

INTRODUCTION
Today's cancer therapy has made substantial progress. Recent success in immuno‐oncology (IO) and combination therapies provide opportunities to enhance activity in the broader population with less drug resistance.1, 2, 3 However, these advances also raise challenges in oncology drug development. At the 2017 American Society of Clinical Pharmacology and Therapeutics (ASCPT) Annual Meeting, these issues were addressed in a symposium with case studies illustrating challenges and opportunities. The presentations and discussion are summarized in this review.
HISTORY
It has been increasingly acknowledged that the conventional maximum tolerated dose (MTD) approach used in oncology drug development may not be ideal, and improving dose selection is needed.4, 5 Historically, the MTD approach has dominated the oncology dose‐finding paradigm. Its origin lies in development of cytotoxic agents based on the underlying assumption that the higher the dose, the greater the likelihood of efficacy and toxicity.6, 7 Dose‐limiting toxicities (DLTs) are predefined in phase I dose escalation trials and, based on DLT criteria, dose is escalated until MTD is reached. Over recent decades, a variety of novel molecular modalities in target and immune therapies have become available that have challenged the traditional MTD‐based dose selection paradigm. Using MTD as the only dose for late‐stage development could be a major contributor to failures of past oncology development paradigms, especially since it results in a large number of dose reductions or discontinuations due to toxicity.8 Therefore, finding the right dose and schedule that best balances risk and benefit is the right direction to shift toward.
Despite the need to improve dose finding in oncology, it is highly challenging due to heterogeneity of the disease, complexity of the biology, high variability of drug response, narrow therapeutic window, development of drug resistance, limitation in study design due to severity of the disease, and urgency to deliver effective treatments to patients. Moreover, oncology clinical trials may not be clearly defined by phases or well controlled. Depending on the novelty of the mechanism, the level of medical need and the promising anticancer activity of a new agent, a phase I study could evolve and grow into a registration trial. For example, the first‐in‐human (FIH) trail of the IO drug pembrolizumab, a monoclonal antibody blocking the interaction between programmed cell death‐1 (PD‐1) receptor and its ligands, served as the primary evidence supporting the initial accelerated approval of its use in melanoma.9 On the other hand, such an opportunity of seamless and accelerated development could present challenges. For instance, clinical data sets may be complex (as they may not have been designed to meet registration needs) or insufficiently powered when analyzed using conventional analysis tools. Moreover, recent success in IO therapy has raised additional challenges such as difficulty to translate from preclinical models and surrogate molecules, complexity of immune systems and their multifaceted impacts on tumors, and heterogeneity of tumors and their microenvironment. Hence, identifying opportunities to develop novel approaches to tackle challenges is key to the success of oncology drug development.
At the ASCPT 2014 Annual Meeting, the MTD paradigm was challenged and expectations to improve dose selection in oncology were discussed.10 During the ASCPT 2017 Annual Meeting, we reviewed what have been accomplished in the oncology dose‐finding paradigm at a symposium: “Finding the Right Dose in the Right Patients for Oncology and Immuno‐oncology: Are We There Yet and How Have Quantitative Pharmacology, Translational and Precision Medicine Been Utilized?”.11 We shared our experience and commentary in challenges and opportunities in oncology and IO dose finding from the perspectives of pharmaceutical industry, clinical practice, and regulatory authority. The opportunities of utilizing modeling and simulation (M&S) during both early‐ and late‐phase oncology drug development were advocated. Multidisciplinary collaboration that integrates quantitative and experimental sciences and translational and precision medicine was emphasized. New direction and advances, especially methods and applications in a broad spectrum of cancer therapy, were also discussed.
CURRENT CHALLENGES
Delaying dose finding to postmarketing
Current challenges in defining the right dose for targeted therapies include failing to find an optimal dose during clinical development and, as a consequence, dose finding continues in postmarketing trials. An example is cabozantinib, which was approved by the US Food and Drug Administration (FDA) for metastatic medullary thyroid cancer (MTC) at a dose of 140 mg administered daily. The approved dose encountered dose reductions in 79% of patients in the pivotal phase III study and resulted in 16% of patients discontinuing due to toxicity. Subsequent studies of cabozantinib, utilizing a revised formulation, have been conducted at a much lower dose of 60 mg.12, 13 Roda et al. reported that dose reductions were prevalent for 34 recently approved targeted agents.14 Seven of these approved agents required dose reductions in more than 50% of patients. This presents a major challenge where the approved dose results in dose reductions in a large number of patients. Many additional oncology drug development challenges still exist, such as defining a tolerable dose for targeted therapies, whether phase I studies are able to identify key toxicity of these agents, and the need for optimization for dosing schedules and drug combinations.
Continued use of 3+3 designs
In MTD determination, the “3+3” approach is widely used in oncology trial design but may not offer the best design. In a review of published articles from 181 evaluable phase I clinical trials, 96% of them utilized a “3+3” design or its variation.15 More study design options, such as model‐based Bayesian approaches, could be considered and explored in oncology clinical trials. The optimal study design in a clinical trial should be evaluated based on the totality of many factors, including scientific basis, end points, sample size, cancer type, development speed, and feasibility in clinical operation. In addition, a large effort is needed to educate clinical teams on the utility of different study designs in drug development.16
Lack of characterization of chronic toxicity
Another challenge in identifying the right dose in a phase I study is the lack of full characterization of chronic toxicity, since typical MTD determination methodologies reflect only Cycle 1 toxicity. This could misrepresent the safety profile for the cancer therapeutic agents as they are administered chronically over multiple cycles, and many long‐term safety events occur much later than in Cycle 1. Additionally, treatment interruptions and dose reductions continue to happen beyond Cycle 1.17 For some targeted therapies, toxicities are delayed and assessment of a tolerable dose in DLT assessment during Cycle 1 is not always feasible. This may result in identification of a dose that is tolerable in Cycle 1 but requires reduced doses or discontinuations in a large number of patients in subsequent cycles due to delayed toxicities, resulting in patients unable to derive meaningful benefit from treatment.16 Approaches have been published where a phase Ib dose expansion cohort can be better designed to inform the recommended phase II dose (RP2D).14 The proposal includes identifying a dose tolerable in 12–20 patients with longer observations (at least in two cycles), which results in dose reductions in less than 30% of patients. Jardim et al. supported the view that challenges of appropriate dose selection from phase I to phase III are reflective of an inadequate number of patients in phase I.18
Adopting novel study designs
Some novel approaches and dosing paradigms are emerging and are worthy of consideration in gaining efficiency in oncology drug development. When higher drug exposure is demonstrated to provide added benefit of efficacy to patients, within‐patient dose escalation strategies have been incorporated as part of clinical development, allowing selection of individualized doses for patients to derive maximal efficacy at the highest tolerable dose possible. Such an approach was adopted and successfully implemented in the drug development of axitinib, a tyrosine kinase inhibitor of vascular endothelial growth factor (VEGFR)‐1, VEGFR‐2, and VEGFR‐3.19 Wages and Tait described novel designs for targeted agents by conducting seamless phase I/II designs that account for both toxicity and efficacy. 20 The method they proposed is a bivariate extension of the continual reassessment method (CRM), combining features of the CRM and order restricted inference. For combination trials, the Clinical Trial Design (CTD) Task Force of the National Cancer Institute (NCI) Investigational Drug Steering Committee recommended selecting dose regimens based on a biological or pharmacological rationale supported by clinical and preclinical data, taking into account potential pharmacokinetic (PK) and pharmacodynamic (PD) interactions.21 Alternative trial designs such as including controls in phase I trials and leveraging mathematical modeling and a Bayesian approach was also suggested.
Testing more than one dose in phase II/III studies
Dose optimization exercise is very much a balancing act in improving efficacy while keeping safety manageable. A major impediment to selection of an optimized dose in oncology lies in a lack of availability of clinical data from multiple dose levels in phase II/III studies, where primary clinical efficacy and safety are assessed. This could limit the ability to conduct robust exposure–response (ER) analyses of long‐term clinical safety as well as efficacy that can help in arriving at the optimized dose(s) or regimen. Recognizing this challenge, establishing a range of phase II doses rather than studying a single phase II dose, as in the current RP2D paradigm, has been recommended.22 There are also multiple recent attempts of testing more than one dose level/regimen in phase II/III studies to maximize the dose optimization opportunity in oncology and IO.23, 24
OPPORTUNITIES FOR MODELING AND SIMULATION
Due to the life‐threatening nature of the disease, oncology clinical trials are often complex and data interpretation is challenging. However, this also offers opportunity of integrating data from different sources such as preclinical data, biomarkers, response end points, multiple trials, and competitors. It also requires multidisciplinary collaboration in pharmaceutical Research and Development. Used proactively and properly, M&S can be a powerful tool to quantitatively inform oncology drug development. Different M&S approaches could be used in order to tackle the primary challenges encountered during different phases of development (Figure 1). Along with the learning and confirming cycles during clinical development,25 models are continuously modified based on data available at the time to inform the next stage of development. Data across molecules can also be integrated to build platform models, such as disease progression, prediction of outcome by early end points, literature meta‐analysis, and quantitative and system pharmacology (QSP) models, to inform the development of molecules acting on the same pathway or in the same indication.
Figure 1.
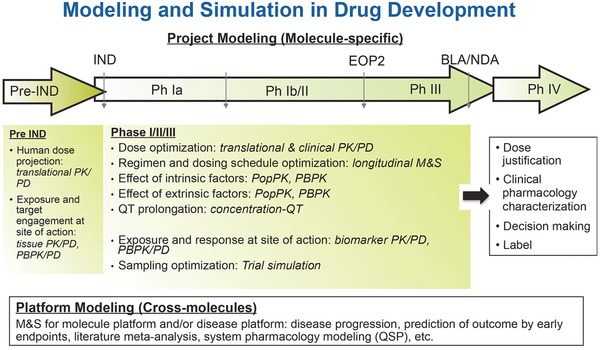
Modeling and simulation in drug development. BLA: Biologics License Applications; EOP2: End of phase II study; IND: Investigational New Drug; M&S: Modeling and Simulation; NDA: New Drug Application; PBPK: Physiologically‐based Pharmacokinetics; PK/PD: Pharmacokinetics/Pharmacodynamics; PopPK: Population Pharmacokinetics.
Prior to an investigational new drug (IND) entering clinical trials, M&S focuses on FIH dose projection and predicting efficacious or biologically effective doses in humans. The primary approaches include PK/PD, physiologically‐based PK (PBPK), and QSP models. Preclinical and competitor data, if available, are used to build the models. During early‐phase clinical development (phase I/II), M&S informs selection of recommended dose for expansion (RDE) and RP2D. It can be used to evaluate the ER relationship and therapeutic window, characterize the time course of biological response and interpatient variability, and identify biomarkers that correlate with pathway modulation. PK/PD and ER models based on clinical data in phase I and II trials are often used and preclinical data can also be leveraged. During late‐phase clinical development (phase III/IV), M&S could be used to further characterize the therapeutic window and the effects of intrinsic and extrinsic factors, evaluate the clinical utility index (CUI), confirm and justify dose selected, and characterize clinical pharmacology properties to inform the drug label. Population PK, PK/PD, ER, and disease progression models are often used to incorporate all the clinical data available. In addition, PBPK models can be used to support clinical pharmacology characterization. Depending on the stage of the development, the right M&S approach(es) should be selected to address the question(s) at hand.
Even though quantitative models have been widely used in many therapeutic areas, their utility and impact in oncology and especially in IO are still evolving. Reviewing recent new drug application (NDA) and biologic license application (BLA) documents revealed that the impact of M&S on oncology dose decision varies. For instance, ER analysis of safety for axitinib was used to support dose titration schemes by the sponsor and was accepted by the FDA.19 In some cases, ER analysis of efficacy and safety was conducted to support dose justification and show no apparent ER relationship. The examples include vismodegib (a hedgehog pathway inhibitor)26 and trametinib (an inhibitor of mitogen‐activated extracellular signal regulated kinase 1 and 2 (MEK1 and MEK2)).27 Although the ER analyses were limited based on data from one dose level in the registration trial, it provides insights on whether patients with low exposure may have a lack of efficacy and whether patients with high exposure may have a higher safety risk within the exposure range at a given dose level, thus justifying the proposed label dose for all patients. In other cases, a postmarketing requirement (PMR) was issued to further optimize the dose. For example, ER analysis of safety was conducted by the sponsor for vandetanib, a tyrosine kinase inhibitors (TKI) that inhibits VEGF with additional activity against epidermal growth factor receptor (EGFR) tyrosine kinase and oncogenic RET kinase. Dose selection was based on MTD, preclinical and phase II data, but a postmarketing trial was required to explore alternative doses and/or regimens to reduce the toxicity but maintain efficacy.28
Modeling & simulation in early‐phase clinical development
To effectively select the right dose in oncology drug development, proactively implementing M&S in early clinical trials is critical. This provides an opportunity to select the right dose range to inform the design of registration trials, so that sufficient data are collected to allow robust analysis that could support dose justification at the registration. Due to limitations in sample size and heterogeneity of patient population at this stage, efficacy data of an investigational oncology drug are limited and inconclusive at this stage. Therefore, efforts should be made to obtain pathway and/or disease biomarker data starting from the phase I trial. This would require multidisciplinary collaboration between research and clinical teams to proactively develop a strategic biomarker plan. Collecting biomarker data could be particularly important in dose finding for IO drugs because the response may be delayed or disguised by pseudo‐progression. While clinical data typically drive dose selection strategy, nonclinical data could also be leveraged in early‐phase clinical development.
One example of using M&S to inform dose selection in early‐phase development is WNT974, a small‐molecule Porcupine inhibitor in clinical investigation, where clinical PK, biomarker, and safety data from the phase I dose escalation were analyzed to inform RDE selection.29 Briefly, ER analyses were conducted for AXIN2, a target‐engagement biomarker, and dysgeusia, the most common adverse event. Based on these analyses, the therapeutic window was estimated. Population PK modeling and simulation were then conducted to predict PK profiles of different dose regimens. By integrating the estimated therapeutic window and predicted PK, a dose regimen that balances biomarker response and safety was selected for the phase I expansion phase. Another example is ABL001, a small‐molecule that inhibits breakpoint cluster region‐abelson (BCR‐ABL), where PK/PD modeling was used to inform RP2D in patients with chronic myeloid leukemia (CML).30, 31 The mRNA level of (%) BCR‐ABL is a molecular response that correlates with efficacy in CML patients. A semiphysiological PK/PD model was developed to describe the kinetics of (%) BCR‐ABL. The average ABL001 concentration for stable disease was then derived based on the parameters estimated from the PK/PD model. PK profiles were simulated and estimated a dose that was predicted to maintain exposure above the estimated threshold concentration for stable disease in ≥95% of patients.
As demonstrated in both examples described, collecting biomarker data is critical to allow robust PK/PD modeling in early clinical trials. Proactive communicating and collaborating within the multidisciplinary team is important, so that the right data analysis strategy and methodology are selected, and the right study design is implemented to collect the right data to allow appropriate modeling. In addition, due to the fast pace and potentially seamless progression of oncology clinical trials, the analyses should be prioritized and performed in real‐time fashion in order to timely impact dose selection in the next stage of clinical trials.
For small‐molecule compounds with high cellular permeability, comparing in vivo exposure of unbound drug at a given dose vs. nonclinical potency data could inform dose selection in early clinical trials. In the examples described above, the minimal concentration at steady state of the selected dose of WNT974 and ABL001 is above the IC50s of in vitro cell proliferation assays after correction for protein binding. However, for small molecules with low membrane permeability or biologics, tissue distribution at the site of action may be important to consider. In general, translation from preclinical to clinical is challenging in oncology and IO. Many factors should be considered, such as the difference in biology and immunology, target expression, and drug disposition. Hence, designing the right clinical studies early on, thoroughly analyzing clinical data, and leveraging nonclinical data are all necessary enablers in early‐phase clinical development.
Modeling & simulation in late‐phase clinical development
M&S has been widely applied in late‐phase clinical development to confirm the right dosing for a broad patient population, fulfil dose adjustment needs in patient subgroups, and characterize the effects of intrinsic and extrinsic factors. Analyses at this stage generally focus on clinical outcomes such as primary efficacy end points and major adverse events. A modeling platform established based on data from phase II/III trials can many times further be applied to inform other projects in the indication, with the same mechanism of action (MOA), and/or in combination therapy. Atezolizumab, a human monoclonal antibody that targets programmed cell death‐1 ligand 1 (PD‐L1), is an example in IO that M&S has contributed to late‐phase clinical development.
Based on the pivotal study IMvigor 210 with atezolizumab 1,200 mg once every 3 weeks (Q3W) in platinum‐treated locally advanced or metastatic urothelial carcinoma patients, ER analyses showed no statistically significant exposure–efficacy relationship for objective response rate (ORR), and no statistically significant exposure–safety relationship for both adverse events of Grade 3–5 and adverse events of special interest (AESI).32 This suggested no improved efficacy would be expected with atezolizumab doses higher than 1,200 mg Q3W, and no improved safety with lower doses, with the caveat that only the 1,200 mg dose level was explored in study IMvigor 210. Collectively with other data, these ER analyses support the atezolizumab labeled dose of 1,200 mg Q3W. Similar analyses were conducted in nonsmall‐cell lung cancer (NSCLC) based on the open‐label randomized‐controlled phase II trial (POPLAR study).33
Longitudinal tumor size models have been proposed to estimate tumor growth inhibition (TGI) metrics based on the sum of longest diameters (SLD) of target lesions per the Response Evaluation Criteria in Solid Tumors (RECIST). A modeling framework with TGI metrics (such as time‐to‐tumor‐growth (TTG), tumor growth rate constant (KG), or Week 8 change in tumor size from baseline) as a predictor for overall survival (OS) has been shown in several tumor types for a variety of treatments of different MOA.34 However, for the recent IO therapies, the dynamics of tumor response to treatment appears to be different with other MOAs. Response patterns with IO are potentially diverse, with delayed responses, and even an initial increase in tumor burden or appearance of new lesions before regression (pseudo‐progression).35 In addition, IO therapies may lead to more long‐term efficacy, as exemplified by more substantial OS separation at late times. The applicability of such a modeling framework for IO therapies was evaluated based on data from the POPLAR study comparing atezolizumab vs. docetaxel in patients with advanced pretreated NSCLC.33, 36 In the POPLAR study, TGI profiles in the atezolizumab and docetaxel arm crossed at about 25 weeks, with more initial shrinkage in docetaxel‐treated patients, and slower on‐treatment KG in atezolizumab‐treated patients. A multivariate parametric model linking baseline patient characteristics (albumin and number of metastatic sites) and on‐treatment KG to OS was developed. This model could well predict the OS hazard ratio (HR) in POPLAR, as well as subpopulations of patients with varying baseline PD‐L1 expression levels, suggesting the validity of such an oncology modeling framework in IO. Tumor dynamic metrics (such as the on‐treatment KG) has strong potential as a model‐based biomarker and an alternative early end point to evaluate efficacy in clinical trials. This approach is being further evaluated and validated for broader application in both the early and late phases of IO development.
FUTURE OPPORTUNITIES
Recent developments in oncology and IO have led to many treatment options available to patients with varying tumor types. This promising evolvement for patients also leads to more opportunity and necessity to optimize dosing for these treatment options and combinations. However, it behooves us as a community to work on optimization solutions, despite additional challenges of a rapidly changing landscape of new treatment options. Working collectively with industry and academia on these challenges, the FDA Office of Hematology and Oncology products along with the American Association of Cancer Research (AACR) recently conducted a series of three dose‐finding workshops (DFWs) on dose optimization in oncology and IO.37, 38, 39 The workshops laid the groundwork to optimize oncology drug development for small molecule kinase inhibitors and biologics, introducing new approaches including the model‐based and adaptive dose‐finding trials, the investigation of multiple doses that have potential for beneficial antitumor effects as opposed to selecting the MTD as the only dose in phase III trials, a systematic and early approach to identifying ER relationships of safety and efficacy as opposed to ad‐hoc ER analyses for safety and efficacy conducted only as an exercise to fulfill regulatory requirements at the time of submission.40
The first DFW (DFW‐1) was conducted in May 2015 with the purpose to provide an interdisciplinary forum to discuss the best practices of dose finding and dose selection in oncology for small molecule kinase inhibitors.37 There were several presentations from academia, industry, and FDA personnel. Recent case studies were presented illustrating the impact of not understanding the dose–response relationship and sources of exposure variability on the benefit–risk profile and regulatory decision‐making. Some successful examples were also presented showcasing prospective selection of optimal dose and dosing schedule by analyzing exposure–safety and/or exposure–efficacy relationships with active cross‐functional collaboration throughout development. One of the take‐home messages of DFW‐1 was that there is a need to have increased importance and urgency in interdisciplinary communications, so that teams are collectively looking at the same data and communicating its implications and revisit/retest when new signals emerge. Another clear message from this conference was that the FDA does not require “3+3” designs for dose‐finding studies and are very open to other model‐based dose escalation study designs.41
The first successful workshop was followed by a second one, which was held in June 2016.38 The discussion was broadened to include large molecules, including IO products, with the discussion focused primarily on efficacy. Data were presented at DFW‐2 about the use of the minimal acceptable biological effect level (MABEL) based on data of receptor occupancy (RO) and pharmacological activity to choose starting doses for large molecules. Strategies such as intrapatient dose escalation and “n” of 1 cohorts are utilized to minimize the exposure to patients at dose levels that are not anticipated to be biologically active.42 These strategies focus on the lower bound of dose ranges to ensure safe starting doses while minimizing investigation of many doses/patients at the lower end of the spectrum, where there would be limited expectation of antitumor activity. However, selection of an optimal dose for efficacy still requires analyses based on PK/PD modeling and the conduct of ER relationships.
A third dose‐optimizing workshop (DFW‐3) was held recently in July 2017 in Washington DC that focused on combination therapy, especially the combination of chemotherapy, targeted therapy, and other IO agents with immune checkpoint inhibitors (ICIs).39 Biomarkers to aid in patient and dose selection and novel end points that can define patient benefit were the focus of DFW‐3. These DFWs offered an invaluable forum for experts from industry, academia, and regulatory agencies across different scientific disciplines to discuss recent advancement and challenges with today's rapidly evolving anticancer therapies. Collaborations across affiliations and across scientific disciplines should be the direction to move forward. This type of collective intelligence in a timely fashion is crucial for faster and smarter oncology drug development to bring more and better treatment options to cancer patients.
There are also certain areas in ER analyses that can be improved to better support dose optimization. For instance, patients doing well on study may encounter many dose reductions that result in having lower exposure. On the other hand, patients who withdraw from the study early (and hence with little clinical benefit) would likely have higher exposure, as they may not encounter many dose reductions. This disparity could confound the interpretation of the ER relationship, thus showing lower exposure has better efficacy. Consideration should be given in assessing any differences in response between the lowest and highest exposure groups due to confounding factors rather than truly due to the observed concentration levels on which data are categorized. Therefore, it is imperative that modeling of such data account for such potential biases or confounding factors. The confounding factors for ER analysis are especially problematic for biologics when there is only one dose level tested. Change of clearance (CL) and disease burden has been uncovered as potentially confounding factors for nivolumab, necessitating the need to study more than one dose.23 Recently, the limitation of ER analysis based on one dose level and a potential “false positive” ER relationship due to confounding factors for biologics have been acknowledged. Various approaches have been explored to address the confounding factors in ER analysis, such as incorporating case–control comparison, time‐varying CL, and TGI.23, 34, 43, 44, 45
To better characterize the ER relationship and support dose optimization, it is more common to test more than one dose level in oncology phase Ib dose expansion and/or phase II studies. Dose ranging is often limited in early‐phase clinical studies by the small sample size and relevance of early clinical efficacy end points (such as ORR and progression‐free survival (PFS)). However, it is also many times limited in late‐phase clinical studies (such as phase III) by ethical considerations as well speed needs to obtain effective treatment approved earlier for patient access. The need for studying more than one dose level in pivotal trials would depend on the therapeutic window and prior understanding of the ER relationship for the molecule. Development of predictive models linking early clinical end points (such as ORR, PFS, biomarker response, observed/predicted tumor metrics) with late efficacy end points (such as OS) could be an effective way of bridging the gap between early and late phases, and maximize the understanding of ER relationships and dose optimization as early as possible.
CONCLUSION
Finding the right dose in the right patients is a hot topic in oncology and IO drug development. Current challenges lead to opportunities to apply novel approaches such as better trial design and advanced M&S to maximize drug development success as well as patient benefit. Application of M&S methodologies to PD surrogates of efficacy and/or to tumor shrinkage can help immensely in optimal dose selection based on efficacy end points.46, 47 Clearly, drug development requires teamwork, and a siloed‐approach to drug development may only optimize individual pieces of the puzzle but not the whole picture. While each function is responsible for generating a certain type of data, that group does not have sole ownership of data. We have to collaborate and work in a cross‐functional way in the interest of the bigger goal of optimizing treatment options for patients to derive maximal efficacy while keeping adverse events as minimal as possible. We believe that adoption of current perspectives regarding innovative designs and analytical approaches that can incorporate key clinical, pharmacologic, PK and PD data, and when appropriate, nonclinical information to guide dose selection can aid immensely in optimizing anticancer therapies.
Conflict of Interest
Y.J. is an employee and stock shareholder of Novartis. J.Y.J. is an employee and stock shareholder of Roche/Genentech. A.S is an employee and stock shareholder of Takeda. D.M.H. has a consulting or advisory role in Atara Biotheapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, and AstraZeneca, and receives research funding from NIH P30 CA008748, AstraZeneca, Puma Biotechnology, and Loxo. G.K. was a federal employee with no conflicts of interest at the time of the ASCPT annual meeting and during the article preparation.
Author Contributions
Y.J., A.S. and J.Y.J, wrote the article. D.M.H. and G.K. contributed to the content and reviewed the article.
Funding
D.M.H. receives research funding from National Institutes of Health P30 CA008748.
Disclaimer
The views described are those of the authors and do not necessarily represent the position of the US Food and Drug Administration or the US government. At the time of the symposium at the ASCPT annual meeting, Dr. Kim was an employee of the US FDA; however, at the time of the publication he was no longer employed by the US government.
References
- 1. Couzin‐Frankel, J . Breakthrough of the year 2013. Cancer immunotherapy. Science 342, 1432–1433 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Spranger, S. & Gajewski, T . Rational combinations of immunotherapeutics that target discrete pathways. J. Immunother. Cancer. 1, 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morrissey, K.M. , Yuraszeck, T.M. , Li, C.C. , Zhang, Y. & Kasichayanula, S . Immunotherapy and novel combinations in oncology: Current landscape, challenges, and opportunities. Clin. Transl. Sci. 9, 89–104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mathijssen, R.H. , Sparreboom, A. & Verweij, J . Determining the optimal dose in the development of anticancer agents. Nat. Rev. Clin. Oncol. 11, 272–281 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Minasian, L. et al Optimizing dosing of oncology drugs. Clin. Pharmacol. Ther. 96, 572–579 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Skipper, H.E. , Schabel, F.M., Jr. & Wilcox, W.S. Experimental evaluation of potential anticancer agents. Xiii. On the criteria and kinetics associated with “curability” of experimental leukemia. Cancer Chemother. Rep. 35, 1–111 (1964). [PubMed] [Google Scholar]
- 7. Skipper, H.E. et al Implications of biochemical, cytokinetic, pharmacologic, and toxicologic relationships in the design of optimal therapeutic schedules. Cancer Chemother. Rep. 54, 431–450 (1970). [PubMed] [Google Scholar]
- 8. Hay, M. , Thomas, D.W. , Craighead, J.L. , Economides, C. & Rosenthal, J . Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Prowell, T.M. , Theoret, M.R. & Pazdur, R. Seamless oncology‐drug development. N. Engl. J. Med. 374, 2001–2003 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Symposium . Challenging the maximum tolerated dosing paradigm in oncology: Threading the needle with targeted agents. American Society of Clinical Pharmacology and Therapeutics Annual Meeting (Atlanta, GA, March 2014).
- 11. Symposium . Finding the right dose in the right patients for oncology and immuno‐oncology: Are we there yet and how have quantitative pharmacology, translational and precision medicine been utilized? American Society of Clinical Pharmacology and Therapeutics Annual Meeting (Washington, DC, March 2017).
- 12. Choueiri, T.K. et al Cabozantinib versus everolimus in advanced renal‐cell carcinoma. N. Engl. J. Med. 373, 1814–1823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drilon, A. et al Cabozantinib in patients with advanced RET‐rearranged non‐small‐cell lung cancer: An open‐label, single‐centre, phase 2, single‐arm trial. Lancet Oncol. 17, 1653–1660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roda, D. , Jimenez, B. & Banerji, U . Are doses and schedules of small‐molecule targeted anticancer drugs recommended by phase I studies realistic? Clin. Cancer Res. 22, 2127–2132 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Le Tourneau, C. , Lee, J.J. & Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 101, 708–720 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janne, P. A. , Kim, G. , Shaw, A. T. , Sridhara, R. , Pazdur, R. & McKee, A. E. Dose finding of small‐molecule oncology drugs: Optimization throughout the development life cycle. Clin. Cancer Res. 22, 2613–2617 (2016). [DOI] [PubMed] [Google Scholar]
- 17. Postel‐Vinay, S. et al Phase I trials of molecularly targeted agents: Should we pay more attention to late toxicities? J. Clin. Oncol. 29, 1728–1735 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Jardim, D.L. , Hess, K.R. , Lorusso, P. , Kurzrock, R. & Hong, D.S. Predictive value of phase I trials for safety in later trials and final approved dose: analysis of 61 approved cancer drugs. Clin. Cancer Res. 20, 281–288 (2014). [DOI] [PubMed] [Google Scholar]
- 19. U.S. Department of Health and Human Services . Food and Drug Administration, Center for Drug Evaluation and Research, NDA 202324 Clinical Pharacology and Biopharmacetuics Review – Axitinib (January 2012).
- 20. Wages, N.A. & Tait, C. Seamless phase I/II adaptive design for oncology trials of molecularly targeted agents. J. Biopharm. Stat. 25, 903–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paller, C.J. et al Design of phase I combination trials: Recommendations of the Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee. Clin. Cancer Res. 20, 4210–4217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ratain, M.J. Targeted therapies: Redefining the primary objective of phase I oncology trials. Nat. Rev. Clin. Oncol. 11, 503–504 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Agrawal, S. , Feng, Y. , Roy, A. , Kollia, G. & Lestini, B . Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. J. Immunother. Cancer. 4, 72 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. https://clinicaltrials.gov. Study to Evaluate the Safety and Efficacy of Two Different Dosing Schedules of Pembrolizumab (MK‐3475) Compared to Ipilimumab in Participants With Advanced Melanoma (MK‐3475‐006/KEYNOTE‐006)
- 25. Sheiner, L.B. Learning versus confirming in clinical drug development. Clin. Pharmacol. Ther. 61, 275–291 (1997). [DOI] [PubMed] [Google Scholar]
- 26. U.S. Department of Health and Human Services . Food and Drug Administration, Center for Drug Evaluation and Research, NDA 203388 Clinical Pharacology and Biopharmacetuics Review – Vismodegib (January 2012).
- 27. U.S. Department of Health and Human Services . Food and Drug Administration, Center for Drug Evaluation and Research, NDA 204114 Clinical Pharacology and Biopharmacetuics Review – Trametinib (April 2013).
- 28. U.S. Department of Health and Human Services . Food and Drug Administration, Center for Drug Evaluation and Research, NDA 22405 Clinical Pharacology and Biopharmacetuics Review – Vandetanib (December 2010).
- 29. Ji, Y. et al Population PK/PD modeling of a first‐in‐class Porcupine inhibitor WNT974 in advanced cancer patients to support dose selection for phase I expansion (Abstract LB‐001). American Society of Clinical Pharmacology and Therapeutics Annual Meeting (San Diego, CA, March 2016).
- 30. Wylie, A.A. et al The allosteric inhibitor ABL001 enables dual targeting of BCR‐ABL1. Nature 543, 733–737 (2017). [DOI] [PubMed] [Google Scholar]
- 31. Meille, C . Pharmacometric analysis of drug‐induced molecular response in chronic phase CML (Presentation). 16th GPCO, Basel M&S Seminar (Basel, Switzerland, September 2016).
- 32. Stroh, M. et al clinical pharmacokinetics and pharmacodynamics of atezolizumab in metastatic urothelial carcinoma. Clin. Pharmacol. Ther. 102, 305–312 (2017). [DOI] [PubMed] [Google Scholar]
- 33. Fehrenbacher, L. et al Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet 387, 1837–1846 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Bruno, R. , Mercier, F. & Claret, L . Evaluation of tumor size response metrics to predict survival in oncology clinical trials. Clin. Pharmacol. Ther. 95, 386–393 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Wolchok, J.D. et al Guidelines for the evaluation of immune therapy activity in solid tumors: immune‐related response criteria. Clin. Cancer Res. 15, 7412–7420 (2009). [DOI] [PubMed] [Google Scholar]
- 36. Claret, L. et al A tumor growth inhibition — Overall survival model built on atezolizumab non‐small cell lung cancer phase II POPLAR data predicts outcome of the phase III study OAK using early tumor kinetics. Submitted. [Google Scholar]
- 37. FDA‐AACR Workshop: Dose‐finding of Small Molecule Oncology Drugs (Washington, DC. May 18–19, 2015).
- 38. FDA‐AACR Workshop: Oncology Dose‐finding Workshop (Washington, DC. June 13, 2016).
- 39. FDA‐AACR Workshop: Oncology Dose Finding Workshop Part III (Washington, DC. July 20, 2017).
- 40. Bullock, J.M. , Rahman, A. & Liu, Q. Lessons Learned: Dose Selection of Small Molecule‐Targeted Oncology Drugs. Clin. Cancer Res. 22, 2630–2638 (2016). [DOI] [PubMed] [Google Scholar]
- 41. Nie, L. et al Rendering the 3 + 3 design to rest: more efficient approaches to oncology dose‐finding trials in the era of targeted therapy. Clin Cancer Res. 22, 2623–2629 (2016). [DOI] [PubMed] [Google Scholar]
- 42. Saber, H. , Gudi, R. , Manning, M. , Wearne, E. & Leighton, J.K. An FDA oncology analysis of immune activating products and first‐in‐human dose selection. Regul. Toxicol. Pharmacol. 81, 448–456 (2016). [DOI] [PubMed] [Google Scholar]
- 43. Yang, J. et al The combination of exposure‐response and case‐control analyses in regulatory decision making. J. Clin. Pharmacol. 53, 160–166 (2013). [DOI] [PubMed] [Google Scholar]
- 44. Li, H. et al Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J. Pharmacokinet. Pharmacodyn. 44, 403–414 (2017). [DOI] [PubMed] [Google Scholar]
- 45. Liu, C. et al Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin. Pharmacol. Ther. 101, 657–666 (2017). [DOI] [PubMed] [Google Scholar]
- 46. Venkatakrishnan, K. & Ecsedy, J.A. Enhancing value of clinical pharmacodynamics in oncology drug development: An alliance between quantitative pharmacology and translational science. Clin. Pharmacol. Ther. 101, 99–113 (2017). [DOI] [PubMed] [Google Scholar]
- 47. Venkatakrishnan, K. et al Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin. Pharmacol. Ther. 97, 37–54 (2015). [DOI] [PubMed] [Google Scholar]