

Abstract
Pathogenic T helper 2 (TH2) cells, which produce increased amounts of the cytokines interleukin-5 (IL-5) and IL-13, promote allergic disorders, including asthma. Thymic stromal lymphopoietin (TSLP), a cytokine secreted by epithelial and innate immune cells, stimulates such pathogenic TH2 cell responses. We found that TSLP signaling in mouse CD4+ T cells initiated transcriptional changes associated with TH2 cell programming. IL-4 signaling amplified and stabilized the genomic response of T cells to TSLP, which increased the frequency of T cells producing IL-4, IL-5, and IL-13. Furthermore, the TSLP- and IL-4–programmed TH2 cells had a pathogenic phenotype, producing greater amounts of IL-5 and IL-13 and other proinflammatory cytokines than did TH2 cells stimulated with IL-4 alone. TSLP-mediated TH2 cell induction involved distinct molecular pathways, including activation of the transcription factor STAT5 through the kinase JAK2 and repression of the transcription factor BCL6. Mice that received wild-type CD4+ T cells had exacerbated pathogenic TH2 cell responses upon exposure to house dust mites compared to mice that received TSLP receptor–deficient CD4+ T cells. Transient TSLP signaling stably programmed pathogenic potential in memory TH2 cells. In human CD4+ T cells, TSLP and IL-4 promoted the generation of TH2 cells that produced greater amounts of IL-5 and IL-13. Compared to healthy controls, asthmatic children showed enhancement of such T cell responses in peripheral blood. Our data support a sequential cytokine model for pathogenic TH2 cell differentiation and provide a mechanistic basis for the therapeutic targeting of TSLP signaling in human allergic diseases.
INTRODUCTION
T helper 2 (TH2) cells are effector T cells that differentiate from naïve CD4+ T cells to produce the cytokines interleukin-4 (IL-4), IL-5, and IL-13. They enable protection against extracellular parasites but also promote allergic inflammation (1). IL-4 is not only produced by TH2 cells but also required for their differentiation in vitro and in vivo (2). IL-4 signaling results in the activation of the transcription factor signal transducer and activator of transcription 6 (STAT6), which, in turn, induces the expression of GATA3, which encodes a key TH2 cell–specific transcription factor. STAT6 and GATA3 (GATA binding protein 3) bind to regulatory regions of the TH2 cytokine locus and activate expression of the IL4, IL5, and IL13 genes. Although IL-4 is produced by activated CD4+ T cells that are differentiating into TH2 cells, the source of IL-4 in vivo during the initial stages of T cell activation remains unresolved. Several studies have identified additional cytokines that promote TH2 cell responses in vivo (1, 3–5). One of these is thymic stromal lymphopoietin (TSLP), which is produced by epithelial cells upon injury, dysfunction, or infection. Furthermore, TSLP is also produced by dendritic cells (DCs) and, thereby, could function during T cell priming in lymph nodes (6, 7).
TSLP is strongly implicated in the pathogenesis of TH2 cell–mediated allergic disorders, including atopic dermatitis, allergic asthma, food allergy, and eosinophilic esophagitis (8). Some studies have reported that TSLP primarily acts on DCs to promote pathogenic TH2 responses (9, 10). However, others have implicated a role for TSLP signaling in CD4+ T cells in TH2 cell–mediated inflammation (11–14). In this regard, ovalbumin (OVA)–sensitized, TSLP receptor (TSL-PR)–deficient (Crlf2−/−) mice fail to develop an inflammatory TH2 cell response in their lungs to inhaled antigen (15). However, transfer of wild-type (WT) CD4+ T cells that express the TSL-PR into OVA- sensitized Crlf2−/− mice promotes allergic inflammation. Similarly, injection of WT CD4+ T cells into Crlf2−/− mice also results in the development of allergic inflammation in the gut to OVA administration (16). Thus, TSLP signaling in CD4+ T cells is required for the generation of robust pathogenic TH2 responses in vivo. However, these analyses have not uncovered a direct role for TSLP in the differentiation of pathogenic TH2 cells.
TSLP signals in mouse and human CD4+ T cells to induce the activation of the transcriptional regulator STAT5 through a pathway requiring the kinases Janus kinase 1 (JAK1) and JAK2 (11, 14). TSLP increases the survival of naïve CD4+ T cells and that of memory TH2 cells; the latter are a form of quiescent TH2 cells that can be rapidly reactivated upon exposure to cognate antigens (17). TSLP induces the secretion of IL-4 and IL-9 in conjunction with T cell receptor (TCR) activation (12, 13, 18). In turn, IL-4 increases the expression of the TSLPR on CD4+ T cells, suggesting a positive feedback loop between the two cytokines (19). We explored whether TSLP could directly promote the differentiation of naïve CD4+ T cells into TH2 cells, particularly a “pathogenic” effector state that produces large amounts of IL-5 and IL-13. Furthermore, we analyzed the interplay between TSLP signaling and IL-4 signaling in the initiation and amplification of the TH2 cell differentiation response and its culmination into a pathogenic state. Finally, we assessed the response of human CD4+ T cells to TSLP signaling and whether such potentially pathogenic TH2 cell responses were enhanced in the context of asthma.
RESULTS
TSLP directly promotes TH2 cell differentiation
To test the role of TSLP signaling in promoting TH2 cell differentiation, we activated naïve CD4+ T cells in vitro with antibodies stimulating CD3 and CD28 and neutralizing interferon-γ (IFN-γ) in the presence of IL-4, TSLP, or both for 3 days. Compared with IL-4–induced TH2 cell differentiation, TSLP statistically significantly increased the frequency of TH2 cells producing IL-4, IL-5, and IL-13 (Fig. 1, A and B) and increased the amount of IL-5 and IL-13 produced per cell (fig. S1A). Increase in both the frequency and amplitude of production resulted in increased secretion of IL-4, IL-5, and IL-13 (fig. S1, B and C). Notably, the addition of exogenous IL-4 during TSLP stimulation did not augment TH2 cytokine production (Fig. 1, A and B). Here-after, we refer to TSLP-derived TH2 cells as TH2TSLP cells and TH2 cells produced by exogenously added IL-4 as TH2IL-4 cells. TSLP did not affect the survival or proliferation of activated CD4+ T cells (fig. S1, D and E) nor the induction of interferon regulatory factor 4 (IRF4) (fig. S1F), which is an essential transcription factor for T cell activation and TH2 cell differentiation (20, 21). Consistent with a previous report (14), TSLP reduced the apoptosis of naïve CD4+ T cells (fig. S1D). These results suggest that TSLP signaling increases the frequency of TH2 cells that produce IL-4, IL-5, and IL-13 through mechanisms that do not involve altered IRF4 activation or enhanced survival or proliferation of activated cells.
Fig. 1. TSLP signaling increases the frequency of TH2 cells expressing IL-4, IL-5, and IL-13.

(A and B) To generate T helper 2 (TH2) cells, naïve CD4+ T cells were activated for 3 days with antibodies against CD3 (αCD3; 5 μg/ml) and CD28 (αCD28; 2 μg/ml) with neutralization of interferon-γ (αIFN-γ; 10 μg/ml) in the absence (TH0) or presence of the indicated cytokines at the following concentrations: interleukin-4 (IL-4) (20 ng/ml; TH2IL-4), thymic stromal lymphopoietin (TSLP) (40 ng/ml; TH2TSLP), IL-4 and TSLP (TH2IL-4+TSLP), or IL-7 (20 ng/ml). (A) The frequency of cytokine-positive cells in a representative experiment is shown. Top: Cells were assessed for IL-13 and IL-4 by flow cytometry. Bottom: Cells were assessed for IL-13 and IL-5. Data in (B) are pooled from 10 independent experiments. ns, not significant. (C and D) Naïve CD4+ T cells were stimulated with indicated cytokines at the same concentrations as those in (A) for 15 min, and then, the phosphorylation of signal transducer and activator of transcription 5 (STAT5) was analyzed by flow cytometry with an antibody against phosphorylated STAT5 (pSTAT5). (C) Geometric mean fluorescent intensity (GMFI; x axes) of pSTAT5 in a representative experiment. Data in (D) are pooled from seven independent experiments. Quantitative data are means ± SEM and were analyzed by one-way analysis of variance (ANOVA). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
TSLP signaling is mediated by a receptor complex containing IL-7 receptor α (IL-7Rα) and TSLPR (22). Given the shared IL-7Rα sub-unit between TSLPR and IL-7R, we tested the effect of IL-7 on TH2 cell differentiation in relation to TSLP. Although IL-7 also stimulated the production of IL-13–positive cells that were indicative of TH2 cell differentiation, IL-7 was less effective than TSLP (Fig. 1, A and B) despite inducing greater STAT5 activation under the culture conditions (Fig. 1, C and D). These results suggest that TSLP may use one or more additional signaling pathways in conjunction with the JAK-STAT5 pathway to efficiently drive TH2 cell differentiation.
TSLP initiates TH2 cell programming in the absence of IL-4
Although TSLP signaling induced an increase in the abundance of IL-4 (fig. S1B) and IL-4Rα (fig. S2A), which promote TH2 cell differentiation (12), the observation that TSLP increased the frequency of IL-5– and IL-13–positive cells to a greater extent than did IL-4 alone suggested that TSLP could function independently of or in synergy with IL-4. To test the action of TSLP in the absence of IL-4 signaling, we included function-blocking antibodies to IL-4 and IL-4Rα during CD4+ T cell differentiation. Even in the presence of the function-blocking antibodies, TSLP stimulated the induction of Il4, Il5, and Il13 expression within 24 hours of CD4+ T cell activation (Fig. 2A). However, expression of these TH2 cytokine genes was not amplified at the later time points in the absence of IL-4. This correlated with the lack of induction of Gata3 expression by TSLP in the absence of IL-4 signaling, which is consistent with a dependence of Gata3 expression on the IL-4/STAT6 pathway. As expected, the function-blocking antibodies abolished IL-5 and IL-13 secretion during IL-4–induced TH2 cell differentiation (Fig. 2B), indicating that this was an effective method for blocking IL-4 signaling. Consistent with the gene expression data (Fig. 2A), TSLP, in the presence of the IL-4– and IL-4Rα (IL-4/IL-4Rα)–blocking antibodies, induced IL-5 and IL-13 secretion but at substantially reduced amounts (Fig. 2B).
Fig. 2. TSLP initiates TH2 cell programming, which is amplified by IL-4.

(A and B) Naïve CD4+ T cells were activated with antibodies, as described in Fig. 1A, in the presence or absence of blocking antibodies against IL-4 and IL-4Rα (αIL-4/4R). TH2 cells differentiated in response to IL-4 (TH2IL-4) served as a control for TH2 cell induction. (A) Kinetics of induction of the indicated genes as determined by real-time polymerase chain reaction (PCR) analysis. Data are representative of three independent experiments and are means ± SD of replicates from a single experiment. (B) The amounts of the indicated cytokines in the cell culture medium were determined by enzyme-linked immunosorbent assay on day 3. Data are means ± SEM of five experiments and were evaluated by one-way ANOVA. (C) Naïve Il4−/− CD4+ T cells were activated as described in Fig. 1A in the presence or absence of TSLP (40 ng/ml), IL-4 (20 ng/ml), or both. The amounts of the indicated cytokines in the cell culture medium were analyzed as described in (B). Data are means ± SEM of eight experiments and were analyzed by one-way ANOVA. (D and E) Naïve Il4−/− CD4+ T cells were activated as described in Fig. 1A in the presence or absence of TSLP (40 ng/ml) and the indicated concentrations of IL-4. Cells were assessed for IL-13 and IL-4 content by flow cytometry. Data in (D) are from a representative experiment. Data in (E) are means ± SEM of seven independent experiments and were analyzed by paired, two-tailed t test. *P ≤ 0.05, **P ≤ 0.01, and ****P ≤ 0.0001.
To complement the antibody-mediated blockade of IL-4 signaling, we performed similar experiments with Il4−/− CD4+ T cells. As we observed with the IL-4/IL-4Rα–blocking antibodies, activation of Il4−/− CD4+ T cells with TSLP induced production of both IL-5 and IL-13, albeit at reduced amounts than in the presence of exogenously added IL-4 (Fig. 2C). Without exogenously added IL-4, the abundance of GATA3 was not increased (fig. S2B). To examine the potential synergistic actions of TSLP with IL-4 on TH2 cell differentiation, we activated Il4−/− CD4+ T cells in the presence of TSLP and increasing concentrations of IL-4. At each concentration of IL-4 used, TSLP in conjunction with IL-4 induced a greater increase in the frequency of IL-13–positive cells (Fig. 2, D and E) and the secretion of IL-5 and IL-13 (fig. S2C) than their sum with the individual cytokines (table S1). Together, these results suggest that TSLP initiates TH2 cytokine gene expression in the absence of IL-4–mediated GATA3 induction and that TSLP functions with IL-4 to amplify TH2 cell differentiation (the frequency of cells producing the cytokines) and enhance TH2 cytokine production (the amount of cytokines produced per cell).
TSLP promotes TH2 cytokine gene expression by repressing Bcl6
We next performed RNA sequencing (RNA-seq) analysis of differentiating TH2IL-4 and TH2TSLP cells to determine whether they manifested distinctive genomic states. Overall, the genome-wide expression profiles of TH2IL-4 and TH2TSLP cells were similar to each other and distinguishable from that of TH0 cells (fig. S3A). TH2IL-4 and TH2TSLP cells displayed the increased expression of 229 genes and the reduced expression of 148 genes by at least twofold when compared with TH0 cells (table S2). TH2IL-4 and TH2TSLP cells showed increased expression of genes required for TH2 cell differentiation and T cell survival, such as Il4, Gata3, Nfil3, Batf3, Atf3, Id2, Rbpj, Il2ra, Bcl2, and Il7r (fig. S3A and table S3). Both IL-4 and TSLP repressed the expression of genes associated with the TH1 and TH17 cell lineages, including Stat4, Rorc, Zbtb32, Bcl6, Cd27, Cd70, and Ccr6.
To analyze genes that were differentially expressed in TH2TSLP and TH2IL-4 cells, we focused separately on the induced and repressed genes. Of the 229 induced genes, 134 were differentially expressed by at least twofold in TH2TSLP versus TH2IL-4 cells at the various time points; 22 of these were increased by TSLP, whereas 112 were increased with IL-4 (Fig. 3A and table S4). Of 22 genes that were preferentially induced in TH2TSLP cells, most encoded TH2 cytokines or other proinflammatory mediators. The increased expression of select genes was validated by quantitative polymerase chain reaction (PCR) analysis (Fig. 3B). These results show that TH2TSLP cells preferentially express a small module of allergy-promoting and proinflammatory cytokine genes and this likely enables their enhanced effector functions.
Fig. 3. TSLP induces the expression of TH2 cytokine genes and represses Bcl6.

(A) Naïve CD4+ T cells were activated as described in Fig. 1A in the presence or absence of IL-4 or TSLP, and transcripts were kinetically profiled by RNA sequencing. The heat map shows genes that were differentially induced by TSLP or IL-4 at the indicated times. Genes shown manifest reads per kilobase of transcript, per million mapped reads (RPKM) of >5 at least one time point and a log2 fold-change value of >2. Genes that were preferentially induced by TSLP at later times are highlighted in the yellow oval and indicated in the right panel. See table S4 for complete data. (B) Real-time quantitative PCR (qPCR) analysis of indicated genes induced in TH0, TH2IL-4, and TH2TSLP cells. Transcript abundance was normalized to that of Eif3k, a housekeeping gene. Data are representative of four independent experiments. Data are means ± SD of three technical replicates. (C) The kinetics of Bcl6 expression were analyzed by qPCR. Data are means ± SEM of four experiments. (D) Flow cytometry analysis of the percentages of BCL6+ cells under the indicated conditions. Data are means ± SEM of six experiments. Data in (C) and (D) were analyzed by two-way ANOVA to compare Bcl6 expression or protein abundance in TH2TSLP cells to that in TH0 cells. (E) Wild-type (WT) naïve CD4+ T cells were preactivated with antibodies against CD3, CD28, and IFN-γ for 1 day and then were transduced with retroviruses expressing green fluorescent protein (GFP) or Bcl6-GFP. After an additional 2 days of activation and 1 day of rest in culture medium, the cells were sorted for GFP expression and then reactivated with antibodies against CD3 and CD28 in the presence or absence of TSLP. RNA was isolated after 14 hours and analyzed for Il4, Il5, and Il13 transcripts by qPCR. Data are means ± SEM of four experiments. Where statistics are indicated, data were analyzed by two-way ANOVA. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Of the repressed genes, Bcl6 was the most differentially affected in TH2TSLP cells, and this was confirmed by real-time PCR analysis (Fig. 3C), which resulted in reduced protein abundance, as analyzed by flow cytometry (Fig. 3D and fig. S3B). To test whether the repression of Bcl6 expression by TSLP signaling was required for TH2 cell differentiation, we transduced activated CD4+ T cells with a retroviral vector encoding Bcl6. Enforced expression of BCL6 in activated CD4+ T cells abolished the TSLP-induced expression of TH2 cytokine genes (Fig. 3E). Our results are consistent with the finding that BCL6 directly represses the Il13 gene in the context of T follicular helper cell differentiation (23). We therefore propose that the antagonism of Bcl6 induction by TSLP (Fig. 3D) promotes the increased frequency and cytokine output of IL-13–producing TH2TSLP cells.
TSLP induces TH2 cytokine expression through JAK2 and STAT5
To determine whether the JAK-STAT5 pathway activated by TSLP (14), in the absence of IL-4 signaling, was required for the induction of TH2 cytokine gene expression, we performed experiments with Il4−/− CD4+ T cells and a JAK2-selective inhibitor (14, 24). We confirmed that JAK2 inhibitor II blocked the phosphorylation of STAT5 induced by TSLP (Fig. 4A) but not the phosphorylation of STAT6 induced by IL-4 (fig. S4A) nor the phosphorylation of STAT5 induced by IL-7 (fig. S4B); signaling by the latter cytokines is dependent on the kinases JAK1 and JAK3 (25). Furthermore, the induction of the Il5 and Il13 genes by TSLP was abrogated by the JAK2 inhibitor (Fig. 4B). As expected, the induction of Gata3 expression by IL-4 signaling was unaffected under these conditions of JAK2 inhibition (Fig. 4B). Surprisingly, the repression of BCL6 by TSLP signaling was not affected by the inhibition of the JAK2-STAT5 pathway (fig. S4C). To validate these results, we used retroviral Cre-mediated deletion of the Stat5a and Stat5b genes in preactivated and rested CD4+ T cells isolated from Stat5a/5bfl/fl mice. In the absence of STAT5 (fig. S4D), TSLP signaling was unable to induce expression of TH2 cytokine genes (Fig. 4C). Note that TSLP still inhibited BLC6 accumulation in the absence of STAT5A/STAT5B (Fig. 4D). We note that, in CD4+ T cells, which were activated and then rested to enable retroviral transduction (“preactivated”), loss of STAT5A/STAT5B resulted in a modest increase in BCL6 abundance. Nevertheless, experiments involving the chemical inhibition of JAK2, when coupled with deletion of the Stat5a/Stat5b locus and constitutive expression of Bcl6, provide evidence that there are distinct pathways by which TSLP signaling promotes robust TH2 cell differentiation.
Fig. 4. TSLP induction of TH2 cytokine genes depends on JAK2 and STAT5 activity.

(A) Naïve Il4−/− CD4+ T cells were preincubated for 1 hour with dimethyl sulfoxide (DMSO) or with Janus kinase 2 (JAK2) inhibitor (inh) II (50 μM). Left: The cells were then stimulated with TSLP for 15 min before the phosphorylation of STAT5 was analyzed by flow cytometry, as described in Fig. 1C. Right: Data are means ± SEM of five experiments analyzed by ANOVA. (B) CD4+ T cells were activated in the presence of DMSO or JAK2 inhibitor II with or without TSLP or IL-4 for 24 hours, and the abundances of the indicated transcripts were then analyzed. Data are means ± SEM of three experiments analyzed by ANOVA. (C and D) Naïve Stat5a/5bfl/fl CD4+ T cells were preactivated for 1 day and then transduced with retroviruses expressing GFP or Cre-GFP, as described in Fig. 3E. The GFP + cells were sorted and restimulated with antibodies against CD3 and CD28 with or without TSLP. (C) The abundances of the indicated transcripts were analyzed 14 hours after activation by qPCR. Data are means ± SEM of three experiments and were analyzed by two-way ANOVA. (D) The percentages of BCL6+ cells were analyzed by flow cytometry 5 hours after reactivation. Data are means ± SEM of three experiments and were analyzed by two-way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
TSLP enhances activating chromatin modifications at TH2 cytokine loci
We next analyzed the chromatin states of TH2TSLP versus TH2IL-4 cells by performing chromatin immunoprecipitation sequencing (ChIP-seq) analysis for the activating histone modification, H3K27ac. Consistent with our RNA-seq analysis, most H3K27ac regions that were induced preferentially under TH2-polarizing conditions (2611 peaks) were shared in TH2IL-4 and TH2TSLP cells (fig. S5A and table S5). However, there were a small number of H3K27ac regions (90 peaks) that were more highly acetylated in the presence of TSLP. Furthermore, seven of these hyper-acetylated chromatin regions occurred within the TH2 cytokine locus that encodes Il4, Il5, and Il13 (Fig. 5A and table S5). Analysis of H3K4me3 by ChIP-seq demonstrated that the hyper-acetylated regions around the Il13 locus were associated with increased H3K4me3 in the presence of TSLP (Fig. 5A). Similar patterns of activating histone modifications, which were dependent on TSLP signaling, were observed near the Il24 gene (Fig. 5B), which encodes an additional TH2-type cytokine (26, 27). As anticipated by the analysis of activating histone modifications, the Il24 gene was more highly expressed in TH2TSLP cells than in TH2IL-4 cells (fig. S5B). Furthermore, STAT5 activated by TSLP bound to regulatory regions within the TH2 cytokine and Faim/Il24 loci, which displayed enhanced H3K27ac and H3K4me3 in TH2TSLP cells (Fig. 5, A and B).
Fig. 5. TSLP augments activating chromatin modifications in TH2 cytokine loci.
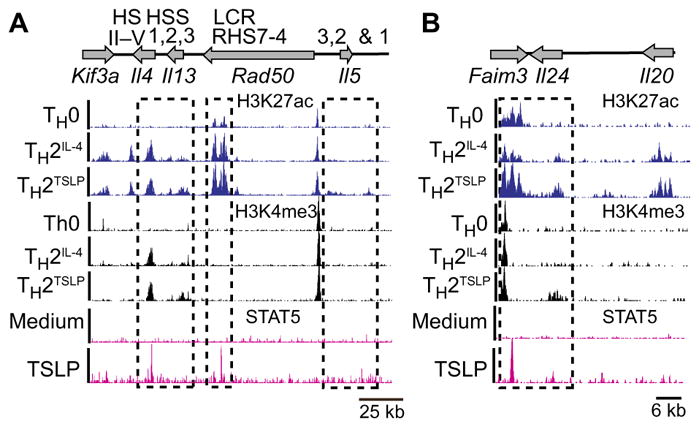
(A and B) Naïve CD4+ T cells were activated as described in Fig. 1A in the presence or absence of IL-4 or TSLP for 3 days and then were analyzed by chromatin immunoprecipitation sequencing (ChIP-seq) for the indicated histone modifications. ChIP-seq tracks for H3K27ac and H3K4me3 are displayed for the TH2 cytokine locus (A) and the Faim/Il24/Il20 locus (B) in the indicated effector states. Results are representative of two independent experiments. For the analysis of TSLP-induced STAT5-binding regions in the genome, naïve CD4+ T cells were preactivated for 3 days with antibodies against CD3, CD28, and IFN-γ and then rested for 3 days. On day 7, the cells were left unstimulated or were stimulated with TSLP (40 ng/ml) for 25 min and then analyzed by ChIP-seq for STAT5 binding to chromatin. Histone ChIP-seq data were superimposed with STAT5 ChIP-seq data from TSLP-activated CD4+ T cells.
Given that TH2TSLP cells depend on an interplay between exogenous TSLP and endogenous IL-4 that is expressed during their differentiation, we examined whether TSLP signaling could induce H3K27ac in the absence of IL-4 at the early time point, when TSLP-mediated expression of TH2 cytokines was observed (Fig. 2A). ChIP-seq analysis of Il4−/− CD4+ T cells activated in the presence of TSLP for 24 hours revealed that TSLP did not induce H3K27ac at focal regions within the TH2 cytokine locus in the absence of IL-4 (fig. S5C). Rather, TSLP enhanced IL-4–induced H3K27ac marks. Given that TSLP induced the expression of the Il5 and Il13 in the absence of IL-4 signaling within 24 hours of T cell activation (fig. S5D) and that this transcriptional programming was not associated with increased H3K27ac, our results suggest a mechanism for TSLP-activated gene expression that is independent of this histone modification. This mechanism may also function to enhance IL-4–mediated gene activation that is associated with increased H3K27ac.
The TSLP-primed effector state is stably programmed in memory TH2 cells
To determine whether the robust TSLP-primed effector state was stably programmed in memory TH2 cells, we used an adoptive transfer approach (28, 29). Naïve CD45.1+ CD4+ T cells were differentiated into TH2IL-4 or TH2TSLP cells (Fig. 1A), and equal numbers were transferred into CD45.2+ recipient mice (Fig. 6A). TH0 cells were used as controls. The frequency and phenotype of the donor cells(CD45.1+CD4+) were assessed 34 days after transfer. TH2IL-4 or TH2TSLP cells were recovered in equivalent numbers that were more abundant than their TH0 counterparts (Fig. 6B). Consistent with their memory phenotype, a large fraction of the TH2 and TH2TSLP cells displayed cell surface expression of CD44 and IL-7Rα (CD127) (Fig. 6C). Consistent with their memory TH2 cell state, the TH2IL-4 and TH2TSLP cells rapidly increased the abundance of the transcription factors GATA3 and IRF4 after stimulation with antibodies against CD3 and CD28 compared with TH0 cells and naïve cells (Fig. 6D). Furthermore, they gave rise to an increased frequency of cells producing IL-4, IL-5, or IL-13 (Fig. 6E). Furthermore, memory TH2TSLP cells manifested a higher frequency of IL-5 and IL-13 producers when compared with conventional memory TH2IL-4 cells. This correlated with increased H3K27ac abundance in regulatory regions downstream of the Il5 and Il13 genes in memory TH2TSLP cells after secondary activation (Fig. 6F). These results suggest that TSLP signaling during a primary CD4+ T cell response can stably program a greater effector cytokine output in memory TH2 cells, which is manifested after secondary activation simply by engagement of the TCR and its co-receptor CD28. Such molecular programming may involve an alteration of chromatin structure that predisposes for increased H3K27 acetylation at the Il5 and Il13 genes upon secondary activation.
Fig. 6. TH2TSLP memory cells manifest increased expression of TH2 cytokines upon reactivation.
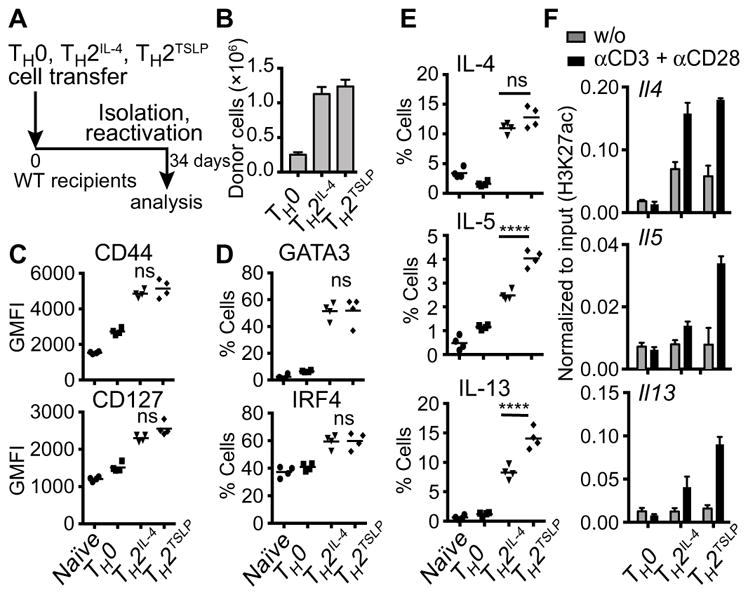
(A) Schematic of the experimental design. Naïve CD45.1+CD4+ T cells activated with antibodies against CD3, CD28, IFN-γ, and IL-4 or TSLP for 3 days and then rested for an additional 3 days in culture medium supplemented with IL-7 (0.5 ng/ml) were transferred into CD45.2+ WT mice. On day 34, total CD4+ T cells were purified from the spleens of the recipient mice and analyzed. (B) Numbers of donor cells in the spleens of recipient mice. (C) Flow cytometric analysis of the expression of the indicated cell surface markers of memory. (D) Analysis of the percentages of cells expressing GATA3 (GATA binding protein 3) or IRF4 after reactivation for 6 hours with antibodies against CD3 and CD28. (E) Analysis of the percentages of cells expressing the indicated cytokines after reactivation with antibodies against CD3 and CD28 for 2 days with phorbol 12, 13-dibutyrate (PDBU) and ionomycin added for the last 5 hours. Data were analyzed by one-way ANOVA. Data in (B) to (E) are from donor cells that were gated on CD45.1+CD4+ cells. Data are means ± SEM of four mice for each group and are from one of three representative experiments. For these experiments, naïve CD4 cells were analyzed 12 days after their transfer into WT recipients because these cells have a shorter half-life. (F) The indicated CD45.1+ CD4+ T cells were sorted from the spleens of CD45.2+ recipient mice 34 days after transfer. The T cells were then activated for 24 hours with antibodies against CD3 and CD28. H3K27ac was analyzed at the promoter of Il4 and at downstream regulatory elements of Il5 and Il13 by ChIP-PCR. Data are from one of three representative experiments. ****P ≤ 0.0001.
TSLP signaling in T cells provokes a hyperinflammatory TH2 response
To test the role of TSLP signaling in CD4+ T cells in enhancing a pathogenic TH2 cell response, we used the house dust mite (HDM) model of allergic airway inflammation that is widely used for the study of allergic asthma (30). Tcra−/− mice, which lack endogenous T cells, were reconstituted with WT or TSLPR-deficient (Crlf2−/−) CD4+ T cells (31). One month later, the mice were sensitized with HDM by intratracheal administration (Fig. 7A). This experimental design enabled us to specifically examine the effects of perturbing TSLP signaling in CD4+ T cells while retaining TSLP responsiveness in other immune cells. Analysis of CD4+ T cells in the blood of control and test mice showed comparable numbers of donor cells (Fig. 7B). Upon HDM administration, there was an equivalent increase in the numbers of donor CD4+ T cells in the lungs of both groups of mice. Compared to HDM-exposed mice reconstituted with WT CD4+ T cells, similarly exposed mice reconstituted with Crlf2−/− CD4+ T cells manifested a weaker TH2 inflammatory response, which was characterized by reduced amounts of IL-4, IL-5, and IL-13 (Fig. 7C) and diminished numbers of eosinophils and macrophages in the bronchoalveolar lavage (BAL) fluid (Fig. 7D). Note that the secretion of IFN-γ and IL-17A was not affected (fig. S6A). Consistent with these findings, mice transplanted with Crlf2−/− CD4+ T cells manifested reduced bronchial hyper-reactivity to challenge with methacholine (Fig. 7E). Together, these results establish that TSLP signaling in CD4+ T cells is required for a potent TH2 cell response and exacerbated airway inflammation in the context of HDM sensitization.
Fig. 7. TSLP signaling in CD4+ T cells promotes hyperinflammatory TH2 response in vivo.
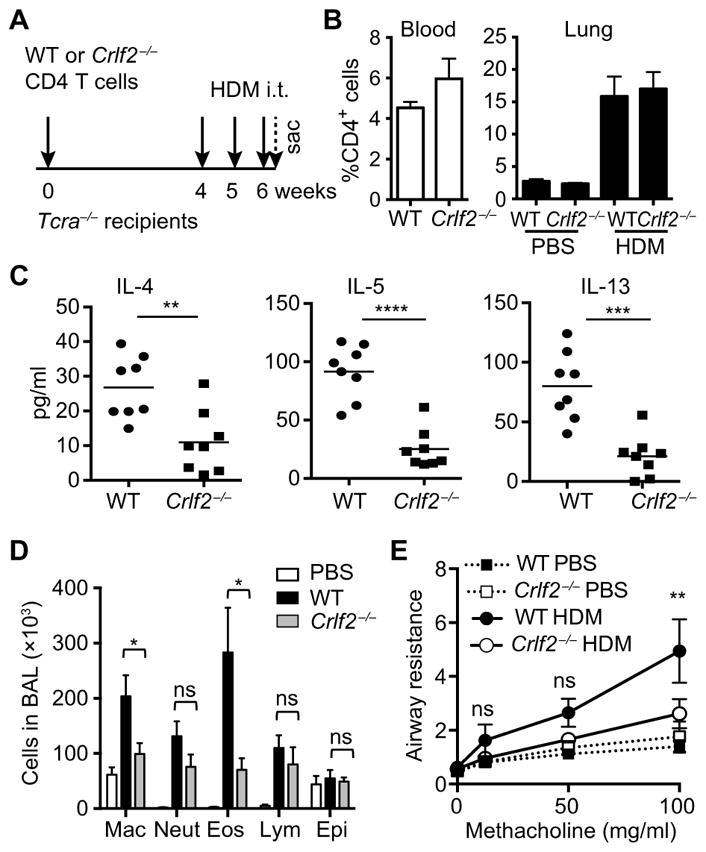
(A to D) WT or Crlf2−/− CD4+ T cells were adoptively transferred into Tcra−/− mice. Four weeks after T cell reconstitution, the mice were sensitized with 100 μg of house dust mite (HDM) administrated intratracheally (i.t.) on a weekly basis and sacrificed on day 2 after the third HDM injection. (A) Schematic of the experimental design. (B) The percentages of CD4+ T cells in the peripheral blood of indicated mice before HDM injection and in the lung after administration of phosphate-buffered saline (PBS) or HDM were determined by flow cytometry. (C) Analysis of the concentrations of IL-4, IL-5, and IL-13 in the bronchoalveolar lavage (BAL) fluid. (D) Cellular counts in the BAL fluid. Mac, macrophage; Neut, neutrophil; Eos, eosinophil; Lym, lymphocyte; Epi, epithelial cell. Data in (C) and (D) are means ± SEM of eight mice per group and were analyzed by two-tailed, unpaired t test. (E) Analysis of the airway resistance in the indicated mice in response to increasing concentrations of methacholine. Data are means ± SEM of 10 mice and were analyzed by two-way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
To directly compare the TH2 effector states of WT CD4+ T cells and Crlf2−/− CD4+ T cells, we used an OVA model of sensitization with a single antigen challenge. In this model, WT and Crlf2−/− CD4+ T cells were transferred into Tcra −/− mice, as described previously (Fig. 7A). One month later, the recipient mice were immunized with OVA protein and aluminum hydroxide (ALUM) to induce TH2 cell differentiation (15). Splenic CD4+ T cells were purified on day 7 after immunization and were restimulated in vitro with irradiated splenocytes loaded with OVA protein. Crlf2−/− CD4+ T cells produced reduced amounts of TH2 cytokines compared to WT CD4+ T cells but were not impaired in their ability to secrete IFN-γ (fig. S6B).
To directly assess the percentage of cytokine-producing CD4+ T cells under conditions in which only these cells could respond to TSLP, WT and Crlf2−/− naïve CD4+ T cells were transferred into Crlf2−/− mice, which were then administered with OVA and ALUM. We found that both donor cell populations proliferated similarly in response to antigen (fig. S6C). Furthermore, WT and Crlf2−/− CD4+ T cells gave rise to similar numbers of TH1 and TH17 cells. Furthermore, the percentage of IL-13–positive TH2 cells was substantially reduced by the loss of Crlf2 in CD4+ T cells (fig. S6D). These results, coupled with our in vitro analyses, suggest that TSLP signaling in CD4+ T cells specifically enhances their ability to undergo robust differentiation into TH2 cells, resulting in the increased production of IL-5 and IL-13.
TSLP functions together with IL-4 to induce the expression of TH2 cytokines by human CD4+ T cells
To assess the effect of TSLP on human CD4+ T cells, such cells were purified from healthy or asthmatic donors and activated in the presence of IL-4, TSLP, or both cytokines (Fig. 8A). Although TSLP and IL-4 alone had modest effects on the expression of TH2 cytokine genes, in combination, they induced robust increases in the expression of IL4, IL5, and IL13 (Fig. 8B), as well as secretion of IL-5 protein (Fig. 8C). Note that, in asthmatic donors, there was more pronounced expression of TH2 cytokine genes and IL-5 protein. These results suggest that human CD4+ T cells also display a dual requirement for TSLP and IL-4 to enable their robust differentiation into TH2 cells. Furthermore, such TH2 cell responses were enhanced in the peripheral T cell compartment of asthmatic children.
Fig. 8. TSLP and IL-4 act in concert to induce the expression of TH2 cytokines by human CD4+ T cells.
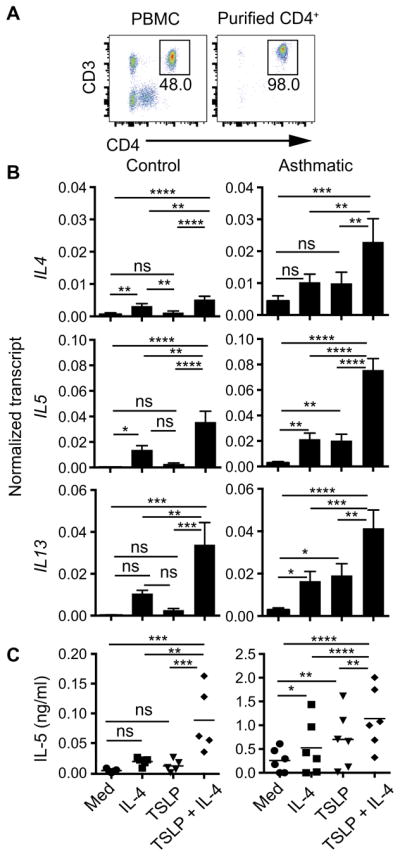
(A to C) Purified human CD4+CD3+ cells isolated from healthy donors (control) or asthmatic donors were activated with anti-CD3 (5 μg/ml), anti-CD28 (2 μg/ml), anti–IFN-γ (5 μg/ml), and IL-4 (40 ng/ml), TSLP (40 ng/ml), or both. (A) Percentage of CD4+CD3+ cells before and after purification. PBMC, peripheral blood mononuclear cell. (B) Analysis of the abundances of RNAs of the indicated genes was measured on day 3 after activation. Data are means ± SEM of six (control) or eight (asthmatic) donors. (C) The amount of IL-5 secreted by the indicated cells was assessed on day 5 after activation. Data are means ± SEM of five (control) or six (asthmatic) donors. The control group contained four children (8 to 9 years of age) and two adults, whereas the asthmatic group contained eight children (5 to 11 years of age). Data were analyzed by one-way ANOVA. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
DISCUSSION
To gain insight into the interplay of TSLP and IL-4 in the generation of TH2 cells, we used an in vitro system in which each signaling pathway could be independently manipulated. Our results suggest that TSLP initiates TH2 cell programming, involving induction of the expression of the genes encoding IL-4, IL-5, and IL-13, in the absence of IL-4 signaling. However, this transcriptional response was not sustained or amplified in the absence of IL-4 signaling. We note that T cells activated in the presence of TSLP induced the production of IL-4 and increased the abundance of IL-4Rα to establish a feed-forward loop for robust TH2 cell differentiation that did not require exogenously supplied IL-4. Nonetheless, TSLP signaling promoted TH2 cell differentiation by pathways that extend beyond the induction of the IL-4 feed-forward loop. This is because, during TH2 cell differentiation, TSLP specifically increased the frequency of IL-4–, IL-5–. and IL-13–expressing effector cells that had enhanced cytokine production. This was not observed when T cells underwent TH2 cell differentiation simply in the presence of saturating amounts of exogenous IL-4. A similar effect of TSLP and IL-4 signaling was observed in human CD4+ T cells. In the human context, IL-4 or TSLP signaling induced the production of low amounts of TH2 cytokines, and the combination of IL-4 and TSLP was necessary for robust TH2 cell differentiation. Thus, we propose a dual cytokine model for robust TH2 cell differentiation, involving an interplay of TSLP and IL-4 signaling that is analogous to the interplay among IL-6, IL-21, and IL-23 in the context of TH17 cell differentiation (32, 33) and that of IL-27 and IL-12 in the context of TH1 cell differentiation (34).
IL-2 and STAT5 are essential for TH2 cell differentiation, both for initiating chromatin accessibility at the Il4 locus and for increasing expression of the Il4ra gene, thereby priming cells for TH2 cell differentiation (5, 35–37). Our current results provide insights into the role of STAT5, suggesting that activation of the STAT5 pathway by TSLP, followed by IL-4 activation of the STAT6-GATA3 pathway, is required for the robust amplification of TH2 cytokine gene expression and enhanced TH2 cell differentiation. Surprisingly, IL-7, whose receptor shares the IL-7Rα chain with TSLPR, induced stronger activation of STAT5 than did TSLP but was less effective in promoting TH2 cell differentiation. A key finding is that TSLP signaling inhibited the induction of Bcl6 expression during T cell activation in a JAK2- and STAT5-independent manner. BCL6 directly represses Il4, Il9, and Il13 expression in CD4+ T cells; thus, its loss leads to increased production of TH2 cytokines (23, 38, 39). Here, we showed that, by antagonizing the induction of BCL6, TSLP promoted the generation of IL-13–expressing cells and magnitude of Il13 gene expression through a STAT5-independent manner. Hence, we propose that TSLP promotes TH2 cell differentiation through two distinct molecular pathways: the activation of STAT5 by JAK2 and the repression of Bcl6 expression, which is not mediated by STAT5. The two pathways appear to function synergistically because STAT5 activation in the presence of sustained Bcl6 expression did not enable induction of TH2 cytokine gene expression. Conversely, the TSLP-mediated repression of Bcl6 expression in the absence of STAT5A and STAT5B was not sufficient to activate the expression of Il5 and Il13.
The genomic consequences of TSLP signaling in the first 24 hours of CD4+ T cell activation are highly focused and include the hallmark TH2 cytokine loci, Il4, Il5, and Il13. Furthermore, this initial transcriptional programming is dependent on the JAK2-STAT5 pathway and can occur in the absence of IL-4 signaling. Note that STAT5 binds to regulatory regions associated with the Il4, Il13, and Il9 genes (39). Thus, we propose that the initial programming of the TH2 cell effector state by TSLP signaling involves focal binding and activation by STAT5. This proposal is consistent with the demonstration that expression of a constitutively activated STAT5 protein in naïve CD4+ T cells, primed in the absence of IL-2 signaling, promotes TH2 cell differentiation even under conditions of IL-4 signaling blockade (35). Note that, under conditions of TSLP signaling, the initial activation of expression of the TH2 cytokine loci by STAT5 was not accompanied by an increase in the activating histone modification H3K27ac at associated regulatory regions. In contrast, the combined action of TSLP and IL-4 signaling at later stages of TH2 cell differentiation resulted in an increase in H3K27ac and H3K4me3 at regulatory regions of the TH2 cytokine loci. These robust histone modifications were not observed with IL-4 alone and were associated with the increased frequency of IL-4–, IL-5–, and IL-13–expressing effector cells with enhanced cytokine production. Thus, underlying our dual cytokine model for robust TH2 cell differentiation is the sequential and synergistic action of TSLP-activated STAT5 and IL-4–activated STAT6, the latter of which results in the induction of the gene encoding the TH2 cell transcription factor GATA3. Consistent with this regulatory model, expression of a constitutively activated STAT5 protein, together with GATA3, in activated CD4+ T cells induces more robust TH2 cell differentiation than does either STAT5 or GATA3 alone (35). The most striking consequence of TSLP signaling in activated CD4+ T cells is that it programs a robust TH2 effector potential in memory cells. Although this TSLP-primed memory TH2 state was not associated with increased H3K27 acetylation at regulatory regions of TH2 cytokine loci, it resulted in enhanced H3K27 acetylation and cytokine gene expression upon secondary activation. The nature of the epigenetic alterations induced by transient TSLP priming that are stably propagated in memory TH2 cells remains to be explored.
Although it is known that TSLP plays a critical role in allergic disorders and asthma through stimulating DCs (3, 9, 40, 41), its direct action on T cells and TH2 cell responses in vivo has been under-appreciated. A few studies previously investigated a direct effect of TSLP on effector and memory CD4+ T cells in the context of OVA models of allergic diseases (15–17, 42). Here, using the HDM model of airway inflammation, which has pathophysiological features similar to those of human allergic asthma, we demonstrated that CD4+ T cells that responded to TSLP generated more pronounced asthma symptoms. Our in vivo findings highlight the importance of TSLP signaling through its direct actions in promoting robust TH2 cell differentiation. Our results are consistent with findings that CD4+ T cells that cannot respond to TSLP and IL-33 are impaired in their potential to produce IL-5 and IL-13 in the context of Nippostrongylus brasiliensis infection (43).
Distinct TH2 cell subsets are generated during allergic responses (18, 44–46). Some of these cells produce large amounts of IL-5, IL-9, and IL-13. These cells have been termed pathogenic TH2 cells. We note that TSLP-primed TH2 cells (TH2TSLP) have characteristics of pathogenic TH2 cells because they produce large amounts of IL-5, IL-9, and IL-13. Furthermore, several genes that are notable in pathogenic TH17 cells were also strongly expressed in TH2TSLP cells, such as Ccl3, Ccl4, Il3, and Csf2. Consistent with this similarity, TSLP increased activating histone modifications in the Faim3-Il24 locus; Faim3 is implicated in the pathogenicity of TH17 cells (33), and IL-24 is suggested to be an additional TH2-type cytokine (27). These observations suggest that pathogenic TH2 cells generated by TSLP priming share some features with their TH17 cell counterparts. We propose that the severity of allergic responses and asthma may be determined by the proportion of conventional TH2 and pathogenic memory TH2 cells, the latter generated by TSLP priming. Furthermore, although memory TH2TSLP cells displayed characteristics shared with IL-4–primed memory TH2 cells, such as the rapid increased production of GATA3 and IRF4, they manifested an increased capacity to produce IL-5 and IL-13 after restimulation in the absence of TSLP (Fig. 6). Together, our findings suggest that TSLP signaling in naïve CD4+ T cells in conjunction with IL-4 signaling increases the pathogenicity of TH2 effector and memory cells. Asthmatic children exhibited more pronounced TH2 cell responses in their peripheral T cell compartment in response to TSLP and IL-4 signaling. It remains to be determined whether this is due to an increase in the number of pathogenic memory TH2 cells. Thus, the blockade of TSLP signaling during the priming of allergic TH2 responses in childhood may prove beneficial in preventing the progression of allergic disorders to severe asthma.
MATERIALS AND METHODS
Study design
The aim of this study was to elucidate the actions and molecular mechanisms by which TSLP induces the differentiation of pathogenic TH2 cells. The experimental design involved in vitro and in vivo experiments with murine T cells, including flow cytometric, RNA-seq, and ChIP-seq analyses. The animal experiments were not randomized. The investigators were not blinded to the allocation of animals during experiments and analyses. Experimental replication and the numbers of animals used are indicated in the figure legends.
Mice
C57BL/6, Ptprca Pepcb/BoyJ (CD45.1), Tcra−/−, and Il4−/− mice were purchased from The Jackson Laboratory. Crlf2−/− mice were described previously (47). These strains were on a C57BL/6 background. Stat5a/5bfl/fl mice have been described previously (48) and were back-crossed for multiple generations to BALB/c mice. Both female and male mice (6 to 12 weeks of age) were used for in vivo and in vitro studies. Mice were housed under specific pathogen–free conditions. All experiments complied with protocols approved by the Cincinnati Children’s Hospital Medical Center (CCHMC) Animal Use and Care Committee.
T cell isolation and stimulation
Murine lymph node and splenic CD4+ T cells were purified with CD4+CD62L+ T Cell Isolation kits (Miltenyi Biotec) or sorted as CD4+CD62LhighCD44lowCD25− naïve cells. Purified naïve CD4+ T cells (95 to 98% purity) were activated with plate-bound anti-CD3 (5 μg/ml), soluble anti-CD28 (2 μg/ml), and soluble anti–IFN-γ (10 μg/ml). Each of these antibodies was obtained from BioXCell. T cells were additionally stimulated with TSLP (40 ng/ml; R&D Systems), IL-4 (20 ng/ml; PeproTech), or IL-7 (20 ng/ml; PeproTech). Cells were cultured in RPMI 1640 (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS), L-glutamine, β-mercapethanol, and antibiotics. To neutralize IL-4, anti–IL-4 (10 μg/ml; 11B11, BioXCell) and anti–IL-4Rα (5 μg/ml; BD Biosciences) were added. The amounts of IL-4, IL-5, and IL-13 secreted by cells into the culture medium were analyzed by enzyme-linked immunosorbent assay (ELISA) with specific kits from R&D Systems. To inhibit JAK2 activation, CD4+ T cells were preincubated for 1 hour with 50 μM JAK2 inhibitor II (CAS 1837-91-8, Calbiochem/Sigma-Aldrich) and then activated for the times indicated in the figure legends in the presence of JAK2 inhibitor II. Dimethyl sulfoxide was used as a control. Human peripheral blood mononuclear cells were isolated by Ficoll gradient centrifugation and then purified with CD4+ T Cell Isolation kits (Miltenyi Biotec) from the whole blood of healthy or asthmatic donors. Purified cells (98% purity) were activated with soluble anti-CD3 (5 μg/ml), anti-CD28 (2 μg/ml; BioXCell), and anti–IFN-γ (5 μg/ml; BioLegend) in the presence or absence of TSLP (40 ng/ml), IL-4 (40 ng/ml), or both.
Retroviral transduction of cells to express Bcl6 and Cre-mediated deletion of Stat5a/Stat5b
Purified CD4+ T cells from WT or Stat5a/5bfl/fl mice were preactivated with antibodies against CD3, CD28, and IFN-γ for 1 day. WT CD4+ T cells were transduced with pMIEG3-BCL6 (a gift from A. Dent; Addgene plasmid 40339), whereas Stat5a/5bfl/fl cells were transduced with CAG-GFP-IRES-CRE (a gift from F. Gage; Addgene plasmid 48201). MSCV-EcoR1-IRES-GFP (Addgene plasmid 66956) and CAG-GFP (Addgene plasmid 16664) were used as controls for the infections of WT and Stat5a/5bfl/fl CD4 T cells, respectively. Transduced cells were incubated under activation conditions for an additional 2 days, rested for 24 hours, and then fluorescence-activated cell–sorted on the basis of green fluorescent protein (GFP) expression. GFP+ cells were reactivated with anti-CD3 (2 μg/ml; plate-bound) and anti-CD28 (2 μg/ml; soluble) antibodies with or without TSLP (40 ng/ml). BCL6 protein was detected by intracellular staining 5 hours after activation. RNA was isolated and analyzed 14 hours after activation.
Flow cytometry analysis
For surface marker expression, cells were incubated in staining buffer for 45 min on ice with anti-Fc blocking antibody (2.4G2, BD Biosciences), BV510–anti-CD4, BV711–anti-CD45.1, fluorescein isothiocyanate (FITC)–anti-CD44 (BioLegend), and eFluor 660–anti-CD127 (eBioscience). For intracellular staining of transcription factors, CD4+ T cells were first stained for 20 min with Fixable Viability Dye eFluor 780 to detect dead cells and then fixed and permeabilized with the FoxP3 fixation Kit (eBioscience). Cells were stained with phycoerythrin (PE)–anti-GATA3, and eFluor 660–anti-IRF4 (eBioscience) or allophy-cocyanin (APC)–anti-BCL6 (BD Biosciences). To detect donor CD45.1 T cells, FITC–anti-CD4 and BV711–anti-CD45.1 monoclonal antibodies (BioLegend) were used. For intracellular staining of cytokines, activated CD4+ T cells were washed and restimulated with phorbol 12, 13-dibutyrate (PDBU) (500 ng/ml; Sigma-Aldrich), 1 μM ionomycin (Sigma-Aldrich), and Brefeldin A (eBioscience) for 5 hours. Harvested cells were fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences) and stained with PE–anti–IL-5, APC–anti–IL-4, eFluor 450–anti–IL-13 (eBioscience), and BV711–anti–IFN-γ (BioLegend). To detect donor CD45.1 T cells, FITC–anti-CD4 and PE–Cy7–anti-CD45.1 (BioLegend) were used. To detect phosphorylated STAT5 (pSTAT5) and pSTAT6, naïve CD4+ T cells were used in the presence of cytokines for 15 min, fixed in Cytofix buffer (BD Biosciences), and permeabilized in 90% methanol on ice. APC anti-pSTAT5 and PE anti-pSTAT6 (BD Biosciences) antibodies were used. Flow cytometry was performed on a Calibur or LSR/Fortessa. To analyze T cell survival, CD4+ T cells were washed in phosphate-buffered saline (PBS), resuspended in 100 μl of annexin V buffer, and stained with FITC–annexin V and 7-amino-actinomycin D (BD Biosciences) for 15 min at room temperature before being analyzed by flow cytometry.
Proliferation assays
Purified naïve CD4+ T cells were labeled for 8 min at room temperature with 2.5 μM carboxyfluorescein diacetate succinimidyl ester (Molecular Probes) and then activated with various concentrations of anti-CD3 antibody in the presence of anti-CD28 and anti–IFN-γ antibodies with or without TSLP for 3 days.
Adoptive transfer and generation of TH2 cells in vivo
Purified naïve polyclonal WT or Crlf2−/− CD4+ T cells were injected intravenously (2 to 5 × 106 cells per mouse) into recipient Tcra−/− or Crlf2−/− mice. On day 34 (Tcra−/−) or on the same day (Crlf2−/−), mice were immunized intraperitoneally with 100 μg of OVA (Pierce Chemical Co.) in 100 μl of PBS that had been mixed with 4 μg of ALUM (Pierce Chemical Co.). One week later, CD4+ T cells were purified from the spleens of the immunized mice and cultured with irradiated (3 grays) splenocytes from WT mice in a 1:2 ratio in the presence of 100 μg of OVA protein for 3 days. Cell culture medium was analyzed by ELISA to detect IL-4, IL-5, IL-13, and IFN-γ (R&D Systems) and IL-9 (BioLegend). Cells were stimulated with PDBU (500 ng/ml) and 1 μM ionomycin in the presence of Brefeldin A for 5 hours before intracellular staining of cytokines was performed, as described previously. To generate memory CD4+ T cells, naïve CD45.1+CD4+ T cells was activated with anti-CD3, anti-CD28, anti–IFN-γ antibodies in the presence or absence of TSLP or IL-4 for 3 days, rested in medium containing IL-7 (0.5 ng/ml) for 3 days, and injected intravenously (107 cells per mouse) into recipient CD45.2+ WT mice. Five weeks later, total CD4+ T cells were isolated from the spleens of recipient mice and stained or activated as indicated in the figure legends. Donor cells were gated on the basis of CD45.1 expression. For ChIP experiments, donor CD45.1+CD4+ T cells were sorted.
Murine HDM asthma model
Tcra−/− mice were injected with 5 × 106 to 6 × 106 WT or Crlf2−/− purified naïve CD4+ T cells. One month later, the mice were anesthetized with ketamine/xylazine, which was followed by intratracheal administration of either 40 μl of PBS or 100 μg of HDM in 40 μl of PBS (Greer Laboratories). HDM was administered two additional times at 1-week intervals. Two days after the last challenge, the mice were sacrificed by pentobarbital injection, and the organs were dissected for analysis. BAL fluid was collected with 1 ml of PBS by cannulation of the trachea and centrifuged at 500g for 5 min. The supernatant was used for the measurement of cytokines by ELISA. The cell pellet was resuspended in PBS containing 2% FBS and centrifuged in a Cytospin at 500g for 5 min. Slides were stained with KWIK-DIFF Stain kit (Thermo Fisher Scientific) to enable morphology-based cell counts. At least 200 cells in random fields were counted for each sample. Lung function analysis was performed using a flexiVent invasive measurement of dynamic resistance, as described previously (49).
RNA isolation and analyses
Naïve CD4+ T cells were activated with antibodies against CD3, CD28, and IFN-γ in the presence or absence of TSLP or IL-4 for 0, 2, 6, 24, 48, and 72 hours. RNA was isolated using the Aurum Total RNA kit (Bio-Rad). For real-time PCR analysis, RNA was reverse-transcribed with the iScript cDNA Synthesis kit (Bio-Rad). All primers and probes were purchased from Integrated DNA Technologies (IDT) (table S6). The ABI 7900HT Fast Sequence Detection System was used to quantitate PCR products. The abundances of the transcripts of interest were determined relative to that of Eif3k complementary DNA (cDNA). For RNA-seq analysis, mRNA was purified from total RNA with the Dynabeads mRNA Purification kit (Invitrogen) and reverse-transcribed with the SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen). The deoxyuridine triphosphate (dUTP) method was used for the preparation of RNA-seq libraries. Next-generation RNA-seq was performed by the CCHMC DNA Sequencing and Genotyping Core with Illumina TruSeq kits and an Illumina HiSeq 2000 sequencing system, which resulted in ~20 million paired-end reads per sample. RNA-seq data are available from the Gene Expression Omnibus (GEO) database (accession number: GSE81384). RNA-seq data analysis was performed with BioWardrobe (50). Briefly, reads were mapped with RNA-STAR 23104886, and reads per kilobase of transcript, per million mapped reads (RPKM) calculations were performed with the BioWardrobe algorithm. Differential gene expression analysis was performed with DESeq2 (51). Genes were considered to be differentially expressed if Padj ≤ 0.05 for at least one time point. Two additional cutoffs were used: a change in expression of more than twofold (higher or lower) and an RPKM value of ≥5 in at least one sample. Biological processes and pathway analyses for gene clusters were assigned with ToppGene software (52).
Chromatin immunoprecipitation sequencing
Cells were cross-linked with 1% formaldehyde for 8 min, and chromatin was prepared as previously described (53). Sonication was performed with a Covaris S220 focused ultrasonicator (Covaris Inc.) at 175-W peak incident power, 10% output, and 200 bursts for 5 min. DNA fragmentation was verified by agarose gel electrophoresis. ChIP was performed with a rabbit polyclonal anti-H3K27ac antibody (Diagenode Inc.), a rabbit monoclonal anti-H3K4me3 antibody (17-614, Millipore), or a rabbit polyclonal anti-STAT5A/5B antibody (AF1584, R&D Systems) and Dynabeads Protein G (Invitrogen) in radioimmunoprecipitation assay buffer (10 mM tris-EDTA, 0.1% SDS, 1% Triton X-100, 150 mM NaCl, and 0.1% sodium deoxycholate). ChIP DNA was converted into indexed libraries with the ThruPLEX DNA-seq kit (Rubicon) and was sequenced on an Illumina HiSeq 2000 sequencing system, which resulted in ~20 to 40 million single-end reads per sample. Data analysis was performed in BioWardrobe (50). Briefly, reads were mapped with BowTie and displayed on a local mirror of the UCSC genome browser as coverage by estimated fragments. Peaks were identified by MACS2, and differential enrichment was analyzed with MAnorm. ChIP-seq data are available from the GEO database (accession number: GSE81384). For ChIP-PCR, IDT primers and probes labeled with fluorescein amidite were used (table S7).
Statistical analyses
All statistical analyses were performed with Prism 6 software (GraphPad Software Inc.), as described in the figure legends: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
Supplementary Material
Fig. S1. TSLP promotes TH2 cell differentiation without affecting the survival or proliferation of activated CD4+ T cells.
Fig. S2. TSLP enhances IL-4Rα abundance and promotes TH2 cell differentiation in synergy with IL-4.
Fig. S3. Comparative transcriptional analysis of TH2IL-4 and TH2TSLP cells.
Fig. S4. JAK2 inhibitor II selectively blocks the TSLP-mediated activation of STAT5.
Fig. S5. Comparative chromatin analysis of TH2IL-4 and TH2TSLP cells.
Fig. S6. TSLP-responsive CD4+ T cells, sensitized in vivo, manifest enhanced production of TH2 cytokines.
Table S1. Quantitative analysis of IL-13–positive CD4+ T cells and the amounts of secreted IL-5 and IL-13.
Table S2. RNA-seq analysis of the gene expression profiles of TH2IL-4 and TH2TSLP cells compared to that of TH0 cells.
Table S3. RNA-seq analysis of gene expression clusters and pathways in TH0, TH2, and TH2TSLP cells.
Table S4. RNA-seq analysis of the gene expression profile of TH2TSLP cells compared to that of TH2IL-4 cells.
Table S5. H3K27ac ChIP-seq tag density coordinates, 2.5-kb intervals around peak centers for shared peaks or peaks specific for TH2 and TH2TSLP cells.
Table S6. Primers for PCR.
Table S7. Primers and probes for ChIP-DNA H3K27ac.
Acknowledgments
We thank M. Shrestha for the assistance with retroviral vectors.
Funding: The study was supported by the Cincinnati Children’s Research Foundation funds (H.S.), the University Research Council (URC) fund from the University of Cincinnati (to Y.R.), Just-In-Time (JIT) award from Center for Clinical and Translational Science and Training (CCTST) (to Y.R.), and T1 (to A.B.) grants from CCTST [Clinical and Translational Science Award (CTSA) 1UL1TR001425-01]. W.J.L. is supported by the Division of Intramural Research, National Heart, Lung, and Blood Institute, NIH. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core in the Division of Rheumatology at CCHMC, which is supported in part by NIH AR-47363, NIH DK78392, and NIH DK90971. I.P.L. was supported by NIH grant R01 HL122300.
Footnotes
Author contributions: Y.R. designed and performed the experiments, analyzed and interpreted the data, and wrote the manuscript. K.D.-S. and P.K.R. provided experimental assistance. I.P.L. provided advice with the asthma model and provided input on the manuscript. G.K.K.H. advised and coordinated the analysis of hCD4 T cells and provided input on the manuscript. A.V.K. and A.B. performed the bioinformatics analysis and provided input on the manuscript. W.J.L. provided input on the manuscript. H.S. directed the studies, interpreted the data, and wrote the manuscript.
Data and materials availability: ChIP-seq and RNA-seq data were deposited in the GEO with the accession number GSE81384.
Competing interests: H.S. is chair of the scientific advisory board of Lycera. W.J.L. is an inventor on NIH patents related to TSLP. A.V.K. and A.B. are cofounders of Datirium LLC, which provides installation and support services for the BioWardrobe data analysis platform. All other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paul WE. History of interleukin-4. Cytokine. 2015;75:3–7. doi: 10.1016/j.cyto.2015.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11:289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Licona-Limón P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. 2013;14:536–542. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 5.Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kashyap M, Rochman Y, Spolski R, Samsel L, Leonard WJ. Thymic stromal lymphopoietin is produced by dendritic cells. J Immunol. 2011;187:1207–1211. doi: 10.4049/jimmunol.1100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elder MJ, Webster SJ, Williams DL, Gaston JSH, Goodall JC. TSLP production by dendritic cells is modulated by IL-1β and components of the endoplasmic reticulum stress response. Eur J Immunol. 2016;46:455–463. doi: 10.1002/eji.201545537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H, Ziegler SF. The multiple facets of thymic stromal lymphopoietin (TSLP) during allergic inflammation and beyond. J Leukoc Biol. 2012;91:877–886. doi: 10.1189/jlb.1211622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 10.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FXF, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rochman I, Watanabe N, Arima K, Liu YJ, Leonard WJ. Cutting edge: Direct action of thymic stromal lymphopoietin on activated human CD4+ T cells. J Immunol. 2007;178:6720–6724. doi: 10.4049/jimmunol.178.11.6720. [DOI] [PubMed] [Google Scholar]
- 12.Omori M, Ziegler S. Induction of IL-4 expression in CD4+ T cells by thymic stromal lymphopoietin. J Immunol. 2007;178:1396–1404. doi: 10.4049/jimmunol.178.3.1396. [DOI] [PubMed] [Google Scholar]
- 13.He R, Oyoshi MK, Garibyan L, Kumar L, Ziegler SF, Geha RS. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc Natl Acad Sci USA. 2008;105:11875–11880. doi: 10.1073/pnas.0801532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rochman Y, Kashyap M, Robinson GW, Sakamoto K, Gomez-Rodriguez J, Wagner KU, Leonard WJ. Thymic stromal lymphopoietin-mediated STAT5 phosphorylation via kinases JAK1 and JAK2 reveals a key difference from IL-7-induced signaling. Proc Natl Acad Sci USA. 2010;107:19455–19460. doi: 10.1073/pnas.1008271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829–839. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blazquez AB, Mayer L, Berin MC. Thymic stromal lymphopoietin is required for gastrointestinal allergy but not oral tolerance. Gastroenterology. 2010;139:1301–1309.e4. doi: 10.1053/j.gastro.2010.06.055. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Du J, Zhu J, Yang X, Zhou B. Thymic stromal lymphopoietin signaling in CD4+ T cells is required for TH2 memory. J Allergy Clin Immunol. 2015;135:781–791.e3. doi: 10.1016/j.jaci.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao W, Zhang Y, Jabeen R, Nguyen ET, Wilkes DS, Tepper RS, Kaplan MH, Zhou B. Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity. 2013;38:360–372. doi: 10.1016/j.immuni.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. Eur J Immunol. 2011;41:1862–1871. doi: 10.1002/eji.201041195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med. 2002;195:1003–1012. doi: 10.1084/jem.20011128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, Pellegrini M, Belz GT, Smyth GK, Febbraio MA, Nutt SL, Kallies A. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol. 2013;14:1155–1165. doi: 10.1038/ni.2710. [DOI] [PubMed] [Google Scholar]
- 22.Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, Ziegler SF, Leonard WJ, Lodish HF. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. doi: 10.1038/76923. [DOI] [PubMed] [Google Scholar]
- 23.Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S, Ichiyama K, Zhang X, Zhang L, Wen X, Tian Q, Bian X-w, Jin W, Wei L, Dong C. Genome-wide analysis identifies Bcl6-controlled regulatory networks during T follicular helper cell differentiation. Cell Rep. 2016;14:1735–1747. doi: 10.1016/j.celrep.2016.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandberg EM, Ma X, He K, Frank SJ, Ostrov DA, Sayeski PP. Identification of 1,2,3,4,5,6-hexabromocyclohexane as a small molecule inhibitor of jak2 tyrosine kinase autophosphorylation. J Med Chem. 2005;48:2526–2533. doi: 10.1021/jm049470k. [DOI] [PubMed] [Google Scholar]
- 25.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, Liang P. Interleukin-24 and its receptors. Immunology. 2005;114:166–170. doi: 10.1111/j.1365-2567.2005.02094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Commins S, Steinke JW, Borish L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol. 2008;121:1108–1111. doi: 10.1016/j.jaci.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 28.London CA, Perez VL, Abbas AK. Functional characteristics and survival requirements of memory CD4+ T lymphocytes in vivo. J Immunol. 1999;162:766–773. [PubMed] [Google Scholar]
- 29.Yamashita M, Shinnakasu R, Nigo Y, Kimura M, Hasegawa A, Taniguchi M, Nakayama T. Interleukin (IL)-4-independent maintenance of histone modification of the IL-4 gene loci in memory Th2 cells. J Biol Chem. 2004;279:39454–39464. doi: 10.1074/jbc.M405989200. [DOI] [PubMed] [Google Scholar]
- 30.Gregory LG, Lloyd CM. Orchestrating house dust mite-associated allergy in the lung. Trends Immunol. 2011;32:402–411. doi: 10.1016/j.it.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohrs M, Shinkai K, Mohrs K, Locksley RM. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 2001;15:303–311. doi: 10.1016/s1074-7613(01)00186-8. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 33.Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H, Regev A. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163:1400–1412. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, Blumenschein WM, Mattson JD, Wagner JL, To W, Zurawski S, McClanahan TK, Gorman DM, Bazan JF, de Waal Malefyt R, Rennick D, Kastelein RA. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 35.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–748. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 36.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, Zhu J, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, Zhao K, Leonard WJ. Priming for T helper type 2 differentiation by interleukin 2–mediated induction of interleukin 4 receptor α-chain expression. Nat Immunol. 2008;9:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–592. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 39.Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, Leonard WJ. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci USA. 2014;111:3508–3513. doi: 10.1073/pnas.1301138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y-J, Soumelis V, Watanabe N, Ito T, Wang Y-H, de Waal Malefyt R, Omori M, Zhou B, Ziegler SF. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. 2007;25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718. [DOI] [PubMed] [Google Scholar]
- 41.Han H, Thelen TD, Comeau MR, Ziegler SF. Thymic stromal lymphopoietin-mediated epicutaneous inflammation promotes acute diarrhea and anaphylaxis. J Clin Invest. 2014;124:5442–5452. doi: 10.1172/JCI77798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jang S, Morris S, Lukacs NW. TSLP promotes induction of Th2 differentiation but is not necessary during established allergen-induced pulmonary disease. PLOS ONE. 2013;8:e56433. doi: 10.1371/journal.pone.0056433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, Gate RE, Haliburton GE, Ye CJ, Marson A, Erle DJ, Locksley RM. A tissue checkpoint regulates type 2 immunity. Nat Immunol. 2016;17:1381–1387. doi: 10.1038/ni.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prussin C, Yin Y, Upadhyaya B. TH2 heterogeneity: Does function follow form? J Allergy Clin Immunol. 2010;126:1094–1098. doi: 10.1016/j.jaci.2010.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Upadhyaya B, Yin Y, Hill BJ, Douek DC, Prussin C. Hierarchical IL-5 expression defines a subpopulation of highly differentiated human Th2 cells. J Immunol. 2011;187:3111–3120. doi: 10.4049/jimmunol.1101283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Endo Y, Hirahara K, Yagi R, Tumes DJ, Nakayama T. Pathogenic memory type Th2 cells in allergic inflammation. Trends Immunol. 2014;35:69–78. doi: 10.1016/j.it.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Al-Shami A, Spolski R, Kelly J, Fry T, Schwartzberg PL, Pandey A, Mackall CL, Leonard WJ. A role for thymic stromal lymphopoietin in CD4+ T cell development. J Exp Med. 2004;200:159–168. doi: 10.1084/jem.20031975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng C-X, Robinson GW, Hennighausen L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewkowich IP, Lajoie S, Stoffers SL, Suzuki Y, Richgels PK, Dienger K, Sproles AA, Yagita H, Hamid Q, Wills-Karp M. PD-L2 modulates asthma severity by directly decreasing dendritic cell IL-12 production. Mucosal Immunol. 2013;6:728–739. doi: 10.1038/mi.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kartashov AV, Barski A. BioWardrobe: An integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 2015;16:158. doi: 10.1186/s13059-015-0720-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rochman M, Kartashov AV, Caldwell JM, Collins MH, Stucke EM, Kc K, Sherrill JD, Herren J, Barski A, Rothenberg ME. Neurotrophic tyrosine kinase receptor 1 is a direct transcriptional and epigenetic target of IL-13 involved in allergic inflammation. Mucosal Immunol. 2015;8:785–798. doi: 10.1038/mi.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. TSLP promotes TH2 cell differentiation without affecting the survival or proliferation of activated CD4+ T cells.
Fig. S2. TSLP enhances IL-4Rα abundance and promotes TH2 cell differentiation in synergy with IL-4.
Fig. S3. Comparative transcriptional analysis of TH2IL-4 and TH2TSLP cells.
Fig. S4. JAK2 inhibitor II selectively blocks the TSLP-mediated activation of STAT5.
Fig. S5. Comparative chromatin analysis of TH2IL-4 and TH2TSLP cells.
Fig. S6. TSLP-responsive CD4+ T cells, sensitized in vivo, manifest enhanced production of TH2 cytokines.
Table S1. Quantitative analysis of IL-13–positive CD4+ T cells and the amounts of secreted IL-5 and IL-13.
Table S2. RNA-seq analysis of the gene expression profiles of TH2IL-4 and TH2TSLP cells compared to that of TH0 cells.
Table S3. RNA-seq analysis of gene expression clusters and pathways in TH0, TH2, and TH2TSLP cells.
Table S4. RNA-seq analysis of the gene expression profile of TH2TSLP cells compared to that of TH2IL-4 cells.
Table S5. H3K27ac ChIP-seq tag density coordinates, 2.5-kb intervals around peak centers for shared peaks or peaks specific for TH2 and TH2TSLP cells.
Table S6. Primers for PCR.
Table S7. Primers and probes for ChIP-DNA H3K27ac.