

Abstract
Purpose
Tumor genomic profiling for personalized oncology therapy is being widely applied in clinical practice even as it is being evaluated more formally in clinical trials. Given the complexities of genomic data and its application to clinical use, molecular tumor boards with diverse expertise can provide guidance to oncologists and patients seeking to implement personalized genetically targeted therapy in practice.
Methods
A multidisciplinary molecular tumor board reviewed tumor molecular profiling reports from consecutive referrals at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins over a 3-year period. The tumor board weighed evidence for actionability of genomic alterations identified by molecular profiling and provided recommendations including US Food and Drug Administration–approved drug therapy, clinical trials of matched targeted therapy, off-label use of such therapy, and additional tumor or germline genetic testing.
Results
One hundred fifty-five patients were reviewed. Actionable genomic alterations were identified in 132 patients (85%). Off-label therapies were recommended in 37 patients (24%). Eleven patients were treated off-label, and 13 patients were enrolled onto clinical trials of matched targeted therapies. Median progression-free survival of patients treated with matched therapies was 5 months ( 95% CI, 2.9 months to not reached), and the progression-free survival probability at 6 months was 43% (95% CI, 26% to 71%). Lack of locally available clinical trials was the major limitation on clinical actionability of tumor profiling reports.
Conclusion
The molecular tumor board recommended off-label targeted therapies for a quarter of all patients reviewed. Outcomes were heterogeneous, although 43% of patients receiving genomically matched therapy derived clinical benefit lasting at least 6 months. Until more data become available from precision oncology trials, molecular tumor boards can help guide appropriate use of tumor molecular testing to direct therapy.
INTRODUCTION
The advent of low-cost, next-generation DNA sequencing (NGS) technologies has led to an explosion in individual tumor molecular profiling with the goal of identifying personalized therapeutic matches for patients with cancer. Case reports and clinical trials attest to the clinical utility of targeting driver gene mutations in cancer types other than those in which the drug is approved for clinical use, such as BRAF V600 mutations in cancers other than melanoma.1 Ongoing efforts, such as the National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH; ClinicalTrials.gov identifier: NCT02465060) and the ASCO Targeted Agent and Profiling Utilization Registry (TAPUR; ClinicalTrials.gov identifier: NCT02693535) trials, aim to identify genomic predictors of response for targeted therapies against diverse genetic variants that may be difficult to study clinically because of their rarity outside of specific disease types.2 However, the use of tumor sequencing in clinical practice has outpaced the implementation and completion of such trials. Surveys have indicated that many oncologists are unsure how to interpret tumor sequencing data and whether their patients will have access to targeted therapies on the basis of the reports (ie, how actionable the findings are in reality).3 In response to these concerns, we established a multidisciplinary Genetic Alterations in Tumors With Actionable Yields (GAITWAY) molecular tumor board at our institution (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, MD) to assist providers with interpretation and use of tumor molecular profiling data. Here, we present our approach to using tumor molecular profiling and our 3-year experience, with associated patient outcomes.
METHODS
The GAITWAY tumor board began meeting weekly in September 2013. Attendees included adult and pediatric medical oncologists representing diverse subspecialties, molecular pathologists with NGS expertise, genetic counselors, phase I clinical trial investigators, research coordinators, a patient advocate, and medical oncology fellows. Tumor testing was ordered by the referring oncologist without the board’s input. The referring oncologist provided the board with the molecular profiling report(s) and the patient’s oncologic and family history. A written summary of the board’s discussion and recommendations was provided to the referring oncologist.
The board reviewed Clinical Laboratory Improvement Amendments–approved NGS cancer gene panel tests from a variety of providers, including Foundation Medicine (Cambridge, MA; FoundationOne, n = 120), Personal Genome Diagnostics (Baltimore, MD; Cancer Select-203 or Cancer Select-88, n = 25), Memorial Sloan Kettering Cancer Center MSK-IMPACT (New York, NY; n = 5), Caris Life Sciences (Irving, TX; MI Profile, n = 3), and several others. Personal Genome Diagnostics and MSK-IMPACT included normal tissue control sequencing and filtered out most germline variants. A small number of reports included multiplatform testing, including NGS, fluorescence in situ hybridization, and protein immunohistochemistry. Only one test used circulating tumor DNA from plasma as the source material (Personal Genome Diagnostics, Lung Select).
Medical records for consecutive patients referred to the board from September 2013 through September 2016 were accessed under a protocol approved by the Johns Hopkins institutional review board. Patient characteristics were analyzed using descriptive statistics. Progression-free survival (PFS) was measured from the first date of treatment with a genomically matched therapy or next nonmatched therapy after tumor board evaluation until the date of disease progression or death, whichever came first. Progression was determined by imaging studies or clinician assessment. Responses were assessed according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Probability of PFS was estimated using the Kaplan-Meier method. Alive patients without progression were censored at the date of last radiographic assessment. Statistical analyses were performed using R software v.3.3.1.
Criteria for Actionability of Genomic Alterations
The board considered a genetic alteration actionable if one of the following conditions was met: it offered a target for a drug approved by the US Food and Drug Administration (FDA) for the patient’s tumor type; it offered a target for a drug approved by the FDA for another tumor type; it offered a target for a drug on a clinical trial; or it was a potential germline mutation for a hereditary cancer predisposition syndrome. The board’s approach to recommending genomically guided therapy is shown in Figure 1. The board had a relatively high threshold for considering a genetic variant as the basis for off-label use of a targeted therapy. Genetic alterations with drugs that specifically targeted the affected protein were of highest priority. In contrast, although many genes could potentially be linked to activation of the mammalian target of rapamycin (mTOR) pathway (eg, NF1, PIK3CA, FBXW7), the board generally did not recommend off-label use of mTOR inhibitors, given that the genetic link to mTOR was indirect or variants in these genes did not predict mTOR inhibitor benefit in clinical correlative studies.4,5 Similarly, CCND1 amplification and CDKN2A/B loss or mutation were not considered a high-level rationale for off-label use of the CDK4/6 inhibitor palbociclib because these alterations had not been shown to predict therapeutic benefit in breast cancer.6 Although multikinase inhibitors like pazopanib are often suggested as a match for FGFR1-3 amplifications,7 the board thought there was insufficient evidence to recommend off-label use and instead preferred clinical trials of fibroblast growth factor receptor (FGFR) inhibitors. The board was more liberal in recommending clinical trials for the alterations discussed earlier, even when the genetic alteration was not directly targeted by the investigational agent (eg, considering KRAS mutations as rationale for an MEK inhibitor trial). The rationale for some recommendations changed over time as new data and literature emerged relevant for a given target. The board’s approach to evaluating specific variants in genes is further described in the Appendix.
Fig 1.
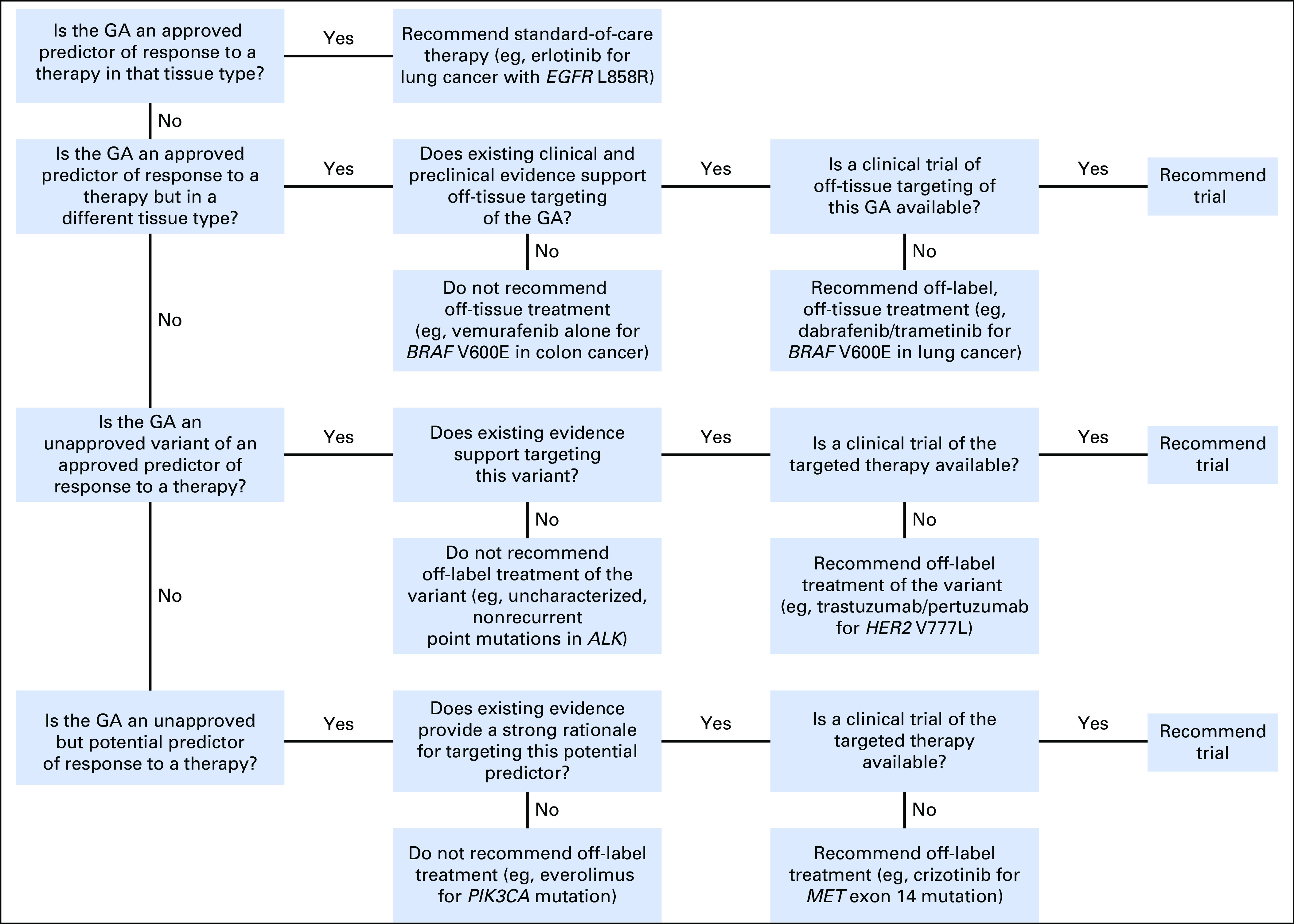
Genetic Alterations in Tumors With Actionable Yields (GAITWAY) tumor board approach to therapeutic recommendations on the basis of tumor genomic analyses. GA, genomic alteration.
RESULTS
From September 2013 through September 2016, 157 cases representing 155 patients were reviewed by the GAITWAY tumor board (two patients had subsequent tumor sequencing and were reviewed twice). Patient demographic characteristics and distribution of tumor types are listed in Table 1. The most frequent tumor types were breast cancer (n = 56), lung cancer (n = 21), and squamous cell carcinoma of the head and neck (n = 11). The breast cancer cases included 20 patients with triple-negative disease previously reviewed as part of Individualized Molecular Analyses Guide Efforts (IMAGE), an institutional precision oncology feasibility trial.8 The mean number of lines of prior systemic therapy for metastatic disease was two lines (range, zero to 11 lines). Follow-up information was available for 129 patients (83%). The main reason for lack of follow-up was that the patient was seen only once for a second opinion.
Table 1.
Patient Demographic Characteristics and Distribution of Tumor Types

The average number of genetic alterations per tumor was 4.8 (range, zero to 16 alterations), excluding variants of uncertain significance or equivocal amplifications. The average number of genetic alterations considered actionable by the board was 1.9 (range, zero to six alterations). Genes recurrently altered across samples were similar to those reported from other series,9-11 although the relative frequencies were influenced by the case mix seen by our tumor board (Fig 2). In terms of targetable pathways, the phosphatidylinositol-3 kinase (PI3K) pathway was most frequently altered (63 alterations), followed by the G1 cell cycle checkpoint (61 alterations) and FGFR pathways (30 alterations).
Fig 2.

Frequency of genomic alterations. Actionable genomic alterations and selected nonactionable alterations are shown. For actionable genes, the number of patients in whom an action was taken is shown. Actions taken included therapy assignment, germline testing, or microsatellite instability evaluation.
The board identified a potentially actionable genomic alteration in 132 patients (85%; Fig 3). Four patients (3%) received therapies approved by the FDA for their tumor type. Thirty-seven patients (24%) had a recommendation for off-label use of an FDA-approved therapy (Table 2 and Appendix Table A1). Twenty-four patients (15%) received a nonstandard, genetically matched therapy off-label (n = 11) or on a clinical trial (n = 13). Reasons patients did not receive recommended off-label therapy are detailed in Figure 3. In no case was the treating oncologist unable to prescribe or obtain a recommended off-label drug as a result of denial by insurance.
Fig 3.

CONSORT diagram of patients discussed at the molecular tumor board. Note, all numbers do not add up because some patients are counted in more than one category (eg, had an actionable alteration for a clinical trial and also were recommended off-label use or had an actionable alteration for therapy and also for germline analysis). (*) One patient with KRAS mutation for whom the board recommended a clinical trial only but who was prescribed off-label trametinib by his primary oncologist. FDA, US Food and Drug Administration; NED, no evidence of disease; PS, performance status.
Table 2.
Patients Who Received Matched Targeted Therapies or for Whom the Board Recommended Off-Label Use of Targeted Therapies

A clinical trial was recommended as an option for 129 patients (83%; including 34 patients who also had a recommendation for off-label therapy), but only 13 patients were enrolled onto a recommended trial (Fig 3). In a number of patients, the trial recommendation was qualified, because there was no clinical evidence that the alterations were predictive of benefit. The most frequent alterations that led to a qualified recommendation were KRAS or MDM2 mutations and alterations activating the G1 cell cycle checkpoint (CDKN2A/B, CCND1, and CDK4). Stronger recommendations were made for trials targeting the FGFR or PI3K pathways, but only four patients were treated on such trials, largely because of lack of availability. Twenty-five patients who investigated clinical trial options were not eligible, could not access a trial for reasons of geographical proximity, or had worsening performance status (Fig 3).
Outcomes of Patients Receiving Matched Targeted Therapy
Follow-up information was available for all 24 patients treated with a matched therapy on a clinical trial or off-label. Two patients had recently initiated therapy, and response assessment was not yet available. With a median follow-up of 7.0 months, the median PFS of patients treated with genomically matched therapy was 5 months (95% CI, 2.9 months to not reached), and the 6-month PFS probability was 43% (95% CI, 26% to 71%; Fig 4A). The median PFS for 54 patients with available follow-up information whose next therapy was nonmatched was 2.97 months (95% CI, 2.4 to 5.13 months), and 6-month PFS was 20% (95% CI, 11% to 35%). Demographic characteristics of the two cohorts are listed in Appendix Table A2. Fifty percent of patients receiving genomically matched therapy had a PFS more than double their PFS on prior therapy (Fig 4B). Two patients with ERBB2 missense mutations, one with triple-negative breast cancer (reported previously8) and one with lung adenocarcinoma, have had prolonged disease control on human epidermal growth factor receptor 2 (HER2) antibody–based therapy. The patient with lung cancer had progression after four cycles of platinum doublet chemotherapy and has achieved a partial response after 12 months on a clinical trial of trastuzumab and pertuzumab. This is notable because there have been few reports of ERBB2-mutant tumors benefiting from HER2 antibody–based therapy in the absence of chemotherapy or small-molecule HER2 kinase inhibitors.14,15 Two patients with lung adenocarcinoma with MET exon 14 skipping splice site mutations have had prolonged stable disease (21 months and 6 months, ongoing) on crizotinib, in line with recent reports.16,17 A patient with BRAF V600E–mutated ameloblastoma experienced a partial response lasting 8 months to dabrafenib and trametinib, again consistent with a previous case report.18
Fig 4.
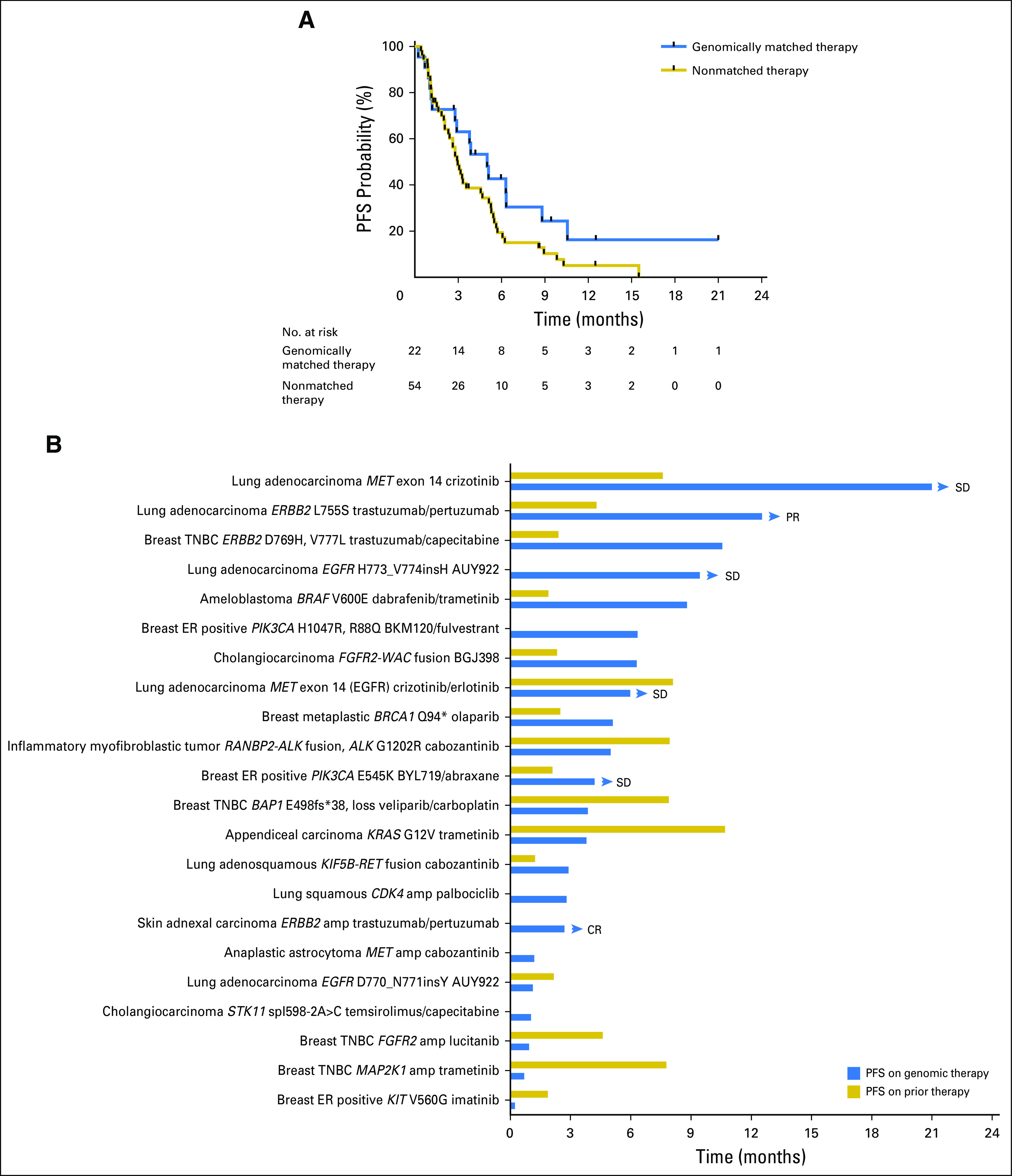
(A) Progression-free survival (PFS) of patients treated with genomically matched therapy (n = 22) or nonmatched therapy (n = 54). All patients with follow-up who were treated with genomically matched therapy are included in the analysis, including one patient who was treated with off-label trametinib for a KRAS mutation when the board had recommended a clinical trial. (B) PFS and PFS on immediate prior therapy for individual patients treated with genomically matched therapies. Eight patients either had no prior systemic therapy for metastatic disease or insufficient documentation of prior therapy. Arrowheads indicate patients continuing on treatment. amp, amplification; CR, complete response; ER, estrogen receptor; PR, partial response; SD, stable disease; TNBC, triple-negative breast cancer.
Evidence for Tumor Evolution and Heterogeneity
Five patients had tumor sequencing performed more than once (in three patients, the multiple reports were reviewed at a single tumor board session). Sequencing of metastatic sites of disease progression on targeted therapy revealed new genetic alterations consistent with pre-existing tumor heterogeneity or acquired drug resistance. A patient with an inflammatory myofibroblastic tumor with an RANBP2-ALK rearrangement at diagnosis was initially treated with crizotinib with an excellent response, followed by ceritinib. Repeat sequencing at the time of progression on ceritinib showed the same RANBP2-ALK fusion as well as an acquired ALK G1202R mutation, which confers resistance to both crizotinib and ceritinib.19 The board reviewed preclinical data on the G1202R mutation and identified activity of cabozantinib as well as newer investigational ALK inhibitors against G1202R or the analogous mutation in ROS1.20,21 The patient was treated with off-label cabozantinib, with stable disease lasting 5 months.
A patient with EGFR exon 21–mutated lung adenocarcinoma with an excellent response to erlotinib developed a new progressing lesion, which was biopsied and sequenced. Sequencing showed a MET exon 14 skipping splice site mutation that had not been present when the tumor was initially sequenced, but no EGFR mutation. The board noted that distinct tumor clones with EGFR and MET mutations could account for the mixed response to erlotinib, and the patient was treated with erlotinib plus crizotinib on the basis of tolerability of the combination in a phase I trial.22 His disease has been stable for 6 months, and treatment is ongoing.
A second patient who had lung cancer with an EGFR exon 19 deletion mutation had tumor sequencing after experiencing progression on many lines of targeted therapy including erlotinib, afatinib and cetuximab, and osimertinib. Sequencing showed the EGFR exon 19 deletion as well as amplification of ERBB2, which has been described as an acquired resistance mechanism in EGFR-mutated lung cancer treated with EGFR inhibitors.23 The board recommended HER2-targeted therapy; unfortunately, the patient’s clinical condition deteriorated, and she died without receiving another line of therapy.
Germline Evaluation Prompted by Tumor Sequencing
The board recommended further clinical or genetic evaluation for a possible germline cancer-predisposing variant for 46 patients. Many of the patients with breast cancer had already had germline testing. The board examined mutant allele frequencies, if available, to determine the likelihood of the alterations being germline. However, because tumor sequencing platforms have not been validated specifically for germline testing, the board recommended genetic counseling to consider dedicated germline analysis. Three patients had additional tumor testing for microsatellite instability, and all were microsatellite stable. Six patients had germline testing for BRCA1/2, PTEN, TP53, and other genes; all were germline nonmutated.
DISCUSSION
This report adds to the growing literature on precision oncology guided by a molecular tumor board. Our series is one of the largest to date to include patient outcomes. Although this was not a prospective clinical trial and treating oncologists were not bound by the board’s recommendations, the molecular tumor board attempted to apply a consistent standard of principles when assessing each patient. Our criteria for evaluating individual mutations and levels of supporting evidence for actionability are similar to those applied in the NCI-MATCH trial.2,24 The board preferred direct rather than indirect drug-gene matches and rarely advocated multikinase inhibitors off-label when the target had not been extensively validated as predictive. We found a high frequency of actionable genomic alterations, similar to previous reports9,11,25,26; however, lack of available clinical trials and a higher threshold for recommending off-label use of targeted therapies reduced the rate of therapeutic use in our patients.
A limitation of our study is that the board reviewed only those patients referred to it. Thirty-eight percent of our patients had genomic variants that could have made them eligible for one of the NCI-MATCH treatment arms, which is higher than the estimated match rate of 23% with the current NCI-MATCH arms open.27 This likely reflects the case mix of patients seen at our cancer center and referred to our tumor board, which was perhaps enriched for variants the referring physician considered actionable.
Lack of locally available trials was a leading reason for lack of application of matched, targeted therapies, as noted by others.8-10 Immunotherapy also emerged as a compelling therapeutic alternative for many patients. Almost all of our patients were reviewed before the NCI-MATCH trial opened at our institution. NCI-MATCH and TAPUR offer access to genomically targeted therapies for a variety of targets that would be difficult for any single cancer center to have in its clinical trial portfolio. Such trials could partially solve the leading barrier to actionability noted here.
Despite these limitations, 15% of our patients were treated with matched targeted therapies on a clinical trial or with off-label drugs. Although the PFS of patients treated with genomically matched therapies compared favorably with that of patients receiving nonmatched therapies, our study was not designed to test the superiority of a genomically guided approach for all patients. The small sample size, retrospective nature of the analysis, and heterogeneous patient population subject to referral bias all limit the utility of such a comparison. The non–genomically treated patients had fewer actionable alterations than the genomically treated cohort and had a different distribution of tumor types. Nonetheless, half of the patients receiving genomically matched therapy had a PFS more than double their prior PFS, and several had prolonged duration of benefit. The patients who benefited most from matched therapy had alterations in genes such as ERBB2, BRAF, EGFR, and MET, for which there is currently patient-level response data from clinical trials, case series, and case reports. As with prospective trials of precision oncology,28 outcomes of series such as ours will be impacted by the criteria used to match targeted therapies to genetic variants found in patients’ tumors. We cannot rule out that our conservative approach toward actionability prevented us from observing greater clinical benefit, but we believe that more liberal off-label use of targeted agents is unlikely to produce better outcomes than we observed. Reasons for the limited benefit of single-agent targeted therapies have been reviewed elsewhere.29
Even when off-label drug use was recommended for a highly actionable mutation, some patients experienced rapid disease progression that precluded them from starting or continuing such therapies. The optimal timing of tumor sequencing has yet to be determined, but the experience of these patients suggest that the review of genomic actionability should take place earlier in the disease course. However, we documented several patients in whom repeat tumor sequencing demonstrated new genomic findings consistent with either pre-existing heterogeneity or clonal evolution. Improvements in the use of NGS panels on cell-free circulating tumor DNA from plasma may allow sampling of tumor heterogeneity and enable serial monitoring of acquired resistance and clonal dynamics in the face of targeted therapy.8,30,31
The board also reviewed patients for whom it disagreed with a prior interpretation and use of tumor profiling data. In one instance, a patient had discontinued an approved chemotherapy drug to which she had been responding (docetaxel) because a tumor profiling biomarker (TUBB3 immunohistochemistry) predicted lack of benefit. A patient with HER2-positive breast cancer was treated ineffectively for several months with enzalutamide on the basis of the tumor staining positive for androgen receptor. The board instead recommended treating the patient with pertuzumab, an FDA-approved therapy, to which she responded. The circumstances of these patients show the value a molecular tumor board can add and highlight that standard-of-care therapies should still be considered as viable and often preferred options, because approved therapies for a given cancer type have high levels of evidence to support their use.
Our standard for recommending off-label use may be more stringent than that used at some other centers or by individual practicing oncologists. Whereas 73% of our patients had a variant for which off-label drug use was suggested by the commercial sequencing provider and 55% had a variant that would have enabled use of an FDA-approved drug on the TAPUR trial, our board recommended off-label use in only 24% of patients. In the absence of more data or practice guidelines, patients and providers have some discretion about where to set the threshold. Large data-sharing efforts and publication of series such as this one will be essential to expand public knowledge about the clinical actionability of genetic variants and outcomes associated with precision oncology as currently practiced.32 We support ongoing trials like NCI-MATCH and TAPUR in the belief that, as much as possible, precision oncology should be carried out in the context of clinical trials so we can replace educated guesswork with evidence.
Appendix
Methods
For uncommon variants in well-established cancer driver genes (eg, non-V600 BRAF mutations), the board considered factors including recurrence of that specific mutation in databases such as Catalogue of Somatic Mutations in Cancer (Zhan F, et al: Blood 108:2020-2028, 2006), sequencing data accessed at cBioPortal (Cerami E, et al: Cancer Discov 2:401-404, 2012), and in published studies; location in a known protein domain; evidence that the variant could be a germline single nucleotide polymorphism; and preclinical functional characterization—to show whether the variant was biologically activating and whether it demonstrated sensitivity or resistance to known drugs. The board generally did not recommend targeting amplifications reported as equivocal or mutations that were clearly subclonal relative to other mutations in the tumor. In some cases, reported variants of uncertain significance were evaluated for potential treatment implications.
Table A1.
Patients Who Received Matched Targeted Therapies or for Whom the Board Recommended Off-Label Use of Targeted Therapies

Table A2.
Demographic Characteristics of the Genomically Matched and Non–Genomically Matched Cohorts of Patients Analyzed for Progression-Free Survival in Figure 4

Footnotes
Supported by National Cancer Institute Grant No. P30 CA006973 to the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center.
W.B.D., P.M.F., and H.K. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: W. Brian Dalton, Patrick M. Forde, Christopher D. Gocke, Jennifer Axilbund, David M. Loeb, Heather A. Parsons, Ben H. Park, Josh Lauring
Financial support: Ben H. Park, Josh Lauring
Administrative support: Ben H. Park, Josh Lauring
Provision of study material or patients: Patrick M. Forde, Hyunseok Kang, Roisin M. Connolly, Vered Stearns, Antonio C. Wolff, David M. Loeb, Christine A. Pratilas, Christian F. Meyer, Heather A. Parsons, Ben H. Park
Collection and assembly of data: W. Brian Dalton, Patrick M. Forde, Hyunseok Kang, Roisin M. Connolly, Vered Stearns, Christopher D. Gocke, Jennifer Axilbund, Cindy Geoghegan, Antonio C. Wolff, Shannon A. Slater, Jennifer Ensminger, Heather A. Parsons, Ben H. Park, Josh Lauring
Data analysis and interpretation: W. Brian Dalton, Patrick M. Forde, Hyunseok Kang, Christopher D. Gocke, James R. Eshleman, Jennifer Axilbund, Dana Petry, Antonio C. Wolff, David M. Loeb, Christine A. Pratilas, Christian F. Meyer, Eric S. Christenson, Heather A. Parsons, Ben H. Park, Josh Lauring
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Personalized Medicine in the Oncology Clinic: Implementation and Outcomes of the Johns Hopkins Molecular Tumor Board
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
W. Brian Dalton
No relationship to disclose
Patrick M. Forde
Research Funding: Bristol-Myers Squibb, AstraZeneca, Novartis, Kyowa
Travel, Accommodations, Expenses: Bristol-Myers Squibb, AstraZeneca, Novartis, Celgene
Hyunseok Kang
Consulting or Advisory Role: AstraZeneca
Research Funding: Boehringer Ingelheim, Merck Sharp & Dohme, Plexxikon, AstraZeneca, VentiRx Pharmaceuticals
Travel, Accommodations, Expenses: AstraZeneca, Pfizer, Eli Lilly
Roisin M. Connolly
Research Funding: Genentech (Inst), Merck (Inst), Merrimack (Inst), Puma Biotechnology (Inst), Novartis (Inst), Clovis (Inst)
Vered Stearns
Research Funding: Abbvie, Celgene, Merck, Medimmune, Novartis, Pfizer, Puma
Christopher D. Gocke
No relationship to disclose
James R. Eshleman
No relationship to disclose
Jennifer Axilbund
Employment: Invitae Corporation
Stock and Other Ownership Interests: Invitae Corporation
Dana Petry
No relationship to disclose
Cindy Geoghegan
Consultant or Advisory Role: Celgene, Pfizer
Antonio C. Wolff
Research Funding: Myriad (Inst)
David M. Loeb
No relationship to disclose
Christine A. Pratilas
Consultant or Advisory Role: Genentech
Christian F. Meyer
Consulting or Advisory Role: Janssen, Eli Lilly, Eisai (Inst)
Travel, Accommodations, Expenses: Plexxicon, Eli Lilly
Eric S. Christenson
No relationship to disclose
Shannon A. Slater
No relationship to disclose
Jennifer Ensminger
No relationship to disclose
Heather A. Parsons
No relationship to disclose
Ben H. Park
Leadership: Horizon Discovery, Foundation Medicine, Genomic Health, Loxo Oncology
Stock and Other Ownership Interests: Loxo Oncology
Consulting or Advisory Role: Horizon Discovery, Genomic Health, Foundation Medicine, Loxo Oncology
Research Funding: Foundation Medicine, Genomic Health
Josh Lauring
Speakers' Bureau: Galderma(I), Abbvie(I)
Patents, Royalties, Other Intellectual Property: Royalty payments from sales of products under a licensing agreement between Horizon Discovery and Johns Hopkins University
REFERENCES
- 1.Hyman DM, Puzanov I, Subbiah V, et al. : Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373:726-736, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conley BA, Doroshow JH: Molecular analysis for therapy choice: NCI MATCH. Semin Oncol 41:297-299, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Kurzrock R, Colevas AD, Olszanski A, et al. : NCCN Oncology Research Program’s Investigator Steering Committee and NCCN Best Practices Committee molecular profiling surveys. J Natl Compr Canc Netw 13:1337-1346, 2015 [DOI] [PubMed] [Google Scholar]
- 4.Hortobagyi GN, Chen D, Piccart M, et al. : Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: Results from BOLERO-2. J Clin Oncol 34:419-426, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jardim DL, Wheler JJ, Hess K, et al. : FBXW7 mutations in patients with advanced cancers: Clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS One 9:e89388, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finn RS, Crown JP, Lang I, et al. : The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol 16:25-35, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Borad MJ, Champion MD, Egan JB, et al. : Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 10:e1004135, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. doi: 10.1158/1078-0432.CCR-16-1543. Parsons HA, Beaver JA, Cimino-Mathews A, et al: Individualized Molecular Analyses Guide Efforts (IMAGE): A prospective study of molecular profiling of tissue and blood in metastatic triple negative breast cancer. Clin Cancer Res 23:379-386, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. doi: 10.1634/theoncologist.2016-0049. Hirshfield KM, Tolkunov D, Zhong H, et al: Clinical actionability of comprehensive genomic profiling for management of rare or refractory cancers. Oncologist . [epub ahead of print on August 26, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meric-Bernstam F, Brusco L, Shaw K, et al. : Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 33:2753-2762, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwaederle M, Parker BA, Schwab RB, et al. : Molecular tumor board: The University of California-San Diego Moores Cancer Center experience. Oncologist 19:631-636, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross JS, Wang K, Chmielecki J, et al. : The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 138:881-890, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J, Axilbund J, Dalton WB, et al. : A polycythemia vera JAK2 mutation masquerading as a duodenal cancer mutation. J Natl Compr Canc Netw 14:1495-1498, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazières J, Barlesi F, Filleron T, et al. : Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann Oncol 27:281-286, 2016 [DOI] [PubMed] [Google Scholar]
- 15.Mazières J, Peters S, Lepage B, et al. : Lung cancer that harbors an HER2 mutation: Epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 31:1997-2003, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Frampton GM, Ali SM, Rosenzweig M, et al. : Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 5:850-859, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Paik PK, Drilon A, Fan PD, et al. : Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov 5:842-849, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaye FJ, Ivey AM, Drane WE, et al. : Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 ameloblastoma. J Natl Cancer Inst 107:378, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Friboulet L, Li N, Katayama R, et al. : The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 4:662-673, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davare MA, Vellore NA, Wagner JP, et al. : Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci USA 112:E5381-E5390, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katayama R, Kobayashi Y, Friboulet L, et al. : Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin Cancer Res 21:166-174, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. doi: 10.1016/j.jtho.2016.09.131. Ou SI, Govindan R, Eaton KD, et al: Phase I results from a study of crizotinib in combination with erlotinib in patients with advanced nonsquamous non-small cell lung cancer. J Thorac Oncol 12:145-151, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takezawa K, Pirazzoli V, Arcila ME, et al. : HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2:922-933, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meric-Bernstam F, Johnson A, Holla V, et al. : A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst 107:djv098, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker BA, Schwaederlé M, Scur MD, et al. : Breast cancer experience of the molecular tumor board at the University of California, San Diego Moores Cancer Center. J Oncol Pract 11:442-449, 2015 [DOI] [PubMed] [Google Scholar]
- 26.Tsimberidou AM, Wen S, Hong DS, et al. : Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: Validation and landmark analyses. Clin Cancer Res 20:4827-4836, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 28.Le Tourneau C, Delord JP, Gonçalves A, et al. : Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 16:1324-1334, 2015 [DOI] [PubMed] [Google Scholar]
- 29.Tannock IF, Hickman JA: Limits to personalized cancer medicine. N Engl J Med 375:1289-1294, 2016 [DOI] [PubMed] [Google Scholar]
- 30.Chabon JJ, Simmons AD, Lovejoy AF, et al. : Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun 7:11815, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwaederle M, Husain H, Fanta PT, et al: Use of liquid biopsies in clinical oncology: Pilot experience in 168 patients. Clin Cancer Res 22:5497-5505, 2016. [DOI] [PubMed]
- 32.Siu LL, Lawler M, Haussler D, et al. : Facilitating a culture of responsible and effective sharing of cancer genome data. Nat Med 22:464-471, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]