

Abstract
RP5063 is a multimodal dopamine (D)‐serotonin (5‐HT) stabilizer with a high affinity for D2/3/4 and 5‐HT1A/2A/2B/7 receptors and moderate affinity for the serotonin transporter. Single‐dose (10 and 15 mg fasting, 15 mg fed) safety in healthy volunteers and multiple‐dose (10, 20, 50, and 100 mg fed, 10 days) safety and pharmacodynamics in patients with stable schizophrenia were defined in two phase I studies. In the single‐dose study, 32 treatment‐emergent adverse events (TEAEs) were observed. Orthostatic hypotension (n = 6), nausea (n = 5), and dizziness (n = 4) were the most common. One serious adverse event (SAE), seen in a patient who should not have been in the study due to a history of seizures, involved brief seizure‐like symptoms. In the multiple‐dose study, 75 TEAEs were reported. Akathisia (n = 20) and somnolence (n = 14) were the most frequent. No clinically significant changes were seen in glucose or prolactin levels, lipid profiles, weight, or electrocardiographic recordings. In both studies, all TEAEs resolved and none led to withdrawal from the study or death. A pharmacodynamic evaluation reflected significant improvements with RP5063 (P < 0.05) over placebo in an analysis of patients with a baseline Positive and Negative Syndrome Scale (PANSS) score ≥50 for positive subscale scores. Improvements of the Trail Making A and Trail Making B test results were observed for patients treated in the 50 mg dose group for days 5, 10, and 16. These findings indicate that RP5063 is well‐tolerated up to 100 mg and displays promising preliminary clinical behavioral and cognition activity signals in patients with stable disease over a 10‐day period.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ RP5063 has no prior data on its safety or clinical activity in humans.
WHAT QUESTION DID THIS STUDY ADDRESS?
The initial safety profile for RP5063 is described in single‐doses normal healthy volunteers and in an multiple doses over 10 days in patients with stable schizophrenia. The multiple‐dose study also evaluated initial clinical activity in the stable population with schizophrenia.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
✓ RP5063 is well tolerated across a dose range from 10–100 mg up to 10 days. Side effects were more common in the 50‐mg and 100‐mg dose groups in the multi‐dose study. Activity signals were seen in patients with stable schizophrenia, particularly PANSS responses seen in those with as baseline score of ≥50 PANSS units. No differences were seen among ethnic groups.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ The data obtained directly corroborate preclinical safety observations and support activity seen in preclinical animal models. They support dose translation from preclinical studies to patients with stable schizophrenia disease.
Schizophrenia, a disorder that affects 1% of the world's population, is a complex, chronic, and debilitating psychiatric condition characterized by a mix of positive, negative, and mood symptoms along with cognitive impairment.1 Management involves antipsychotic therapy, with all approved treatments blocking dopamine (D) receptors, particularly D2, and second‐generation products blocking serotonin (5‐hydroxytryptamine, 5‐HT), notably 5_HT2A.2 Present treatments are limited by side effects, such as extrapyramidal symptoms (EPS), metabolic and cardiovascular issues, hyperprolactinemia, as well as sexual and endocrine dysfunctions, that undermine compliance.3, 4, 5, 6, 7
RP5063, a D/5‐HT stabilizer, is a promising candidate for the treatment of schizophrenia. This compound possesses high‐binding affinity with partial agonist activity for D2/3/4 and 5‐HT1A/2A, and antagonist activity for 5‐HT2B/7 receptors, and moderate‐binding affinity for the serotonin transporter (SERT).8 Rodent models of pharmacologic‐induced behaviors associated with schizophrenia have demonstrated that RP5063 was active in limiting both psychosis and cognitive symptoms.8, 9
Pharmacokinetic (PK) studies showed that RP5063 was well absorbed, widely distributed, highly protein bound, and dose dependent in its systemic exposure.10 Metabolic studies found that RP5063 was mainly metabolized by the hepatic enzymes—primarily through CYP450 3A4 (64%) and minimally by CYP2D6 (17%) (unpublished).
Preclinical safety pharmacology studies indicated that RP5063 was well tolerated in toxicity evaluations ranging from 1–9 months and produced no metabolic, cardiovascular, pulmonary, or central nervous system adverse effects.8 The no observable effect level for cardiovascular, respiratory, and central nervous system effects in rats were 45, 100, and 10 mg/kg, respectively (human equivalent dose, HED): 24, 16, and 1.6 mg/kg or 1,440, 960, and 96 mg/60 kg patient, respectively). Acute toxicity studies in rodents did not report of any signs of catalepsy, convulsions, or seizures (unpublished). Canine studies did not reveal any marked changes in blood glucose, electrocardiographic (ECG), cardiac troponin I levels, or blood pressure measurements (unpublished).
This article reports the initial clinical experience with RP5063. The primary focus involves the safety of single doses (10 and 15 mg) in normal healthy men using an ascending‐dose study design and of multiple doses (10, 20, 50, and 100 mg) over 10 days in male patients with stable schizophrenia. The secondary focus examines the initial clinical activity of multiple doses in the stable schizophrenia population over a 10‐day period.
MATERIALS AND METHODS
Study conduct
Both studies were institutional review board approved investigations and conducted per US Food and Drug Administration (FDA) guidelines.11 Informed consent was obtained per the institutional review board and Helsinki Declaration requirements. A Data Safety Committee established a priori stopping criteria, which was used to ensure safety. It evaluated study data after each cohort to make dosing, sampling, and monitoring recommendations for the next cohort.
Single‐dose study in healthy men (fasting and food effect)
This single‐dose study involved escalating single doses of RP5063 in normal healthy men. Excluded were individuals with pre‐existing neurological, medical, or laboratory abnormalities, or family history of neurological disorders.
In this randomized, double‐blind, placebo‐controlled, ascending‐dose study, the dose‐escalation portion involved two cohorts (10 and 15 mg) of eight recruited volunteers each. Although the planned dose escalation scheme included 10, 15, 25, 50, and 100 mg doses, the Data Safety Committee review limited doses to 10 and 15 mg based on that sufficient data were obtained related to: (i) a predictable PK profile with dose‐dependent increase in drug exposure; and (ii) dose exposure dependent tolerability (data on file). Participants were randomized 3:1 (active: placebo). A sentinel pair of participants, randomized 1:1 (active: placebo), were entered first, followed by the remaining participants of the cohort randomized 5:1 (active: placebo) after a 2‐day safety evaluation. A third cohort of eight participants was evaluated using a crossover design for the effect of fed and fasting states on drug pharmacokinetics and safety.
The study schedule involved: (i) screening (Day ‐28 to Day ‐2); (ii) admission (Day ‐1); (iii) in‐house days (Days 1–3) at the study clinic (PAREXEL Early Phase Clinical Unit, Los Angeles, CA). Study duration ranged from 31 to 33 days.
Multiple‐dose study in stable male patients with schizophrenia
This randomized, double‐blind, placebo‐controlled, ascending‐dose study included four cohorts (10, 20, 50, and 100 mg dosed q.d.). Each was comprised of eight male patients with stable schizophrenia (chronic, all types with Total Positive and Negative Syndrome Scale (PANSS) score ≤90 points). Excluded were patients with pre‐existing neurological, medical, or laboratory abnormalities, unstable or active exacerbation of schizophrenia, a family history of neurological disorders, or a history of hypersensitivity to products with a mechanism of action similar to RP5063 (e.g., aripiprazole).
The escalated dose schedule to once‐daily 100 mg was based upon the single ascending‐dose PK, dose‐dependent drug exposure and safety, preclinical no observable effect level HED data, and the observed tolerability with antipsychotic medications in patients was higher than seen in healthy subjects.12
The study proceeded through the dose‐ascension cohorts as planned. Each cohort was randomized 6:2 (active: placebo). The participants remained within the clinic (PAREXEL Early Phase Clinical Unit) for the duration of the study and were administered RP5063 30 min following a standardized, FDA‐approved high‐fat meal once daily for 10 days based on single‐dose fed/fasting data, indicating a slight increase in the extent of absorption and longer half‐life with food.
The study schedule involved: (i) screening (Day ‐35 to Day ‐6); (ii) admission (Day ‐5); (iii) antipsychotic washout (Day ‐5 to Day ‐1); and (iv) in‐house days (Days 1–17). The study duration was 57 days.
Study drug
Participants were administered oral capsules of RP5063 based on the dose and randomization schedule. Both studies used 5 mg and 10 mg capsules of RP5063 and placebo. The multiple‐dose study also used RP5063 25 mg capsules.
Safety and pharmacodynamic parameters
Safety was evaluated throughout both studies. Parameters were recorded at regular intervals: adverse events; concomitant medications; physical examination; vital signs (supine blood pressure and heart rate, oral body temperature, and respiratory rate, with orthostatic measurements on Day 1 at the time bed restriction was lifted for the single‐dose study); 12‐lead ECG (PR, RR, QT, QTcB, and QRS intervals); electroencephalography; clinical laboratories (hematology, clinical chemistry [including prolactin], urinalysis); and suicidal ideation (Columbia Suicide Severity Rating Scale [C‐SSRS] score).
The multiple‐dose study incorporated several standardized assessments: (i) the Simpson Angus Scale; (ii) the Abnormal Involuntary Movement Scale; (iii) the Barnes Akathisia Scale; and (iv) the Calgary Depression Scale. Preliminary clinical activity was assessed using the Clinical Global Impression (CGI) scale (CGI‐Severity (CGI‐S) for baseline (Day ‐1) and CGI‐Improvement (CGI‐I) (Day 17) for change from baseline, PANSS (Days ‐1, 2, 3, 5, 7, 10, 12, and 16), the Digital Symbol Substitution Test (Days ‐1, 5, 10, and 16, the Trail Making Test A and B or Trails A and B (Days ‐1, 5, 10, and 16), and the Bond and Lader visual analogue scale for subjective effects (Days ‐1, 2, 5, 10, and 16).
Statistical plan and analysis
Formal sample size calculations were not performed. The number of study participants was chosen based on feasibility and was considered sufficient to meet the study objectives.
All demographic, safety, and pharmacodynamic data were summarized in tabular format by descriptive statistics. Adverse events were classified according to the Medical Dictionary for Regulatory Activities. Change from baseline was calculated for vital signs and ECG findings.
Analysis of activity in the multidose study used the intent‐to‐treat analysis data set. The primary analysis of PANSS is a standardized 30‐item drug‐sensitive instrument with a balanced representation of positive and negative symptoms that gauges their relationship to one another and to global psychopathology.13 Analysis involved the evaluation of the change from baseline of Total and Subscale Scores, and PANSS Marder Factor Scores (a validated analysis of five PANSS factors of schizophrenia).14, 15, 16 Least‐squares adjusted means were calculated for the active treatment group compared with placebo. An analysis of covariance with group (treatment or placebo) as factors and baseline score as a continuous covariate was utilized.
A secondary evaluation of the PANSS data, as a post hoc analysis, evaluated participants (N = 19 total: treatment = 13; placebo = 6) who had a total score of ≥50 or greater at baseline (with 50 as the median baseline score; range, 39–69] for the total data set. This analysis involved multiple statistical tests, including repeated measures analysis, to assess whether patients with greater symptomatology at baseline were more likely to show change with treatment.13 The analysis of this population considered Marder factors and included an assessment of dose (≤20 mg, ≥50 mg) using a repeated measures analysis of covariance and a Tukey post hoc analysis.
A formal dose response analysis was not undertaken due to limited patient numbers. The PK and exposure analyses were reported in a separate publication.10
RESULTS
Disposition, extent of exposure, and demographics
The single‐dose study recruited 24 individuals (Table 1), and 23 completed the trial. Participants were white or Japanese men (age, 20–45 years; weight, 18.5–30.0 kg/m2), with no differences seen between these two groups in the PK evaluation.10 They received one dose of either the placebo (n = 4), 10 mg (n = 6), or 15 mg (n = 7) formulations. Seven participants in the food‐effect cohort received two 15‐mg doses.
Table 1.
Demographics for phase I single‐dose study population
| Parameter | Placebo (N = 4) | 10 mg (N = 6) | 15 mg (N = 6) | Food effect: 15 mg (N = 8) |
|---|---|---|---|---|
| Age, years | ||||
| Mean (SD) | 31.8 (4.35) | 31.2 (5.74) | 26.2 (4.26) | 26.1 (5.99) |
| Height, cm | ||||
| Mean (SD) | 177.5 (14.47) | 179.3 (6.70) | 171.6 (7.83) | 175.8 (3.19) |
| Weight, kg | ||||
| Mean (SD) | 80.60 (12.312) | 81.78 (9.902) | 68.97 (11.008) | 73.04 (6.719) |
| Body mass index, kg/m2 | ||||
| Mean (SD) | 25.55 (1.542) | 25.43 (2.839) | 23.53 (4.408) | 23.63 (2.197) |
| Race | ||||
| White, no. (%) | 3 (75.00) | 5 (83.33) | 4 (66.67) | 6 (75.00) |
| Japanese, no. (%) | 1 (25.00) | 1 (16.67) | 2 (33.33) | 2 (25.00) |
| Ethnicity (part of white) | ||||
| Hispanic, no. (%) | 0 (0.00) | 1 (16.67) | 1 (16.67) | 0 (0.00) |
| Non‐Hispanic, no. (%) | 4 (100.00) | 5 (83.33) | 5 (83.33) | 8 (100.00) |
| Sex | ||||
| Male, no. (%) | 4 (100.00) | 6 (100.00) | 6 (100.00) | 8 (100.00) |
In the multiple‐dose study, all 32 randomized male patients (Table 2; aged 18–65 years; weight, 19.5–38.0 kg/m2) completed the 10‐day schedule, with six patients per treatment group (n = 24) and two patients receiving placebo (n = 8). Patients in the 10‐mg, 20‐mg, and 50‐mg groups received 100, 200, and 500 mg, respectively. Five patients in the 100‐mg treatment group received 1,000 mg; one patient, who discontinued prematurely, received 400 mg.
Table 2.
Demographics for phase I multiple‐dose study population
| Parameter | Placebo (N = 8) | 10 mg (N = 6) | 20 mg (N = 6) | 50 mg (N = 6) | 100 mg (N = 6) |
|---|---|---|---|---|---|
| Age, years | |||||
| Mean (SD) | 44.9 (10.56) | 40.7 (12.5) | 47.7 (7.61) | 38.8 (12.3) | 47.3 (4.37) |
| Height, cm | |||||
| Mean (SD) | 179.13 (11.34) | 176.78 (6.00) | 173.78 (6.86) | 179.47 (9.52) | 175.23 (6.34) |
| Weight, kg | |||||
| Mean (SD) | 97.4 (9.59) | 87.67 (19.69) | 85.57 (14.84) | 100.75 (20.44) | 83.12 (11.97) |
| Body mass index, kg/m2 | |||||
| Mean (SD) | 30.54 (3.77) | 28.25 (7.06) | 28.33 (4.58) | 31.13 (4.72) | 27.07 (3.75) |
| Race | |||||
| White, no. (%) | 2 (25.00) | 2 (33.33) | 1 (16.67) | – | 1 (16.67) |
| Black no. (%) | 6 (75.00) | 4 (66.67) | 5 (83.33) | 6 (100.00) | 5 (83.33) |
| Sex | |||||
| Male, no. (%) | 8 (100.00) | 6 (100.00) | 6 (100.00) | 6 (100.00) | 6 (100.00) |
Patients in the multiple‐dose study met the definition for stable patients with schizophrenia based on showing minimal symptoms, as defined by low PANSS and CGI‐S scores.12 Baseline scores for PANSS and CGI‐S were comparable between the pooled (all doses) for RP5063 and placebo groups; no statistical evaluation was conducted due to the small numbers. Mean total PANSS score (±SD; range) at baseline in the pooled RP5063 group (n = 24) was 50.86 (±6.55; range, 39–69) and in the placebo group (n = 8) was 53.75 (±7.17; range, 43–65) and fell below the threshold of ≤60 at enrollment. Mean CGI‐S for the pooled RP5063 group at baseline was 3.04 and placebo was 3.00 and fell below the threshold of ≤4 within 4 weeks of study entry. Baseline PANSS and CGI‐S were not significantly different between treatment and placebo.
Single‐dose safety
Tables 3 and 4 summarize the breakdown of treatment‐emergent adverse events (TEAEs) for the single‐dose study. Twelve participants and 32 TEAEs were reported, with orthostatic hypotension (n = 6; 5 participants), nausea (n = 5; 4 participants), and dizziness (n = 4; 4 participants) being the most frequent. No TEAEs led to study withdrawal or death and all resolved by the study's end.
Table 3.
Summary of TEAEs in single‐dose and multiple‐dose studies
| TEAEs in single‐dose study: no. of events (% of total for all events) | ||||||
|---|---|---|---|---|---|---|
| Placebo (N = 4) | 10 mg (N = 6) | 15 mg (N = 6) | 15 mg fed (N = 7) | 15 mg fasting (N = 8) | Category total | |
| TEAEs | 0 (0%) | 7 (22%) | 8 (25%) | 3 (9%) | 14 (44%) | 32 (100%) |
| SAEs | 0 (0%) | 1 (100%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (100%) |
| TEAEs in multiple‐dose study: no. of events (% of total for all events) | ||||||
|---|---|---|---|---|---|---|
| Placebo (N = 8) | 10 mg (N = 6) | 30 mg (N = 6) | 50 mg (N = 6) | 100 mg (N = 8) | Category total | |
| All AEs | 10 (13%) | 9 (12%) | 12 (16%) | 24 (32%) | 20 (27%) | 75 (100%) |
| All TEAEs | 10 (13%) | 9 (12%) | 12 (16%) | 24 (32%) | 20 (27%) | 75 (100%) |
| Related TEAEsa | 9 (12%) | 5 (7%) | 10 (13%) | 19 (25%) | 18 (24%) | 61 (81%) |
| Not related TEAEsb | 1 (1%) | 4 (5%) | 2 (3%) | 5 (7%) | 2 (3%) | 14 (19%) |
| Mild TEAEs | 10 (13%) | 8 (11%) | 12 (16%) | 23 (31%) | 14 (19%) | 67 (89%) |
| Moderate TEAEs | 0 (0%) | 1 (1%) | 0 (0%) | 1 (1%) | 4 (5%) | 6 (8%) |
| Moderate to severe TEAEs | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 2 (3%) | 2 (3%) |
AE, adverse event; SAEs, serious adverse event; TEAE, treatment‐emergent adverse event.
aSame participants as in the fasting group who experienced TEAE.
bRelated TEAEs includes all TEAEs considered “related,” “possibly related,” and “probably related” to study drug by the study investigator.
cNot related TEAEs includes all TEAEs considered “unlikely to be related” and “unrelated” to study drug by the study investigator.
Table 4.
TEAEs by system organ class and preferred term in the single‐dose study
| TEAE by system organ class preferred term | Placebo (N = 4) no. (%), E | 10 mg (N = 6) no. (%), E | 15 mg (N = 6) no. (%), E | Fed 15 mg (N = 7) no. (%), E | Fasting 15 mg (N = 8) no. (%), E |
|---|---|---|---|---|---|
| Cardiac disorders | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 1 (12.50) 1 |
| Tachycardia | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 1 (12.50) 1 |
| Gastrointestinal disorders | 0 (0.00) 0 | 1 (16.67) 3 | 2 (33.33) 3 | 0 (0.00) 0 | 1 (12.50) 1 |
| Nausea | 0 (0.00) 0 | 1 (16.67) 2 | 2 (33.33) 2 | 0 (0.00) 0 | 1 (12.50) 1 |
| Retching | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Vomiting | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 |
| General disorders and administrative site conditions | 0 (0.00) 0 | 2 (33.33) 2 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Fatigue | 0 (0.00) 0 | 2 (33.33) 2 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Nervous system disorders | 0 (0.00) 0 | 2 (33.33) 2 | 1 (16.67) 3 | 2 (28.57) 2 | 3 (37.50) 5 |
| Dizziness | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (14.29) 1 | 2 (25.00) 2 |
| Dizziness postural | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (12.50) 1 |
| Other | 0 (0.00) 0 | 2 (33.33) 2 | 2 (33.33) 2 | 1 (14.29) 1 | 2 (25.00) 2 |
| Headache | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 1 (12.50) 1 |
| Hypoesthesia | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 |
| Somnolence | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (14.29) 1 | 0 (0.00) 0 |
| Syncope | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (12.50) 1 |
| Tension headache | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Tonic convulsion | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Skin and subcutaneous tissue disorders | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 2 (25.00) 2 |
| Hyperhidrosis | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 2 (25.00) 2 |
| Vascular disorders | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (14.29) 1 | 5 (62.50) 5 |
| Hot flush | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (12.50) 1 |
| Orthostatic hypotension | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (14.29) 1 | 4 (50.00) 4 |
E, number of adverse events; N, number of volunteers receiving at least 1 dose; no., number of volunteers with adverse events; TEAE, treatment‐emergent adverse event.
Percentages calculated using the number of volunteers receiving at least 1 dose (N) in each cohort as the denominator.
One serious adverse event (SAE), involving brief seizure‐like symptoms, was reported in one participant (10 mg group). Follow‐up revealed that this individual should not have been in the study due to pre‐existing history of seizures.
For laboratories, mean prolactin values were slightly decreased from baseline to 24 h, with maximum decreases of 28.7%, 16.9%, 39.5%, and 30.3% for 10‐mg fasting, 15‐mg fasting, 15‐mg fed (food effect), and 15‐mg fasting (food effect), respectively. These observations with prolactin were not different from those with placebo. No clinically significant postdose abnormalities, trends, or TEAEs in any laboratory or urinalysis observations were noted.
Assessment of cardiovascular safety did not reveal any significant effects. Five participants reported six events of orthostatic hypotension and two participants reported tachycardia. No clinically significant changes in PR, RR, QRS, QT, or QTcB intervals were observed; one participant had a QTcB outlier at day 5 that increased by at least 60 ms.
Further safety, physical examination, or electroencephalographic findings were not clinically significant. No participants displayed suicidal ideation or behavior per the C‐SSRS.
Multiple‐dose safety
Tables 3 and 5 summarize the TEAEs in the multidose evaluation. None led to study withdrawal or death. Twenty‐seven patients reported 75 TEAEs across all treatment groups. The majority included nervous system (n = 43), psychiatric (n = 11), and gastrointestinal disorders (n = 10). The most common TEAEs included somnolence (n = 20; 19 patients), akathisia (n = 14; 12 patients), extrapyramidal effects (n = 5; three patients), and dystonia (n = 3; two patients). All TEAEs resolved within the study period, and did not result in death, significant mortality, or study withdrawal.
Table 5.
TEAEs by system organ class and preferred term in the multiple‐dose study
| TEAE by system organ class, no. (%) E | Placebo N = 8 | 10 mg N = 6 | 20 mg N = 6 | 50 mg N = 6 | 100 mg N = 6 |
|---|---|---|---|---|---|
| Eye disorders | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Vision blurred | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Gastrointestinal disorders | 2 (25.00) 2 | 0 (0.00) 0 | 1 (16.67) 1 | 4 (66.67) 4 | 2 (33.33) 3 |
| Abdominal distension | 1 (12.50) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Constipation | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 2 (33.33) 2 | 0 (0.00) 0 |
| Dry mouth | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 |
| Dyspepsia | 1 (12.50) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Nausea | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (16.67) 1 | 1 (16.67) 1 |
| Vomiting | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (16.67) 1 |
| Infections and infestations | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 |
| Furuncle | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 |
| Metabolism and nutrition disorders | 1 (12.50) 1 | 1 (16.67) 1 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Decreased appetite | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Increased appetite | 1 (12.50) 1 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Musculoskeletal and connective tissue disorders | 1 (12.50) 1 | 0 (0.00) 0 | 1 (16.67) 1 | 1 (16.67) 1 | 1 (16.67) 1 |
| Back pain | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 |
| Muscle twitching | 1 (12.50) 1 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 |
| Myalgia | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Nervous system disorders | 4 (50.00) 4 | 3 (50.00) 3 | 4 (66.67) 7 | 5 (83.33) 14 | 6 (100.00) 15 |
| Akathisia | 0 (0.00) 0 | 0 (0.00) 0 | 3 (50.00) 3 | 3 (50.00) 3 | 6 (100.00) 8 |
| Dizziness postural | 1 (12.50) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Dystonia | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 2 (33.33) 3 | 0 (0.00) 0 |
| Extrapyramidal disorder | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 1 (16.67) 2 | 1 (16.67) 2 |
| Somnolence | 3 (37.50) 3 | 2 (33.33) 2 | 4 (66.67) 4 | 5 (83.33) 6 | 5 (83.33) 5 |
| Psychiatric disorders | 1 (12.50) 1 | 2 (33.33) 5 | 2 (33.33) 2 | 2 (33.33) 2 | 1 (16.67) 1 |
| Anxiety | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Insomnia | 1 (12.50) 1 | 1 (16.67) 1 | 1 (16.67) 1 | 1 (16.67) 1 | 0 (0.00) 0 |
| Nightmare | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 |
| Psychotic disorder | 0 (0.00) 0 | 1 (16.67) 2 | 1 (16.67) 1 | 1 (16.67) 1 | 0 (0.00) 0 |
| Tension | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Skin and subcutaneous tissue disorders | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Hyperhidrosis | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 1 (16.67) 1 | 0 (0.00) 0 |
| Vascular disorders | 1 (12.50) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
| Thrombophlebitis | 1 (12.50) 1 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 | 0 (0.00) 0 |
E, number of TEAEs; N, number of volunteers exposed to treatment; no., number of volunteers with at least 1 TEAE; %, (n/N * 100); TEAE, treatment‐emergent adverse event.
The incidence of TEAEs appeared to increase with dose, with little difference between the 50‐mg and 100‐mg doses (Table 3). More patients reported somnolence at these higher doses. Two moderate‐to‐severe adverse events involved akathisia at the 100‐mg dose.
Except for an elevated mean creatinine phosphokinase level of 654.5 units/L on day 21 with the 50‐mg dose, no other clinically significant abnormal blood chemistry, hematology, or urine measurements, or differences among treatments and with placebo, were observed. No treatment‐related changes or trends were identified in glucose, prolactin, or lipid levels.
For cardiovascular safety, 12‐lead ECG values did not report any dose‐related and clinically significant changes. In an outlier analysis (including Bazett's and Fridericia's corrections according to the FDA Guidance to Industry) of QTcB values, several isolated prolongations (>30 to ≤60 ms) were observed in all groups (more frequently in the 10‐mg and 20‐mg groups and placebo), and one change higher than 60 ms was observed in the 100‐mg group at Day 21.17
Physical examination findings and vital sign changes or abnormalities were unremarkable.
Results from the Simpson Angus Scale, Abnormal Involuntary Movement Scale, C‐SSRS, and Calgary Depression Scale did not display any meaningful changes or group differences. The Barnes Akathisia Scale scores were higher in the 50‐mg and 100‐mg groups.
Pharmacodynamic activity of multiple doses in patients with stable schizophrenia
Clinical Global Impression assessment
At baseline, CGI‐S scores for all patients in the 10‐mg, 50‐mg, and 100‐mg groups, the pooled placebo groups, and five patients (83.3%) in the 20‐mg group were 3 (mildly ill) and in one patient (16.7%) in the 20‐mg group was 4 (moderately ill). A change to minimal improvement in the CGI‐I at Day 17 was only seen in the 20‐mg and 100‐mg dose groups. At Day 17, only one patient (16.7%) in the 20‐mg group and five patients in the 100‐mg group (83.3%) scored a 3 (minimal improvement).
Positive and negative syndrome scale
Treatment with RP5063 (pooled doses) did not show significance in PANSS Total Score or individual subscale related to change from baseline to each visit or vs. placebo. A trend was noted with treatment at Day 7 in the Positive subscale score, a 0.61 reduction from baseline. No dose dependency was observed.
Analysis of PANSS Marder Factor Scores for the total population showed significance vs. placebo with two subscales. For the Marder Anxiety/Depression Factor, treatment increased by 0.46 and placebo reduced by 0.25 (P = 0.002). For the Marder Impulsivity/Hostility Factor, treatment decreased by 0.12 and placebo increased by 0.76 (P = 0.05).
In patients with a PANSS ≥50, those treated with RP5063 (pooled doses) trended toward a reduced PANSS Total Score from baseline (P = 0.85) and toward a change from baseline in the General Psychopathology Score subscale (P = 0.074).
The most notable finding in this pooled RP5063 population was with the repeated measures analysis for PANSS Positive Subscale Scores (Figure 1 a). This analysis revealed that treatment was significant from placebo with respect to scores across time (P = 0.005) and after controlling for baselines (P = 0.004). Analysis of change from baseline showed that treatment provided a significant difference in producing improvements on Days 5, 7, 10, and 16 (P = 0.008, P = 0.010, P = 0.036, and P = 0.017, respectively).
Figure 1.
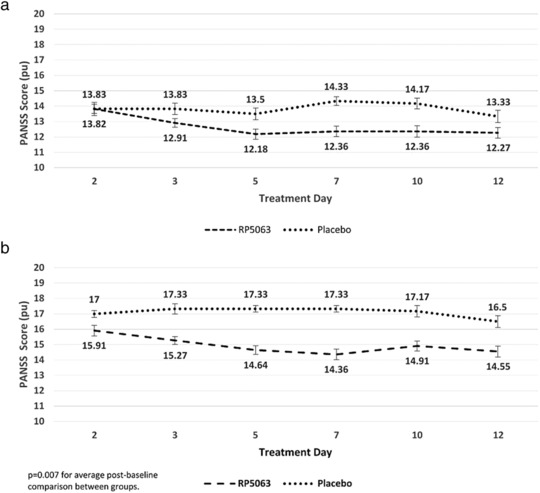
Positive and Negative Syndrome Scale (PANSS) Repeated Measures Analysis of Patients with PANSS >50 for (a) Positive Subscale Scores and (b) Positive Subscale Scores with Marder Factors for RP5063 (pooled data, all doses) as compared with placebo A.
Similar observations were seen with a repeated measures analysis of Positive Subscale Scores that considered Marder Factors in patients with baseline PANSS ≥50 (Figure 1 b). Treatment was significant from placebo (P = 0.007) for the Marder Positive Factor Score. Repeated measures analysis revealed a significant reduction in the treatment group across time (P = 0.038) and after controlling for baseline scores (P = 0.027). With respect to subscales, treatment produced a significant reduction in the Marder Anxiety/Depression Factor (P = 0.009).
For the Marder‐derived Positive Factor Score in this population, patients receiving ≥50 mg showed a 4.4 decrease, ≤20 mg showed a 1.01 decrease, and placebo showed a 1.00 decrease in scores from baseline (P = 0.024). The Tukey post hoc analysis was significant between the ≤20 mg and ≥50 mg groups (P = 0.001) and between the ≥50 mg and placebo groups (P = 0.001).
TRAIL MAKING TESTS A AND B
Test A
At baseline, patients in the 50‐mg and 100‐mg groups and the pooled placebo group showed a similar duration time (34.0–35.0 s). The 10‐mg and 20‐mg groups were 25.5 and 43.8 s, respectively. The mean number of errors on Days 5, 10, and 16 during treatment in the Trail Making Test A were 0 (10‐mg and 100‐mg and the pooled placebo groups) or close to 0 with a maximum of 0.5 errors in the 50‐mg group. For executive function, the 50‐mg group showed mean reductions of 6.3, 7.8, and 13 s on Days 5, 19, and 16, respectively. The 20‐mg group showed a slight decrease from baseline on Days 10 and 16 of 2.8 and 5.5 s, respectively. The 100‐mg group trended toward baseline by Day 16, with a peak improvement of 8.8 s on this day (Figure 2 a).
Figure 2.
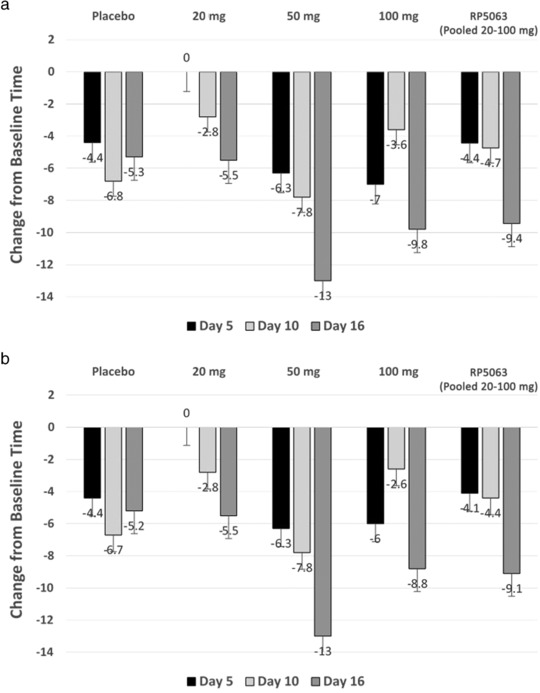
Changes in (a) trails A and (b) trails B test times from baseline at days 5, 10, and 16 with RP5063 20 mg, 50 mg, 100 mg, and pooled (20–100 mg) doses.
Test B
At baseline, the mean duration to perform Trail Making Test B ranged from 67.5 s in the 20‐mg group to 103.3 s in 100‐mg group. The 50‐mg group exhibited reductions of 6.3, 7.8, and 13 s at Days 5, 10, and 16. The 20‐mg group improved by 2.8 and 5.5 s at Days 10 and 16, respectively. The 100‐mg group was inconsistent from Days 5–16, with the greatest improvement of 9.8 s on Day 16. No dose dependency was noted in either test A or B (Figure 2 b).
Bond and Lader visual analog scale for subjective effects
Changes from baseline between the groups did not reveal dose dependency or any consistent trend. In the 10‐mg, 20‐mg, and 100‐mg groups, minor changes were observed and in the 50‐mg group more significant changes were seen between baseline and Day 10.
DISCUSSION
Safety considerations
These studies begin to define the clinical safety profile of RP5063. They provided valuable insight that guided dose planning for the phase II clinical study dose range of 15–50 mg.8 Doses in both studies seemed to be safe and generally well tolerated. No safety signals were seen in vital signs, patient weight, and metabolic parameters. The TEAEs in the multidose study were more prevalent in the 50‐mg and 100‐mg dose groups. A moderate‐to‐severe TEAE involving akathisia occurred at the 100‐mg dose. The TEAEs did not threaten life, lead to significant morbidity or mortality, or result in study withdrawal. A maximum tolerated dose was not identified in either study.
These initial safety observations were clarified further in the phase II evaluation of RP5063.8 In this study, RP5063 dosed between 15 and 50 mg dosed once daily for 28 days had a TEAE rate of 54.8% (similar to positive control of aripiprazole at 55%). Insomnia (23%) and agitation (9.1%) were the most common events. The akathisia rate was 5%, and discontinuation rate was 17%, lower than typically seen for antipsychotics.8, 18 Good tolerability was seen in all ethnic groups.
These initial safety observations are encouraging as related to the management of schizophrenia. Patients with schizophrenia experience multiple comorbidities at a higher rate than normal healthy individuals due to lifestyle, personal care neglect, and treatment barriers for ongoing conditions.7, 19, 20 Between 40% and 60% of patients with schizophrenia are overweight, and their baseline cardiometabolic and cardiovascular risks are higher than those of the general population.7, 19, 20 Coupled with other existing or treatment‐induced comorbidities (e.g., EPSs or akathisia [>80%], QT interval prolongation [40%], and hyperprolactinemia [>25%]), these underlying conditions can quickly limit the clinician's choice when considering the side effects with available medications.4
Furthermore, approved treatments are associated with varying degrees of EPS and hyperprolactinemia,7, 21 leading to endocrine disruptions (e.g., breast enlargement, increase the risk of breast cancer, and a decrease in or loss of sexual interest) directly through hyperprolactinemia and indirectly through D2 receptor antagonism. Although reduced sexual activity is associated with the onset of psychosis, antipsychotic‐induced hyperprolactinemia is linked with reductions in sexual interest, activity, and satisfaction.21, 22, 23 Increased prolactin levels (amisulpride and risperidone) have also resulted in sexual side effects (e.g., retrograde ejaculation and priapism seen with clozapine, risperidone, and olanzapine).3, 7 These side effects can limit compliance, reported between 40% and 50%.24, 25
Second‐generation agents are limited by cardiometabolic effects.6, 7 In a meta‐analysis, standard doses of clozapine, olanzapine, and risperidone over a 10‐week period resulted in mean unintentional weight increases of 4.45 kg, 4.15 kg, and 2.10 kg, respectively.26 Another study reported that 78.8% of patients experienced unintentional weight gain of >7%, as compared with baseline.27 Furthermore, type 2 diabetes mellitus has been linked to use of these agents, leading to the FDA to require a warning related to hyperglycemia and diabetes in the package inserts for agents of this class.28 Hyperlipidemia is another cardiometabolic factor. Studies have shown that dibenzodiazepine‐derived atypical agents are associated with higher serum triglyceride levels (clozapine, olanzapine, and quetiapine) and increased cholesterol levels (clozapine and olanzapine) after 8 weeks of treatment.29, 30 These metabolic risks are compounded by the potential for direct cardiovascular effects including increase in QTc interval, myocarditis, and worsening hypertension control.31, 32, 33, 34, 35
Pharmacodynamic activity
Because the multiple‐dose study used patients with stable schizophrenia, the data from this study provided an early assessment of the pharmacodynamic behavior and activity of RP5063 in this population. However, detecting a significant signal for clinical efficacy is limited by multiple factors: (i) the patient population; (ii) low baseline PANSS for overall analysis; (iii) small size of the treatment groups; and (iv) short duration of treatment. Evaluation of longer‐term effects would have required a longer treatment period, such as in the phase II study over 28 days or the planned phase III study over 42 days.8 Nevertheless, considering the half‐life of RP5063, 10 days of dosing could translate to 2 weeks of exposure, which was not evaluated in this multidose phase 1 study.8, 10
Several tests (Trails A and B, PANSS) identified some early clinical efficacy signals. Notable was the secondary analysis to explore the PANSS observations. A baseline score of 50 (study population PANSS score range, 39–69) was the median. It represented a natural point for analysis. Typical PANSS scores range in stable schizophrenia from 30 to 70, a score of ∼50 is defined as “moderately ill.”31, 36 Thus, assessing patients with higher PANSS scores (≥50) at baseline would be more likely to detect change. This analysis found statistical significance for the Positive Subscale scores, as well as favorable trends with the reduction of PANSS Total Score from baseline and in the General Psychopathology Score from baseline vs. placebo. The Marder responses for the Positive Subscale Score reflected similar observations and were significant relative to the anxiety/depression subscale. Further analysis of the 19 (RP5063 pooled doses, n = 13; placebo, n = 6) patients with respect to Positive Subscale Scores and Marder Factors indicated significant differences in the higher dose ≥50‐mg group, as compared with both placebo and the ≤20‐mg dose groups (P = 0.001). Although these subanalysis findings are encouraging in reflecting a signal of positive clinical efficacy, it is important to recognize its power limitations due to the relative small sample size.
Nonetheless, these observations were used to aid in the design and dosing to include a 50‐mg group for the phase II trial, in which patients with acute exacerbations of schizophrenia showed much higher baseline levels of PANSS total score.8 This study showed that the 15‐mg and 50‐mg doses improved the PANSS total score, the primary study end point, as compared with placebo (P < 0.05), with median rates of improvement over baseline of 23% and 22%, respectively. Both these doses were superior to placebo with respect to PANSS subscales and CGI scale scores. These results have set the stage for a phase III evaluation in the acute population, in which the 15‐mg, 30‐mg, and 50‐mg doses will be assessed over a 42‐day period.8
CONCLUSIONS
Tolerability is key for effectiveness beyond short‐term efficacy for an antipsychotic agent in neuropsychiatric disorders. These phase I studies found that RP5063 appears to be safe and generally well tolerated at doses ranging from 10–100 mg administered over 10 days. Most adverse events were mild and occurred at the higher doses of 50 mg and 100 mg. All TEAEs resolved and none led to study withdrawal or death. Notable was the lack of clinically significant changes in glucose or prolactin levels, lipid profiles, weight, or ECG findings. The multiple‐dose pharmacodynamic evaluation provided early insight regarding clinical activity of RP5063 relevant to psychosis, along with mood and cognitive comorbidities, in patients with stable schizophrenia over a 10‐day period. This activity with RP5063, combined with tolerability for improved compliance, offers the potential to fulfill unmet needs in psychiatry.
Conflict of Interest
L.B. and M.C. are employees of Reviva Pharmaceuticals, Inc, and R.I. is a consultant to Reviva Pharmaceuticals, Inc.
Supporting information
SUPPLEMENTAL INFO
Acknowledgments
Editorial support was provided by John M. York, PharmD, MBA. This assistance was funded by Reviva Pharmaceuticals, Inc. PAREXEL, Inc. performed the research under the supervision of Drs. Bhat and Cantillon.
Author Contributions
L.B., M.C., and R.I. wrote the manuscript. L.B., M.C., and R.I. designed the research. L.B., M.C., and R.I. analyzed the data.
Source of Funding
These studies were supported by a grant to PAREXEL International from Reviva Pharmaceuticals, Inc.
References
- 1. van Os, J. & Kapur, S. Schizophrenia. Lancet 374, 635–645 (2009). [DOI] [PubMed] [Google Scholar]
- 2. The American College of Neuropsychopharmacology (ACNP) . Neuropsychopharmacology – 5th Generation of Progress Section 6, Schizophrenia. http://www.acnp.org/publications/neuro5thgeneration.aspx (2010).
- 3. Baldwin, D. & Mayers, A. Sexual side‐effects of antidepressant and antipsychotic drugs. Adv. Psychiatr. Treat. 9, 202–210 (2003). [Google Scholar]
- 4. Citrome, L. et al Lack of tolerable treatment options for patients with schizophrenia. Neuropsychiatr. Dis. Treat. 11, 3095–3104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meltzer, H.Y. et al A randomized, double‐blind, placebo‐controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J. Clin. Psychiatry 76, 1085–1090 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Panariello, F. , De Luca, V. & de Bartolomeis, A. Weight gain, schizophrenia and antipsychotics: new findings from animal model and pharmacogenomic studies. Schizophr. Res. Treatment, 459284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Üçok, A. & Gaebel, W. Side effects of atypical antipsychotics: a brief overview. World Psychiatry 7, 58–62 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cantillon, M. , Prakash, A. , Alexander, A., Ings, R., Sweitzer, D. & Bhat, L . Dopamine serotonin stabilizer RP5063: a randomized, double‐blind, placebo‐controlled multicenter trial of safety and efficacy in exacerbation of schizophrenia or schizoaffective disorder. Schizophr. Res. 189, 126–133 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Rajagopal, L. et al RP5063, an atypical antipsychotic drug with a unique pharmacologic profile, improves declarative memory and psychosis in mouse models of schizophrenia. Behav. Brain Res. 332, 180–199 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Cantillon, M. , Ings, R. & Bhat, L. Pharmacokinetics of RP5063 following single doses to normal healthy volunteers and multiple doses over 10 days to stable schizophrenic patients. Clin. Transl. Sci. (2017). [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. FDA Guidance: Guidance to Industry: Bioanalytical Method Validation. Draft Guidance. September 2013. https://www.fda.gov/downloads/Drugs/Guidances/ucm368107.pdf (2013). Accessed May 13, 2017.
- 12. Cutler, N.R. Pharmacokinetic studies of antipsychotics in healthy volunteers versus patients. J. Clin. Psychiatry 62 Suppl 5, 10–13 (2001); discussion 23–24. [PubMed] [Google Scholar]
- 13. Kay, S.R. , Fiszbein, A. & Opler, L.A. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull. 13, 261–276 (1987). [DOI] [PubMed] [Google Scholar]
- 14. Marder, S.R. , Davis, J.M. & Chouinard, G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J. Clin. Psychiatry 58, 538–546 (1997). [DOI] [PubMed] [Google Scholar]
- 15. Marder, S.R. et al The Mount Sinai conference on the pharmacotherapy of schizophrenia. Schizophr. Bull. 28, 5–16 (2002). [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez‐Jimenez, R. et al Cognition and the five‐factor model of the positive and negative syndrome scale in schizophrenia. Schizophr. Res. 143, 77–83 (2013). [DOI] [PubMed] [Google Scholar]
- 17. FDA Guidance: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs. October 2005. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073153.pdf (2005). Accessed January 9, 2018. [PubMed]
- 18. Kane, J.M. et al Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J. Clin. Psychiatry 63, 763–771 (2002). [DOI] [PubMed] [Google Scholar]
- 19. Schatzberg, A.F. & Nemeroff, C.B. The American Psychiatric Publishing Textbook of Psychopharmacology 4th edn (American Psychiatric Publishers, Washington, D.C., 2009). [Google Scholar]
- 20. Meyer, J.M. Antipsychotics and metabolics in the post‐CATIE era. Curr. Top. Behav. Neurosci. 4, 23–42 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Rozan, G.H. , Tuchin, T. & Kurland, M.L. Some implications of sexual activity for mental illness. Ment. Hyg. 55, 318–323 (1971). [PubMed] [Google Scholar]
- 22. Sartorius, N. Physical illness in people with mental disorders. World Psychiatry 6, 3–4 (2007). [PMC free article] [PubMed] [Google Scholar]
- 23. Lyketsos, G.C. , Sakka, P. & Mailis, A. The sexual adjustment of chronic schizophrenics: a preliminary study. Br. J. Psychiatry 143, 376–382 (1983). [DOI] [PubMed] [Google Scholar]
- 24. Hansen, T.E. , Casey, D.E. & Hoffman, W.F. Neuroleptic intolerance. Schizophr. Bull. 23, 567–582 (1997). [DOI] [PubMed] [Google Scholar]
- 25. Birnbaum, M. & Sharif, Z. Medication adherence in schizophrenia: patient perspectives and the clinical utility of paliperidone ER. Patient Prefer. Adherence 2, 233–240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lacro, J.P. , Dunn, L.B. , Dolder, C.R., Leckband, S.G. & Jeste, D.V . Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J. Clin. Psychiatry 63, 892–909 (2002). [DOI] [PubMed] [Google Scholar]
- 27. Allison, D.B. et al Antipsychotic‐induced weight gain: a comprehensive research synthesis. Am. J. Psychiatry 156, 1686–1696 (1999). [DOI] [PubMed] [Google Scholar]
- 28. Alvarez‐Jiménez, M. et al Attenuation of antipsychotic‐induced weight gain with early behavioral intervention in drug‐naive first‐episode psychosis patients: a randomized controlled trial. J. Clin. Psychiatry 67, 1253–1260 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Berg, J. , Stajich, G. & Zdanowicz, M. Atypical antipsychotic‐induced type 2 diabetes. Pharmacy Times. (2012). http://www.pharmacytimes.com/publications/issue/2012/march2012/olanzapine-and-clozapine-atypical-antipsychotic-induced-type-2-diabetes (2012). Accessed May 22, 2017.
- 30. Meyer, J.M. A retrospective comparison of weight, lipid, and glucose changes between risperidone‐ and olanzapine‐treated inpatients: metabolic outcomes after 1 year. J. Clin. Psychiatry 63, 425–433 (2002). [DOI] [PubMed] [Google Scholar]
- 31. Wu, R.R. et al Effects of typical and atypical antipsychotics on glucose‐insulin homeostasis and lipid metabolism in first‐episode schizophrenia. Psychopharmacology (Berl). 186, 572–578 (2006). [DOI] [PubMed] [Google Scholar]
- 32. Wilton, L.V. , Heeley, E.L. , Pickering, R.M. & Shakir, S.A . Comparative study of mortality rates and cardiac dysrhythmias in post‐marketing surveillance studies of sertindole and two other atypical antipsychotic drugs, risperidone and olanzapine. J. Psychopharmacol. 15, 120–126 (2001). [DOI] [PubMed] [Google Scholar]
- 33. Killian, J.G. , Kerr, K. , Lawrence, C. & Celermajer, D.S . Myocarditis and cardiomyopathy associated with clozapine. Lancet 354, 1841–1845 (1999). [DOI] [PubMed] [Google Scholar]
- 34. La Grenade, L. , Graham, D. & Trontell, A. Myocarditis and cardiomyopathy associated with clozapine use in the United States. N. Engl. J. Med. 345, 224–225 (2001). [DOI] [PubMed] [Google Scholar]
- 35. Yasui‐Furukori, N. & Fujii, A. Worsened hypertension control induced by aripiprazole. Neuropsychiatr. Dis. Treat. 9, 505–507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leucht, S. , Kane, J.M. , Etschel, E., Kissling, W., Hamann, J. & Engel, R.R . Linking the PANSS, BPRS, and CGI: clinical implications. Neuropsychopharmacology 31, 2318–2325 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL INFO