

Abstract
Objective:
The aim of the present study was to investigate the role of ionic homeostasis in cisplatin (cisdiamminedichloroplatinum (II), CDDP)-induced neurotoxicity. CDDP is a severely neurotoxic antineoplastic agent that causes neuronal excitotoxicity. According to some studies, calcium influx increases, whereas potassium efflux decreases neuronal death. Nimodipine and glibenclamide were used to analyze the role of ionic flows in CDDP-induced neurotoxicity in rat primary cerebellar granule cell (CGC) culture.
Materials and Methods:
CGC culture was prepared from the cerebella of Sprague Dawley 5-day-old pups. The submaximal concentration of CDDP was determined and then given with 1, 10, or 50 µM of drugs into culture. Neurotoxicity was investigated using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) assay. One-way analysis of variance, Kruskal–Wallis H test, and Tukey test were applied for statistical analysis.
Results:
CDDP induced neurotoxicity in a concentration-dependent manner. Neither nimodipine nor glibenclamide was able to protect CGCs against CDDP neurotoxicity.
Conclusion:
By blocking L-type voltage-gated calcium channels, nimodipine did not prevent CDDP neurotoxicity in CGCs. Ca2+ influx via these channels seemed to be insufficient to cause a change in CDDP-induced neurotoxicity. Similarly, glibenclamide failed to prevent CDDP neurotoxicity. Further studies are needed to elucidate the mechanisms of these preliminary results.
Keywords: Cerebellar granule cells, glibenclamide, KATP channels, nimodipine
Introduction
Cisplatin (cis-diamminedichloroplatinum (II), CDDP) is a platinum-based antineoplastic agent widely used against a variety of cancers [1]. However, its clinical benefits are limited owing to its adverse effects, such as ototoxicity, nephrotoxicity, cardiotoxicity, hepatotoxicity, and central and peripheral neurotoxicity [2]. Neurotoxicity is an important dose-limiting adverse effect that may cause not only dose reduction or drug cessation during therapy but also reduction of the quality of life of patients. The neurotoxicity induced by CDDP is found to have active neuronal cell death features and also has glutamate-dependent excitotoxic features [3]. CDDP affects calcium homeostasis and causes increased intracellular calcium concentrations [4]. However the knowledge about the neurotoxicity of cerebellar granule cells (CGCs) induced by CDDP is limited.
The ion movements are crucial for neurotoxic or neuroprotective mechanisms in antineoplastic agent therapy. Particularly, calcium and potassium ions may have critical roles in neuronal cell death [5]. Calcium ions are fundamental for performing normal cellular functions and for cellular survival. However, calcium concentration may reach critical levels in pathological conditions, such as ischemia or brain damage, leading to cellular damage or cell death [6, 7]. Increased intracellular calcium levels are caused by release from internal stores and calcium influx via channels in the membrane. The dysregulation of calcium homeostasis and calcium signaling is known to contribute to the neurotoxic side effects of CDDP. Particularly, voltage-gated calcium channels are believed to play a role in CDDP-induced neurotoxicity [4]. Thus, the aim to buffer excess intracellular calcium may be achieved by using calcium antagonists that block voltage-gated calcium channels [8].
KATP channels belong to Kir channels characterized by inward rectification by allowing more current to flow inward than outward. These channels are important ion channels that regulate many cellular functions, neuronal signaling, and membrane excitability according to both electrical activity and metabolic status of the cell. In many cell types, it was shown that they may have a role in neuroprotective mechanisms [9]. However, there is a controversy in the results of studies about the contribution of these channels to neuroprotective strategies. In previous studies, the blockade of KATP channels protects the dopamine neurons from degeneration; however, the activation of KATP channels can also protect mesencephalic neurons from MPP+ cytotoxicity [10, 11]. Moreover, KATP channel activation was found to protect CGCs from oxidative stress [11].
In previous CGC neurotoxicity studies, neuronal death was shown to arise from several reasons, such as excess glutamate release, changes in Ca2+ homeostasis, deficiency of K+, reactive oxygen species production, and caspase activation [8, 12]. Any change in the concentrations of these molecules may cause cell death. Although molecular mechanisms of pathological cell loss have been extensively investigated in many studies, the contribution of the disruption of ionic homeostasis to cell death is still unknown. However, excitotoxicity was found to be increased by calcium influx and decreased by potassium efflux in different cell types [13].
The evaluation of neurotoxicity can be achieved by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) assay, which is widely used in cytotoxicity studies. The MTT assay is a colorimetric viability test based on the enzymatic reduction of the MTT (thiazolyl blue tetrazolium bromide) molecule to formazan crystals in the presence of viable cells. This enzymatic reduction concludes with a change in color that can be easily detected by an ELISA reader system in terms of absorbance values [14].
CDDP was found to activate excitotoxic mechanisms and also trigger active neuronal death [3]. It is widely known that calcium influx increases, whereas potassium efflux decreases excitotoxicity [13]. The aim of this preliminary study was to evaluate the potential roles of the channels that regulate calcium and potassium ions in the neurotoxicity of CDDP. To determine the effect of calcium ions, the cells were treated with nimodipine (L-type calcium channel blocker). The cells were also treated with glibenclamide (KATP channel blocker) to evaluate the potential therapeutic effect of KATP channels in CDDP neurotoxicity.
Materials and Methods
Primary cultures of CGCs were prepared from 5-day-old newborn Sprague–Dawley rats with modifications in the method described by Xu and Wojcik [15]. Approval was obtained from the local ethics committee for animal experimentations. In addition, informed consent was obtained from the participants of the study, and the institution’s ethics committee approved the study (protocol number: 273/2012). The animals were procured from the Medical and Surgical Research Center in Eskişehir Osmangazi University. Briefly, newborn rats were decapitated, and the cerebellum was removed. To prevent contamination, the cerebellum was washed twice with calcium-free Hank’s Balanced Salt Solution (HBSS) (HyClone Thermo Scientific, Germany). Then, it was suspended in 5 ml calcium-free HBSS containing 2 ml of trypsin–EDTA (0.25% trypsin–0.02% EDTA; Sigma Aldrich, USA) at 37°C for 40 min. Trypsin digestion was attenuated by adding 5 ml of fresh Dulbecco’s Modified Eagle Medium (DMEM) (Sigma Aldrich, Germany). DMEM solution comprised 10% fetal calf serum (FCS) (HyClone Thermo Scientific, Germany), 50 µg/mL penicillin, and 0.2 mM l-glutamine (Sigma Aldrich, Germany). After centrifugation at 1000 rpm for 5 min, the resulting pellets were suspended. After enzymatic disintegration, the cell suspension was pipetted up and down using Pasteur pipets with soft-headed tips that provide mechanic disintegration called trituration.
The bases of 25 cm2 polypropylene tissue culture flasks were covered with poly-D-lysine (Sigma Aldrich, Germany). Twenty-four hours before the experiment, 0.1 mg/mL poly-D-lysine (30,000–70,000 MW) was dissolved in phosphate-buffered saline, and the bases of 96-well plates were also covered with the same solution. After keeping at room temperature (25°C) for 5 min, excess poly-D-lysine was vacuumed and left to dry at 4°C overnight. Then, the cell suspension was transferred into covered flasks to allow adherence to the surface, and culture flasks were incubated at 37°C and humidified at 95% air and 5% CO2. After 30 min, the media was changed to eliminate non-adherent cells, and fresh DMEM containing 10% FCS was added into the flasks. Culture media was changed twice a week, and neurons were used for neurotoxicity experiments following an 8-day in vitro incubation.
The dye exclusion method was applied to stain the cells with 0.4% trypan blue for counting live cells. An inverted light microscope was used to examine stained and non-stained cells immediately. Approximately 10,000 cells per well were plated in 96-well culture plates in drug-free DMEM medium overnight. CDDP (100, 200, and 500 µM) (Sigma Aldrich, USA) was applied to wells at gradually increasing concentrations. After applying the drug into cell suspensions, the plates were incubated overnight at 37°C and humidified at 95% air and 5% CO2. The toxic effects of CDDP were evaluated using the drug alone or with nimodipine (Sigma Aldrich, USA) or glibenclamide (Sigma Aldrich, USA) at concentrations of 1, 10, and 50 µM.
The MTT assay was used for cytotoxicity determination. MTT was dissolved in HBSS at a final concentration of 1 mg/mL. Briefly, at the end of the incubation period, the cells inside the 96-well culture plates were incubated with MTT solution at 37°C for 4 h. After vacuuming the incubation solution, the cells were lysed with 150 μL dimethyl sulfoxide and subjected to the measurement of optical density at 540 nm as reference on an ELISA system (Thermo Lab Systems Multiscan EX, USA) [16, 17].
Results were evaluated using the ELISA reader system (Thermo Lab Systems, Germany). Drug applications to 96-well plates were handled at different times so each plate was evaluated according to its control data. Data were analyzed using SPSS 15.0 statistical package program (IBM SPSS Inc., Chicago, IL, USA). The percentage of cell death scores was calculated, and the results were statistically analyzed using Kruskal–Wallis H and Tukey tests. A p-value<0.05 was considered significant.
Results
The percentage of cell death according to each group was calculated using the following formula: [relative cell death = 1 − (n / nK ort)]. As expected, CDDP induced neuronal cell death concentration dependently (Figure 1). The minimum inhibitor concentration of CDDP was determined as 200 μM (p<0.01) (Table 1, Figure 1). Nimodipine and glibenclamide combinations were performed together with 200 μM of CDDP.
Figure 1.
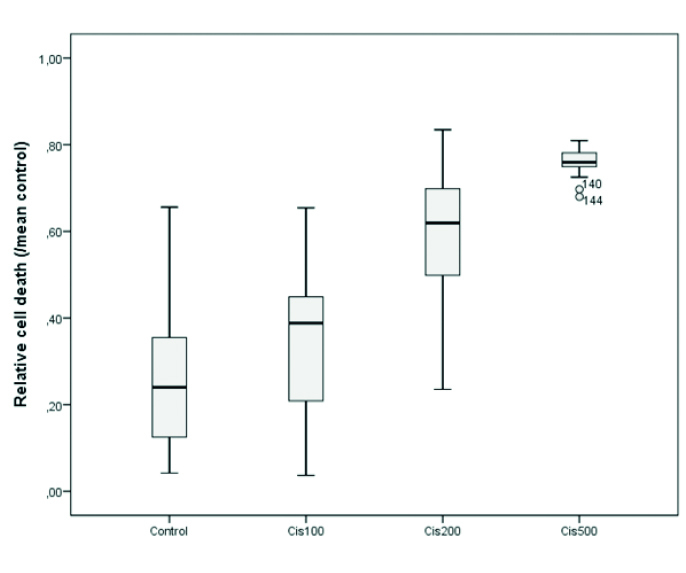
Cisplatin neurotoxicity at 100, 200, and 500 μM concentrations
cis: cisplatin
Table 1.
Concentration-dependent cytotoxicity induced by cisplatin (Cis100, Cis200, and Cis500: cisplatin 100 μM, 200 μM, and 500 μM, ***p<0.001)
| n | Mean±SE | Median (25%–75%) | p* | Pairwise comparisons | ||
|---|---|---|---|---|---|---|
| 1 | Control | 33 | 0.265±0.028 | 0.240 (0.115–0.360) | <0.001 | 1–3***, 1–4*** |
| 2 | Cis100 | 23 | 0.351±0.036 | 0.388 (0.193–0.450) | ||
| 3 | Cis200 | 72 | 0.591±0.017 | 0.619 (0.497–0.701) | ||
| 4 | Cis500 | 24 | 0.759±0.006 | 0.760 (0.749–0.782) |
Kruskal–Wallis one-way analysis of variance on ranks (median 25%–75%)
The difference between the CDDP group and the CDDP–nimodipine combination groups was not significant. In addition, nimodipine did not induce any toxicity at all concentrations (1, 10, and 50 μM). Thus, nimodipine neither increased nor decreased the neurotoxicity of CDDP (Table 2, Figure 2). CDDP neurotoxicity did not significantly change when it was applied together with glibenclamide. However, 10 μM glibenclamide increased CDDP neurotoxicity (p<0.001). In addition, glibenclamide alone was not toxic to the CGCs at all concentrations applied (Table 2, Figure 3).
Table 2.
Relative cell death values for the cisplatin/nimodipine combination groups (Cis200: cisplatin 200 μM; N1, N10, and N50: nimodipine 1, 10, and 50 μM, *p<0.05, **p<0.01, ***p<0.001)
| n | Mean±SE | Median (25%–75%) | p* | Pairwise comparisons | ||
|---|---|---|---|---|---|---|
| 1 | Cis200 | 72 | 0.591±0.017 | 0.619 (0.497–0.701) | <0.001 | 1–5**, 1–6**, 1–7**, 2–5*,3–6***, 4–7*** |
| 2 | Cis200+N1 | 12 | 0.657±0.014 | 0.646 (0.626–0.698) | ||
| 3 | Cis200+N10 | 47 | 0.676±0.014 | 0.696 (0.603–0.746) | ||
| 4 | Cis200+N50 | 24 | 0.658±0.030 | 0.680 (0.648–0.727) | ||
| 5 | N1 | 8 | 0.167±0.040 | 0.177 (0.053–0.261) | ||
| 6 | N10 | 11 | 0.206±0.060 | 0.120 (0.027–0.412) | ||
| 7 | N50 | 18 | 0.272±0.032 | 0.270 (0.196–0.367) |
Kruskal–Wallis one-way analysis of variance on ranks (median 25%–75%)
Figure 2.
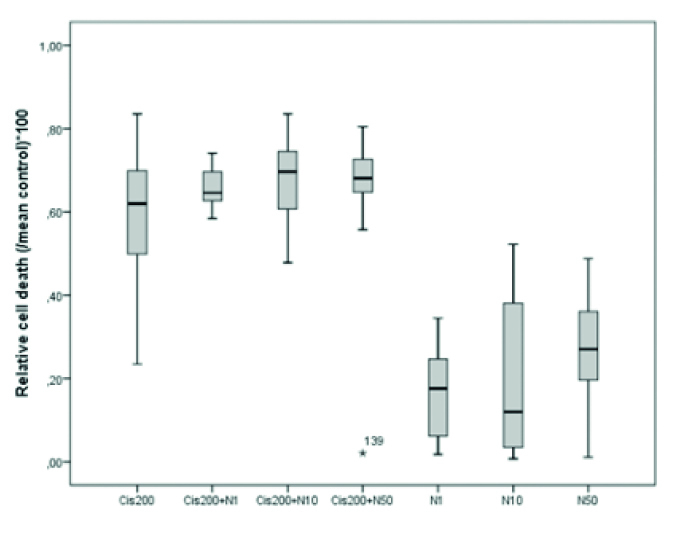
Cisplatin neurotoxicity at 100, 200, and 500 μM concentrations
cis: cisplatin
Figure 3.
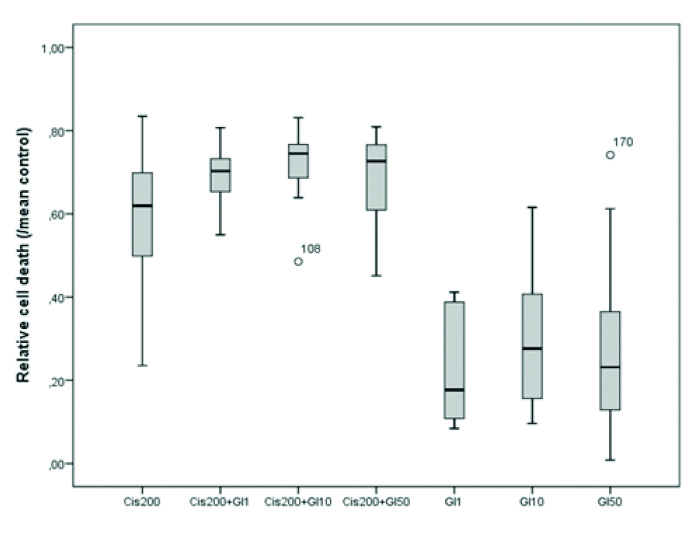
The relative cell death values of cisplatin and glibenclamide combination groups
cis: cisplatin, Gl: glibenclamide
Discussion
The present study investigated the effects of L-type Ca2+ channel blocker nimodipine and KATP channel blocker glibenclamide in CDDP-induced neurotoxicity using primary CGCs. Neither nimodipine nor glibenclamide significantly changed CDDP neurotoxicity.
An increase in intracellular calcium concentration is a common mechanism for the neurotoxic side effects of CDDP. Calcium channels are mainly of L-type (60%–70%) in 1-week old CGC culture [18]. Thus, calcium influx via these channels may be believed to be critical for neuronal cell death. However, our results were quite the opposite. The blockade of L-type calcium channels via nimodipine could not protect the cells. Neuronal L-type calcium channels have different gating properties identified in a variety of neurons, including CGCs [19]. This difference in gating properties of L-type calcium channels may be due to nimodipine ineffectiveness. Moreover, nimodipine-sensitive L-type current contributed to 15% of the total current, and the prevention of calcium influx via nimodipine-sensitive calcium channels may be inadequate to prevent CDDP neurotoxicity [20].
The other calcium influx routes may be responsible for CDDP neurotoxicity rather than the L-type calcium channels. In the past, an increase in intracellular calcium was known to be responsible for neurotoxicity, whatever way calcium goes into the cell; however, recent studies focused on the way of calcium influx and the involved second messenger systems [21]. Similarly, in hippocampal neurons, calcium influx via L-type calcium channels was found to be harmless, but calcium influx via N-methyl-d-aspartate (NMDA) receptors was toxic for the cells [22]. Thus, the blockade of L-type calcium channels was suggested to be unable to prevent toxic calcium influx in our study. The increase in toxicity may be relevant to other calcium routes (e.g., NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, kainate receptors, and Na+/Ca2+ exchanger) rather than L-type calcium channels as shown in a previous study [22].
Table 3.
Relative cell death values for the cisplatin/glibenclamide combination groups (Cis200: cisplatin 200 μM; G1, G10, and G50: glibenclamide 1, 10, and 50 μM, **p<0.01, ***p<0.001)
| n | Mean±SE | Median (25%–75%) | p* | Pairwise comparisons | ||
|---|---|---|---|---|---|---|
| 1 | Cis200 | 72 | 0.591±0.017 | 0.619 (0.497–0.701) | <0.001 | 1–5**, 2–5***, 1–7***, 4–7***, 1–6**, 3–6***, 1–3*** |
| 2 | Cis200+G1 | 12 | 0.695±0.020 | 0.703 (0.648–0.737) | ||
| 3 | Cis200+G10 | 36 | 0.726±0.011 | 0.745 (0.685–0.769) | ||
| 4 | Cis200+G50 | 12 | 0.687±0.033 | 0.727 (0.590–0.770) | ||
| 5 | G1 | 9 | 0.233±0.048 | 0.177 (0.100–0.388) | ||
| 6 | G10 | 12 | 0.296±0.048 | 0.276 (0.153–0.439) | ||
| 7 | G50 | 23 | 0.274±0.038 | 0.231 (0.126–0.385) |
Kruskal–Wallis one-way analysis of variance on ranks (median 25%–75%)
L-type calcium channels were shown to couple with Ca2+/CaM kinase II (CaMKII), enabling the channels to play a primary role in CaMKII activation [23]. The inhibition of CaMKII was shown to cause neurotoxicity via dysregulation of glutamate/calcium signaling and enhanced neuronal excitability [24]. CaMKII can also regulate cAMP response element-binding protein (CREB) phosphorylation, which is crucial for neuronal survival. Thus, blockade of L-type calcium channels with nimodipine may cause inactivation of CaMKII and disruption of CREB phosphorylation, leading to neurotoxicity and neuronal cell death [25]. Future studies should aim to investigate the roles of these molecules.
The physiology of calcium signaling also differs from cell to cell. Both Purkinje neurons and cerebellar granule neurons may have different signaling mechanisms [22]. Hence, unsuccessful inhibition of neurotoxicity by voltage-gated channels may have originated from their different calcium signaling mechanisms. However, previous studies have shown that nimodipine protects CGCs from intracellular calcium increase induced by NMDA and hydrogen sulfur neurotoxicity [26, 27]. In our previous study, we also studied CDDP neurotoxicity with different types of neuronal cells, including dorsal root ganglion neurons. CDDP also induced physiological alterations related to calcium dynamics in the cell. Nimodipine was able to protect the dorsal root ganglion neurons from the toxic effects of CDDP [28]. However, CGCs were different from the dorsal root ganglion neurons because the cerebellum is one of the regions of the central nervous system that develops post-natally [29]. CGC progenitors still migrate into the internal granular layer of the cerebellum even during early postnatal period [30]. Thus, at that time, CGCs undergo rapid growth and may be more vulnerable to the devastating effects of drugs. Because injury may generate some structural changes in this region. For example, Piccolini et al. [31] noticed that CDDP induces alterations in matrix metalloproteinase expression in the developing rat cerebellum. The information related to such physiological changes in CGCs in the presence of a neurotoxic drug CDDP is limited.
In addition, the capability of nimodipine to block channels can be based on the state of the channels, and channels bind nimodipine with different affinities according to their state [32]. The nimodipine-sensitive current is most probably responsible for the increase of calcium as determined by prolonged depolarizations in these cells. Thus, the sensitivity of voltage-gated calcium channels to nimodipine may be different based on the type of cells and condition.
In excitable cells, such as neurons, the regulation of potassium ion movements is fundamental for the regulation of membrane potential and electrical activity. Any change in the concentration of potassium ions that occurred in intracellular or intercellular compartments contributes to the activation of cell death mechanisms. KATP channels deliver potassium inside or outside of the cell according to the membrane potential. KATP channels decrease membrane excitability by regulating K+ ion flow direction. Thus, they may be the candidate ion channels for the protection of cells from excitotoxicity [33].
At normal physiological status, KATP channels are closed; however, in the presence of a toxic agent, the membrane is depolarized [33]. This causes an excitation in the cells. KATP channels open, and K+ ions flow through the outside of the membrane to defeat excitation and then hyperpolarization occurs. If KATP channels are exogenously closed, excitation cannot be blocked and excitotoxicity occurs in the cells.
In the study, to investigate the relationship between the neurotoxicity of CDDP and K+ ion movement across the membrane through KATP channels, glibenclamide (blocker of KATP channel) at 1, 10, and 50 µM concentrations was applied with CDDP. Glibenclamide did not cause any significant change in CDDP neurotoxicity.
First, in the presence of a neurotoxic agent, KATP channels are opened to suppress excitation; however, later, the loss of excess potassium ion from the cell may contribute to the toxicity of CDDP. If the level of potassium decreases, some enzymes, such as caspases and endonucleases, may get activated, leading to induction of apoptosis [34].
Similarly the efflux of K+ ions and the loss of intracellular potassium are also critical in apoptosis, like in case of Ca2+ influx and the accumulation of intracellular calcium [5]. Physiological concentrations of intracellular potassium act as a suppressor on proapoptotic molecules. If the loss of potassium is greater than the cell can tolerate, caspases, cytochrome C, and endonucleases would be activated, and then apoptotic cascade starts for these cells. Moreover, the cells lose water with potassium efflux, and this may cause shrinkage of the cells by decreasing cell volume, which is one of the characteristic features of apoptosis. Potassium ion is abundant in the cells, and even if tens of mM loss can be tolerated, a 50% loss cannot be tolerated by the cells. Thus, high potassium loss must be considered for causes of cell death. Therefore, the contribution of KATP channels to CDDP neurotoxicity was so complex. Mitochondrial KATP channels may be more effective to decrease toxicity as shown in a previous study [13]. KATP channels of neonatal rats in our study might be inadequate to protect the neurons from cell death. In an earlier study, Xia and Haddad showed that the formation of KATP channels and sulfonylurea receptors (subunit of KATP channels) occurs postnatally, and it is the highest level at maturation. Thus, KATP channel activation in the mature period may be more protective against the neurotoxic reaction [35].
The present study has some limitations. First, the content of the study was restricted to only one type of calcium and potassium channels. In addition, only the cytotoxicity test was performed to investigate the effects of drugs. MTT assay is an absorbance-based assay that provides information about the viability of the cells and how well they metabolize the component. It is not always comparable to % growth inhibition. In future studies, electron microscopy techniques may also be included in the study to detect the histopathological features of CDDP-induced neurotoxicity.
Finally, this preliminary study provides an insight for future studies in CDDP-induced neurotoxicity. It is difficult to explain the exact mechanisms of the interaction between the neurotoxicity of CDDP and calcium/potassium ion movements in CGCs. The results of the previous studies are also complicated to understand the missing points related to ionic homeostasis and neurotoxicity. The mechanisms of the interaction between the effects of CDDP and intracellular ionic homeostasis are complex and multidirectional phenomena that must be comprehensively investigated.
Footnotes
Ethics Committee Approval: Ethics committee approval was received for this study from the ethics committee of Eskişehir Osmangazi University (273/2012).
Informed Consent: Written informed consent was obtained from patients who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - K.E.; Design - K.E., C.C.U.; Supervision - K.E.; Resources - K.E., C.C.U.; Materials - K.E., C.C.U.; Data Collection and/or Processing - C.C.U.; Analysis and/or Interpretation - K.E., C.C.U.; Literature Search - K.E., C.C.Ü.; Writing Manuscript - C.C.U.; Critical Review - K.E.
Conflict of Interest: Authors have no conflict of interest to declare.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Desoize B, Madoulet C. Particular aspects of platinum compounds used at present in cancer treatment. Crit Rev Oncol Hematol. 2002;42:317–25. doi: 10.1016/S1040-8428(01)00219-0. [DOI] [PubMed] [Google Scholar]
- 2.Florea AM, Busselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011;3:1351–71. doi: 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rzeski W, Pruskil S, Macke A, et al. Anticancer agents are potent neurotoxins in vitro and in vivo. Ann Neurol. 2004;56:351–60. doi: 10.1002/ana.20185. [DOI] [PubMed] [Google Scholar]
- 4.Tomaszewski A, Busselberg D. Cisplatin modulates voltage gated channel currents of dorsal root ganglion neurons of rats. Neurotoxicology. 2007;28:49–58. doi: 10.1016/j.neuro.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Yu SP. Regulation and critical role of potassium homeostasis in apoptosis. Prog Neurobiol. 2003;70:363–86. doi: 10.1016/S0301-0082(03)00090-X. [DOI] [PubMed] [Google Scholar]
- 6.Penn RD, Loewenstein WR. Uncoupling of a nerve cell membrane junction by calcium-ion removal. Science. 1966;151:88–9. doi: 10.1126/science.151.3706.88. [DOI] [PubMed] [Google Scholar]
- 7.Simon RP, Griffiths T, Evans MC, Swan JH, Meldrum BS. Calcium overload in selectively vulnerable neurons of the hippocampus during and after ischemia: an electron microscopy study in the rat. J Cereb Blood Flow Metab. 1984;4:350–61. doi: 10.1038/jcbfm.1984.52. [DOI] [PubMed] [Google Scholar]
- 8.Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- 9.Yamada K, Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol. 2005;38:945–9. doi: 10.1016/j.yjmcc.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 10.Uchida S, Yamada S, Nagai K, Deguchi Y, Kimura R. Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 1997;61:2083–90. doi: 10.1016/S0024-3205(97)00881-3. [DOI] [PubMed] [Google Scholar]
- 11.Xie J, Duan L, Qian X, et al. K(ATP) channel openers protect mesencephalic neurons against MPP+-induced cytotoxicity via inhibition of ROS production. J Neurosci Res. 2010;88:428–37. doi: 10.1002/jnr.22213. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- 13.Teshima Y, Akao M, Li RA, et al. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34:1796–802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- 14.Ganot N, Meker S, Reytman L, Tzubery A, Tshuva EY. Anticancer metal complexes: synthesis and cytotoxicity evaluation by the MTT assay. J Vis Exp. 2013:e50767. doi: 10.3791/50767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Wojcik WJ. Gamma aminobutyric acid B receptor-mediated inhibition of adenylate cyclase in cultured cerebellar granule cells: blockade by islet-activating protein. J Pharmacol Exp Ther. 1986;239:568–73. [PubMed] [Google Scholar]
- 16.Scheuber PH, Mossmann H, Beck G, Hammer DK. Direct skin test in highly sensitized guinea pigs for rapid and sensitive determination of staphylococcal enterotoxin B. Appl Environ Microbiol. 1983;46:1351–6. doi: 10.1128/aem.46.6.1351-1356.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scudiero DA, Shoemaker RH, Paull KD. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–33. [PubMed] [Google Scholar]
- 18.Parri HR, Lansman JB. Multiple components of Ca2+ channel facilitation in cerebellar granule cells: expression of facilitation during development in culture. J Neurosci. 1996;16:4890–902. doi: 10.1523/JNEUROSCI.16-16-04890.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koschak A, Obermair GJ, Pivotto F, et al. Molecular nature of anomalous L-type calcium channels in mouse cerebellar granule cells. J Neurosci. 2007;27:3855–63. doi: 10.1523/JNEUROSCI.4028-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–9. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–21. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep. 2013;33:97–101. doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashpole NM, Song W, Brustovetsky T, et al. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. J Biol Chem. 2012;287:8495–506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell KF, Bent RJ, Meese-Tamuri S, et al. Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. J Neurochem. 2013;126:274–87. doi: 10.1111/jnc.12176. [DOI] [PubMed] [Google Scholar]
- 26.Duzenli S, Bakuridze K, Gepdiremen A. The effects of ruthenium red, dantrolene and nimodipine, alone or in combination, in NMDA induced neurotoxicity of cerebellar granular cell culture of rats. Toxicol In Vitro. 2005;19:589–94. doi: 10.1016/j.tiv.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 27.García-Bereguiaín MA, Samhan-Arias AK, Martín-Romero FJ, Gutiérrez-Merino C. Hydrogen sulfide raises cytosolic calcium in neurons through activation of L-type Ca2+ channels. Antioxid Redox Signal. 2008;10:31–42. doi: 10.1089/ars.2007.1656. [DOI] [PubMed] [Google Scholar]
- 28.Erol K, Yiğitaslan S, Ünel Ç, Kaygısız B, Yıldırım E. Evaluation of cisplatin neurotoxicity in cultured rat dorsal root ganglia via cytosolic calcium accumulation. Balkan Med J. 2016;33:144–51. doi: 10.5152/balkanmedj.2016.161110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernocchi G, Fanizzi FP, De Pascali SA, et al. Neurotoxic effects of platinum compounds: studies in vivo on ıntracellular calcium homeostasis in the ımmature central nervous system. Toxics. 2015;3:224–248. doi: 10.3390/toxics3020224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biran V, Verney C, Ferriero DM. Perinatal cerebellar injury in human and animal models. Neurol Res Int. 2012;2012:858929. doi: 10.1155/2012/858929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piccolini VM, Avella D, Bottone MG, Bottiroli G, Bernocchi G. Cisplatin induces changes in the matrix metalloproteinases and their inhibitors in the developing rat cerebellum. Brain Res. 2012;1484:15–28. doi: 10.1016/j.brainres.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 32.Marchetti C, Usai C. High affinity block by nimodipine of the internal calcium elevation in chronically depolarized rat cerebellar granule neurons. Neurosci Lett. 1996;207:77–80. doi: 10.1016/0304-3940(96)12492-7. [DOI] [PubMed] [Google Scholar]
- 33.Sun XL, Zeng XN, Zhou F, et al. KATP channel openers facilitate glutamate uptake by GluTs in rat primary cultured astrocytes. Neuropsychopharmacology. 2008;33:1336–42. doi: 10.1038/sj.npp.1301501. [DOI] [PubMed] [Google Scholar]
- 34.Bortner CD, Hughes FM, Jr, Cidlowski JA. A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem. 1997;272:32436–42. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 35.Xia Y, Haddad GG. Major differences in CNS sulfonylurea receptor distribution between the rat (newborn, adult) and turtle. J Comp Neurol. 1991;314:278–89. doi: 10.1002/cne.903140206. [DOI] [PubMed] [Google Scholar]