- 45.Nebbioso A, Carafa V, Benedetti R, et al. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012;6:657–82. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.