

Abstract
Unsuppressed hepatic glucose production (HGP) contributes significantly to glucose intolerance and diabetes, which can be modeled by genetic inactivation of hepatic insulin receptor substrate (Irs) 1 and Irs2 (LDKO-mice). We previously showed that glucose intolerance in LDKO-mice is resolved by hepatic inactivation of the transcription factor FoxO1 (i.e., LTKO-mice)—even though the liver remains insensitive to insulin. Here, we report that insulin sensitivity in the white adipose tissue (WAT) of LDKO-mice is also impaired, but is restored in LTKO-mice in conjunction with normal suppression of HGP by insulin. To establish the mechanism by which WAT insulin signaling and HGP were regulated by hepatic FoxO1, we identified putative hepatokines—including excess follistatin (Fst)—that were dysregulated in LDKO-mice but normalized in LTKO-mice. Knockdown of hepatic Fst in the LDKO-liver restored glucose tolerance, WAT insulin signaling, and the suppression of HGP by insulin; however, expression of Fst in the liver of healthy LTKO-mice had the opposite effect. Of potential clinical significance, knockdown of Fst also improved glucose tolerance in high-fat fed obese mice, and serum FST was reduced in parallel with glycated hemoglobin in obese individuals with diabetes who underwent therapeutic gastric bypass surgery. We conclude that follistatin is a pathological hepatokine that might be targeted for diabetes therapy during hepatic insulin resistance.
Keywords: insulin resistance, insulin receptor substrate, follistatin, hepatic glucose production, diabetes
Introduction
The complex pathophysiology of type 2 diabetes (T2D) arises from cell-autonomous consequences of insulin resistance that can propagate among heterologous tissues through dysregulated lipid flux1–3, bile acids or circulating intermediary metabolites4, as well as dysregulated production and secretion of cytokines, adipokines and hepatokines3,5,6. Although T2D is characterized by systemic insulin resistance, disruption of hepatic insulin signaling alone recapitulates many aspects of T2D7, including enhanced endogenous glucose production (EGP)8. Though EGP includes renal and intestinal contributions9, it is mainly a function of hepatocytes. Hepatic glucose production (HGP) is suppressed directly by insulin via inhibition of hepatic glycogenolysis and gluconeogenesis, and indirectly by insulin-mediated suppression of adipose tissue lipolysis, glucagon secretion, and neuronal signals8.
The proteins insulin receptor substrate (Irs) 1 and Irs2 link the activated insulin receptor kinase (IRK) to the PI3K→Akt cascade, which regulates the expression of hundreds of hepatic genes by inactivating Forkhead box O (FoxO) family transcription factors10,11. We previously investigated the consequences of hepatic Irs1 and Irs2 deletion in LDKO (Irs1L/L•Irs2L/L•CreAlb)-mice and the complementary LTKO (Irs1/2L/L•FoxO1L/L•CreAlb)-mice12. In addition to dysregulated hepatic metabolism and mitochondrial dysfunction13, LDKO-mice develop systemic metabolic disease—including glucose intolerance, hyperinsulinemia and dysregulated energy homeostasis12,14. Remarkably, hepatic and systemic dysregulation is largely corrected upon further genetic deletion of hepatic FoxO1 in the LDKO-mice—despite continued persistent and complete hepatic insulin resistance12,13. These findings are confirmed and extended by others using compound hepatic-specific IRK•FoxO1- or Akt1•Akt2•FoxO1-knockout mice1,15–17. The normalization of HGP in these models owes, at least partially, to the re-sensitization of white adipose tissue (WAT) to insulin, which indirectly reduces hepatic gluconeogenesis by decreasing substrate availability and down-regulating hepatic pyruvate carboxylase activity1.
In models of complete hepatic insulin resistance (including LTKO-mice), genetic disruption of hepatic FoxO1 substantially normalizes the expression of hundreds of dysregulated hepatic genes12,16. Within such mice, FoxO1-dependent gene expression, or metabolic signals generated in hepatocytes, could reasonably contribute to dysregulation of peripheral insulin sensitivity and promote the delivery of metabolic intermediates to the liver—including excess glycerol and free fatty acids (FFAs) from adipose tissue3. Thus, while failing to restore hepatic insulin signaling per se, disruption of hepatic FoxO1 in LDKO-mice (i.e., LTKO-mice) and similar models effectively restores glucose tolerance12,15,17,18. Just how hepatic FoxO1 disruption might prevent the manifestation of peripheral insulin resistance has remained unclear.
Comprehensive analyses of gene expression reveals various genes encoding secreted proteins expressed in the human liver (so-called hepatokines), some of which might affect glucose and lipid metabolism5,6,19. We hypothesized that FoxO1-dependent dysregulation of hepatokines in LDKO-liver might promote peripheral insulin resistance—and substantially underlie the restoration of metabolic homeostasis in LTKO-mice. In this report, we use the LDKO- and LTKO-mice to identify follistatin (Fst)—best known for its modulation of TGF-β superfamily members20,21—as a key FoxO1-dependent hepatokine that can dysregulate WAT insulin sensitivity and hepatic gene expression to propagate systemic metabolic disease during hepatic insulin resistance.
Results
Hepatic Foxo1 is required for excess HGP and glucose intolerance during hepatic insulin resistance
Lipolysis of WAT triglycerides—which is suppressed by insulin—indirectly promotes hepatic gluconeogenesis1. To establish whether WAT insulin resistance alone dysregulates systemic glucose tolerance, we produced FDKO-mice by intercrossing Irs1L/L•Irs2L/L-mice (Cntr) with adipose-specific CreAdipo-mice (adiponectin promoter-driven Cre mice) to delete Irs1 and Irs2 specifically in adipocytes (Supplemental Fig. 1a). Compared to floxed Cntr or LDKO-mice (Irs1L/L•Irs2L/L•CreAlb), the FDKO-mice displayed normal glucose tolerance (Supplemental Fig. 1b), a result that is similar to mice lacking the adipocyte insulin receptor (FIRKO-mice)22 or adipocyte Akt1 and Akt223. Thus, dysregulation of the IR→IRS1/2→PI3K→Akt cascade in WAT alone was insufficient to dysregulate systemic glucose homeostasis during the intraperitoneal glucose tolerance test (GTT).
To understand the requirement of hepatic insulin signaling for systemic glucose homeostasis, we investigated glucose tolerance and HGP in LDKO-mice and LTKO-mice12. As shown previously, LDKO-mice developed glucose intolerance, which was corrected in the LTKO-mice (Supplemental Fig. 1b,c). Since excess HGP contributes to systemic glucose dysregulation, we used a hyperinsulinemic-euglycemic clamp to compare HGP in LDKO- and LTKO-mice. The glucose infusion rate (GIR) required to maintain euglycemia during insulin infusion (4 mU/kg/min) was significantly greater in Cntr-mice than in LDKO-mice (Fig. 1a); however, the GIR was normal in the LTKO-mice, despite the absence of hepatic Irs1 and Irs2 (Fig. 1b). Moreover, insulin failed to suppress HGP in LDKO-mice, whereas HGP was suppressed normally by insulin in LTKO-mice (Fig. 1c,d). Thus, hepatic insulin signaling was dispensable for glucose tolerance and regulation of HGP when hepatic FoxO1 was inactivated.
Figure 1.

Hepatic FoxO1 dysregulates WAT insulin signaling. (a,b) Glucose infusion rates (GIR) before and at steady-state (bar graphs) during hyperinsulinemic-euglycemic clamp of four-month old LDKO and Cntr mice (n = 7) (a), or LTKO and Cntr3 mice (n = 8) (b). (c,d) Hepatic glucose production (HGP) in LDKO and Cntr mice (n = 7) (c) or LTKO and Cntr3 mice (n = 8) (d) before and during the hyperinsulinemic-euglycemic clamp; inset shows the calculated suppression (%) of HGP by insulin. (e) Insulin-stimulated Irs1•p110PI3K complex formation in eWAT of four-month old LDKO, LTKO and control mice (n = 4–5). (f) Insulin-stimulated Irs1•p110PI3K complex formation in eWAT of three-month old Irs1L/L•Irs2L/L mice three weeks after injection with CreAAV•TBG or GFPAAV•TBG (n = 4–6). (g) Insulin-stimulated Irs1•p110PI3K complex formation in eWAT of eighteen-week old LTKO and Cntr3 mice two weeks after injection with FoxO1AdV (n = 3–5). (h) Fasting blood glucose levels in two-month old FDKO and Irs1L/L•Irs2L/L mice two weeks after injection with CreAAV•TBG or control GFPAAV•TBG (n = 7–8). (i) Glucose tolerance of FDKO and Irs1L/L•Irs2L/L mice two weeks after CreAAV•TBG or GFPAAV•TBG injection; inset, areas under glucose curves (n = 5–6). Data were analyzed by one-way ANOVA (c,d,e,h,i) and unpaired Student’s t-test (a,b,c,d,f,g). All data are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Hepatic FoxO1 dysregulates WAT insulin signaling
Normal suppression of HGP in LTKO-mice suggested that insulin indirectly suppressed HGP through its effects on white adipose tissue or other tissues. To investigate insulin signaling in the extra-hepatic tissues of LDKO-mice, we measured Akt phosphorylation (pS473Akt and pT308Akt) in the skeletal muscle, epigonadal white adipose tissue (eWAT), inguinal WAT (iWAT) and brown adipose tissue (BAT) upon completion of the hyperinsulinemic-euglycemic clamp. Compared with Cntr-mice, LDKO-mice showed reduced pS473Akt and pT308Akt in eWAT, iWAT and BAT, with insignificant changes in skeletal muscle (Supplemental Fig. 1d–f). By contrast, pS473Akt and pT308Akt phosphorylation were normal in eWAT and iWAT isolated from LTKO-mice (Supplemental Fig. 1g–i). Moreover, by comparision against Cntr-mice, insulin-stimulated Irs1•p110PI3K complex formation significantly decreased (by 49%) in eWAT of LDKO-mice, whereas the association was normal in eWAT of LTKO-mice (Fig. 1e). These results suggest that hepatic FoxO1 promoted WAT insulin resistance in LDKO-mice.
To determine if adult-onset hepatic insulin resistance promoted FoxO1-dependent WAT insulin resistance, LDKO-mice were created acutely by infecting Irs1L/L•Irs2L/L-mice with CreAAV•TBG (adeno-associated virus encoding Cre recombinase, driven by the hepatocyte-specific thyroid-binding globulin (TBG) promoter) or control GFPAAV•TBG. Like LDKO-mice, Irs1L/L•Irs2L/L•CreAAV•TBG-mice displayed low levels of hepatic Irs1 and Irs2 proteins and developed glucose intolerance (Supplemental Fig. 2a,b). Conversely, glucose-tolerant adult LTKO-mice infected with FoxO1AdV (adenovirus encoding FoxO1) developed glucose intolerance (Supplemental Fig. 2c,d). Consistently, insulin-stimulated Irs1•p110PI3K complex formation decreased significantly in eWAT of both Irs1L/L•Irs2L/L•CreAAV•TBG-mice and LTKO•FoxO1AdV-mice (Fig. 1f,g). Thus, hepatic FoxO1 promoted adipose tissue insulin resistance following acute loss of hepatic insulin signaling.
Next, we investigated the activation state of hormone-sensitive lipase (Hsl) in eWAT by measuring the phosphorylation of this protein. Compared with Cntr-mice, pS660Hsl was higher in the eWAT of LDKO-mice during fasting and insulin-stimulated conditions; however, pS660Hsl was indistinguishable in eWAT from LTKO-mice and the litter-matched controls (Cntr3) during insulin stimulation (Supplemental Fig. 2e). Consistent with these results, insulin failed to suppress circulating FFAs in LDKO-mice but suppressed FFAs normally in LTKO-mice (Supplemental Fig. 2f,g). Moreover, compared to control mice, H&E staining revealed smaller adipocytes in the eWAT from LDKO-mice and LTKO•FoxO1AdV-mice (Supplemental Fig. 2h). These results suggest that hepatic FoxO1 suppressed indirectly Irs1→PI3K→Akt─┤Hsl signaling in the eWAT of LDKO-mice.
Finally, we compared RNA expression profiles of eWAT obtained from Cntr- and LDKO-mice, and Cntr3- and LTKO-mice. Compared against the controls, the expression of 82 genes decreased, and 83 genes increased more than 2-fold in eWAT of LDKO-mice, most of which returned to normal expression in eWAT of LTKO-mice (Supplemental Fig. 2i and Supplemental Table 1). These results suggest that disruption of hepatic Irs1 and Irs2 promotes FoxO1-dependent gene expression changes in eWAT, which might integrate with hepatic dysfunction to dysregulate systemic glucose homeostasis. Indeed, CreAAV•TBG -mediated deletion of hepatic Irs1 and Irs2 in the FDKO-mice caused fasting hyperglycemia and severe glucose intolerance (Fig. 1h,i), suggesting that hepatic and adipose insulin resistance synergistically disrupt glucose homeostasis (Supplemental Fig. 2j).
Identification of FoxO1-dependent hepatokines in LDKO-mice
Previous work suggests that inflammatory cytokines might dysregulate systemic glucose metabolism24. We measured the circulating concentrations of several cytokines in LDKO- and LTKO-mice—including TNFα, IL-6, IL-1β, IL-2, IL-4, IL-5, KC and MIP1a; however, only IL-6 displayed significant FoxO1-dependent elevation (Supplemental Fig. 3a–h). We generated LDKO•IL-6−/−-mice to test whether IL-6 contributed to glucose intolerance and WAT insulin resistance; however, glucose intolerance was not normalized in LDKO•IL-6−/−-mice compared against Cntr•IL-6−/− (Supplemental Fig. 3i). Furthermore, insulin-stimulated formation of the Irs1•p110PI3K complex remained low in eWAT from LDKO•IL-6−/−-mice (Supplemental Fig. 3j). We conclude that elevated IL6 was not responsible for dysregulated glucose homeostasis and eWAT insulin resistance in LDKO-mice.
To test whether serum from LDKO-mice contains hepatic-derived circulating factors that modulate peripheral insulin action, cultured 3T3-L1 adipocytes were treated for 24 hours with 5% mouse serum obtained from LDKO- or Cntr-mice and then incubated briefly with insulin. Serum from the LDKO-mice significantly reduced (by 51%) the formation of the Irs1•p110PI3K complex, whereas heat-inactivated serum had no effect (Fig. 2a). These data suggest that heat-labile circulating protein(s) or other factor(s) in LDKO-mice might inhibit insulin signaling in adipose tissues.
Figure 2. Identification of Fst315 as a hepatic FoxO1-regulated hepatokine regulating systemic glucose homeostasis.

(a) Insulin-stimulated Irs1•p110PI3K complex formation in 3T3-L1 adipocytes cultured with serum from overnight fasted four-month old LDKO (vs Cntr) mice without (left, n = 8) or with heat inactivation of the serum (56°C for 45 min.) (right, n = 4). (b,c) Identified genes encoding putative hepatic secreted proteins that decreased (b) or increased (c) >3-fold in LDKO mice (Benjamini-Hochberg FDR<0.05). Several of these secreted proteins seemed likely to have effects upon peripheral metabolism (marked as *) were selected for functional screening in mice. (d–i) Five-week old C57BL6 mice were challenged with high fat diet (45% fat) for 2 months, then infected with GFPAAV•TBG or hepatokineAAV•TBG (2×1011 Genome Copy (GC)/mouse) encoding the indicated genes: fasting blood glucose (d) and fasting serum insulin (e) were measured one week after infection and used to calculate (f) HOMA2 IR as a measure of systemic insulin resistance (n = 5–10). (g) Insulin tolerance tests (ITT) were performed four weeks after infection with GFPAAV•TBG or hepatokineAAV•TBG and summarized by the areas under glucose curves (AUC) (n = 4–10). (h,i) Glucose tolerance tests were performed one week after AAV infection (h) and summarized (i) by the areas under glucose curves (AUC) (n = 4–10). (j) Fasting serum Fst levels measured four weeks after infection of HFD-fed C57BL6 mice with Fst315AAV•TBG or GFPAAV•TBG (n = 5). Data were analyzed by two-way ANOVA (h), one-way ANOVA (d,e,f,g,i) and unpaired Student’s t-test (a, j). Data are reported as the mean±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
To find hepatokines that promote systemic insulin resistance in LDKO-mice, we used published Affymetrix microarray data to identify the liver genes that were higher or lower (≥ 3-fold, FDR<0.05) in LDKO-mice compared to Cntr-mice (Array Express accession #E-MEXP-1649)12. We further selected the genes that changed in LDKO-mice but were expressed normally in LTKO-mice, and filtered this subset against a curated list of secreted proteins that was confirmed by Ingenuity Pathway Analysis. This strategy revealed 19 putative hepatokines that were ≤ 3-fold lower, and 66 that were ≥ 3-fold higher in LDKO-mice (Fig. 2b,c). Some of these hepatokines might have effects upon peripheral metabolism—including higher lipoprotein lipase (Lpl; facilitates triglyceride uptake), follistatin (Fst; inhibits TGFβ→Smad signaling25), or insulin-like growth factor binding protein 1(Igfbp1; inhibits Igf1 signaling); or lower insulin-like growth factor 1 (Igf1; reduced body growth). Thus, we infected wild-type (WT) mice maintained on a high fat diet (HFD) with AAVTBG vectors encoding several candidate hepatokines or a control GFPAAV•TBG. The first cohort—including growth hormone receptor (Ghr), glutathione peroxidase 3 (Gpx3), serine peptidase inhibitor, clade B, member 6a (Serpinb6a), myelin protein zero like 1(Mplz1) or neurotrophin 3 (Ntf3)—had no effects on fasting glucose levels, insulin levels, glucose tolerance or insulin tolerance (Supplemental Fig. 4a–f). In the second cohort leukocyte elastase inhibitor A (Serpinb1a) and serum amyloid a4 (Saa4) decreased glucose without an effect upon serum insulin, whereas Igf1 and inhibin E (Inhbe) increased serum insulin levels without effects on glucose (Fig. 2d,e). However, Fst315 (circulating follistatin) increased circulating glucose and insulin, and the homeostatic model assessment of insulin resistance (HOMA-IR) suggested that Fst315 promoted systemic insulin resistance (Fig. 2d–f). Fst315 also promoted significant insulin resistance during the insulin tolerance test (ITT) (Fig. 2g). Most of the tested hepatokines had an insignificant effect upon glucose tolerance; however, Fst315 impaired glucose tolerance significantly and Lpl significantly improved glucose tolerance (Fig. 2h,i). Finally, we confirmed expression of the virus-encoded hepatokine genes in liver by qPCR (Supplemental Fig. 4g,h). Fst315AAV•TBG infection of Cntr-mice on a HFD increased circulating Fst315 23-fold (Fig. 2j).
To determine whether a physiological 2-fold increase circulating Fst could dysregulate glucose tolerance, C57BL6 mice on a HFD for 2 months were infected with a lower dose of Fst315AVG•TBG or control GFPAAV•TBG. Ten days after Fst315AVG•TBG infection, circulating Fst was 2.2-fold higher than in controls, which still elevated fasting glucose and impaired glucose tolerance (Supplemental Fig. S4i–k). Together these results suggest that Fst315 contributed—at least in part—to the hepatic FoxO1-dependent WAT insulin resistance and glucose intolerance in LDKO-mice.
FoxO1 regulates Fst expression in LDKO-mice
Two Fst isoforms are generated by alternative mRNA splicing, including membrane-bound (autocrine) Fst288 and the longer circulating (endocrine) Fst315 (ref. 21). Compared to fed Cntr-mice, hepatic Fst mRNA (total and Fst315) and circulating Fst protein was 2-fold higher during fasting, and 6-fold higher during STZ-induced type 1 diabetes (Supplemental Fig. 5a–e). This pattern of expression was consistent with previous work showing that hepatic Fst is upregulated by an elevated glucagon/insulin ratio25. Although this ratio was relatively low in LDKO-mice—glucagon was elevated 4-fold while insulin was elevated 20-fold (Supplemental Fig. 5f,g)—the LDKO-liver fails to respond to insulin, so the effect of glucagon might be exaggerated. Compared to Cntr-liver, mRNA in LDKO-liver encoding Fst288 and Fst315 increased 10- and 30-fold (Fig. 3a). Moreover, the concentration of circulating Fst in fasted LDKO-mice increased 20-fold, and immunostaining revealed more Fst protein in liver sections (Fig. 3b,c). Fst mRNA was also higher in the liver of hepatocyte-specific insulin receptor knockout mice (LIRKO-mice), confirming that hepatic insulin signaling suppressed Fst expression (Supplemental Fig. 5h). Consistent with these results, serum Fst was ~2-fold higher in genetically obese mice (ob/ob) or Cntr-mice fed a HFD (Supplemental Fig. 5i), which can be expected to impair glucose tolerance (Supplemental Fig. 4j).
Figure 3. Fst expression and secretion is positively regulated by FoxO1, and down-regulated by gastric bypass surgery in obese diabetic individuals.

(a) Relative expression of Fst315 and Fst288 mRNAs, or total Fst mRNA, measured by qPCR on liver RNA from four-month old LDKO and LTKO mice (n = 3–5). (b) Serum total Fst concentration in four-month old LDKO and LTKO mice (n = 7–10). (c) Representative images of liver sections from four-month old LDKO and control mice immunostained using antibody against Fst and DAPI to visualize nuclei (n = 2 from each group). Scale bars, left (500 µm); middle and right (50 µm). (d) Relative Fst gene expression in primary hepatocytes treated for 12 hours with or without glucagon (Gcg, 100nM) 48 hours after infection with shFoxO1AdV or control shGFPAdV (n = 3). (e) Total Fst mRNA expression and protein concentration in cell lysates or culture medium of primary hepatocytes infected with FoxO1AdV or GFPAdV for 24 hours (n = 3). (f) Total Fst mRNA expression and protein concentration in cell lysates or medium of primary hepatocytes infected with shGFPAdV and shFoxO1AdV for 48 hours (n = 3). (g) Chromatin-immunoprecipitation (ChIP) with FoxO1 antibody showing FoxO1 binds directly to the Fst promoter. (h,i) Serum HbA1c% (h) and total Fst concentration (i) in obese diabetic individuals before and 6 months after Roux-en-Y gastric bypass surgery (n = 9) (see Supplemental Table 2 for individual data). Data were analyzed by one-way ANOVA (a,b,d), unpaired Student’s t-test (e,f,g) and paired Student’s t-test (h,i). Data are reported as the mean±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Circulating and hepatic Fst levels were normal in the LTKO-mice suggesting that FoxO1 promoted its expression during hepatic insulin resistance (Fig. 3a,b). Moreover, infection of primary hepatocytes from C57BL6 mice with shFoxO1AdV decreased glucagon-stimulated Fst, compatible with a role for FoxO1 in stimulation by a high glucagon/insulin ratio (Fig. 3d). Consistent with these results, FoxO1AdV increased Fst mRNA and protein concentration in cultured hepatocytes and increased the concentration of Fst secreted into the medium (Fig. 3e). By contrast, infection with shFoxO1AdV decreased Fst mRNA and the concentration of Fst protein in cultured hepatocytes and in the medium (Fig. 3f).
Finally, we used chromatin-immunoprecipitation (CHIP) with a FoxO1 antibody to determine whether FoxO1 binds directly to the Fst promoter. By comparison with strong FoxO1 binding to the Pck1 promoter (positive control), or weak background binding to the Gapdh promoter (negative control), FoxO1 bound significantly between −525 to −2175 of the Fst promoter (Fig. 3g). These regions contain several consensus FoxO1 binding sites that are homologous among human, mouse and rat supporting the hypothesis that nuclear FoxO1 is an important regulator of Fst expression (Supplemental Fig. 5j).
Bariatric surgery reduces FST in obese individuals with diabetes
Previous reports show that plasma Fst is elevated in individuals with type 2 diabetes25,26. To investigate the relation between glucose dysregulation and FST in humans, we measured circulating FST concentration in a group of obese individuals with diabetes (BMI: 45.5±3, %HbA1c: 8.4±1) before and after treatment by Roux-en-Y gastric bypass surgery (RYGB) (Supplemental Table 2).
Six months after RYGB, %HbA1c decreased significantly to 6%, confirming a beneficial effect upon glycemic control (Fig. 3h). Moreover, the serum FST concentration correlated positively with declining %HbA1c (Pearson correlation: r = 0.5977, P = 0.0088) as it decreased significantly by 48% six months after RYGB (Fig. 3i). Importantly, the FST concentration changed within the biologically active range (Supplemental Fig. 4i–k). Thus, the beneficial effect of RYGB might be related, at least in part, to reduced circulating FST.
Knockdown of hepatic Fst promotes WAT insulin sensitivity and reduces HGP
To establish whether excess endogenous hepatic Fst promotes WAT insulin resistance, we deleted hepatic Fst alleles by infecting LDKO-liver with CRISPR/Cas9-based AAV bearing two different guide RNAs targeting Fst (designated sgFst1AAV•TBG or sgFst2AAV•TBG). Compared to the control nullAAV•TBG vector, sgFst2AAV•TBG significantly reduced total hepatic and circulating Fst 2-fold, whereas sgFst1AAV•TBG produced an intermediate effect that did not reach significance (Fig. 4a,b). Consistent with the differential reduction of Fst in liver using these two different guide RNAs, glucose tolerance reached the normal range in LDKO•sgFst2AAV•TBG–mice, whereas LDKO•sgFst1AAV•TBG–mice displayed an intermediate but insignificant reduction (Fig. 4c). A similar differential effect on serum insulin levels was also observed with the two guide RNAs versus control (Fig. 4d). Compared to nullAAV•TBG-mice, eWAT from LDKO•sgFst2AAV•TBG-mice displayed greater Irs1•p110PI3K complex formation and Akt phosphorylation (pT308Akt and pS473Akt) (Fig. 4e–g); however, pS660Hsl was significantly lower in both LDKO•sgFst1AAV•TBG-mice and LDKO•sgFst2AAV•TBG-mice compared to the control (Fig. 4f,g).
Figure 4. Knockdown of Fst improves WAT insulin sensitivity, reduces HGP and improves glucose tolerance in LDKO mice.

Panels a–g: Two to three-month old LDKO mice were infected with ‘nullAAV•TBG’ (encoding luciferase) or with sgFstAAV•TBG (encoding small guide RNA sgFst1 or sgFst2) to effect partial hepatic Fst knockout (n = 7). (a,b) Hepatic Fst protein content (a) and serum total Fst concentration (b) were measured two weeks after infection. (c) GTTs were performed 10 days after infection and summarized by the areas under curves (AUC). (d) Fasting serum insulin levels were measured two weeks after infection. (e) Insulin-stimulated Irs1•p110PI3K complex formation in eWAT, measured two weeks after infection. (f,g) Insulin regulated phosphorylation of Akt and HSL in eWAT was analyzed two weeks after infection by Western blotting (f) and quantitated by densitometry (g). Panels h–k: Five-month old LDKO mice were infected with ‘nullAAV•TBG’ or shFstAAV•TBG (encoding a miRNA-like hairpin RNA against Fst) and hyperinsulinemic-euglycemic clamps were conducted four weeks later (n = 5). (h and i) Blood glucose concentrations (h) and glucose infusion rates (i) before and at steady-state during hyperinsulinemic-euglycemic clamp. (j,k) Basal and clamped hepatic glucose production (j) and calculated suppression of HGP by insulin during the clamp (k). Panels l–n: Five weeks of C57BL6 mice maintained on high fat diet for two months were infected with shFstAAV-TBG or NullAAV-TBG (n = 7). Serum Fst levels were measured two weeks after infection (l); GTTs (m) and fasting blood glucose levels (n) were measured ten days after infection. Data (b,d,e) were reported as the median±95%CI, and Kruskal-Wallis test was used to identify significant differences between treatments of LDKO mice. Other data were reported as the mean±SEM. Data were analyzed by One-way ANOVA (a,c,g,j), two-way ANOVA (i) and unpaired Student’s t-test (k,l,m,n). P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
The intermediate response to sgFst1AAV•TBG infection appears to arise from its variable infection or effect upon Fst levels. To confirm the relation between circulating Fst and the biological responses, we calculated Pearson correlation coefficients for the combined groups of mice. Significant correlations were calculated in all cases: Fst vs Ins: r = 0.93, P < 0.0001; Fst vs GTT-AUC: r = 0.763, P < 0.0001; and Fst vs PI3K: r = −0.408, P = 0.0310. Thus, the Fst concentration measured across each group (Cntr, null, sgFst1 and sgFst2) correlated significantly with each biological response. Taken together, these results suggest that lowering hepatic Fst improved insulin sensitivity of eWAT and glucose tolerance in LDKO-mice.
Since CRISPR/Cas9-based gene editing can produce off-target effects, we used hepatic shFstAAV•TBG infection to validate the benefit of reducing hepatic Fst mRNA in LDKO-mice (Supplemental Fig. 6a). Compared against the nullAAV•TBG infection, eWAT from LDKO•shFstAAV•TBG-mice displayed significantly less hepatic and circulating Fst (Supplemental Fig. 6b,c), improved glucose tolerance (Supplemental Fig. 6d), increased Akt phosphorylation (pT308Akt and pS473Akt), and decreased pS660Hsl (Supplemental Fig. 6e,f). Together these results confirm that hepatic-derived Fst promotes glucose intolerance and insulin resistance of eWAT in LDKO-mice.
To establish the relationship between hepatic Fst and HGP, hyperinsulinemic-euglycemic clamp was conducted on LDKO-mice infected with shFstAAV•TBG or nullAAV•TBG. The GIR required to maintain euglycemia was significantly greater in LDKO•shFstAAV•TBG-mice versus LDKO•nullAAV•TBG-mice, suggesting that reducing hepatic Fst increased systemic insulin sensitivity (Fig. 4h,i). Compared to the control LDKO•nullAAV•TBG-mice, HGP decreased in LDKO•shFstAAV•TBG-mice during the insulin clamp and the suppression of HGP by insulin was restored (Fig. 4j,k). Thus, knockdown of hepatic Fst in LDKO mice provided a benefit similar to that conferred by genetic disruption of hepatic FoxO1.
To establish whether Fst contributes to glucose intolerance in a non-genetic model of diabetes, C57BL6 mice maintained on high-fat diet for two months were infected with shFstAAV•TBG or NullAAV•TBG. Serum Fst levels were 37% lower in shFstAAV•TBG mice than in the control NullAAV•TBG-mice (Fig. 4l). Suppression of hepatic Fst resulted in significantly lower circulating glucose before and during the GTT compared to control-treated mice (Fig. 4m, n). Thus, hepatic Fst promoted glucose intolerance during genetic or physiological diabetes.
Fst promotes WAT insulin resistance and HGP in WT mice
Next, we investigated directly whether circulating Fst in LDKO-mice can dysregulate adipose insulin signaling. Cultured 3T3-L1 adipocytes were incubated with mouse serum obtained from Cntr- or LDKO-mice, then treated briefly with insulin. Compared with serum from Cntr-mice, serum from LDKO-mice resulted in significantly lower (by 37%) insulin-stimulated p110PI3K•Irs1 complex formation; moreover, addition of an antibody to neutralize Fst in the LDKO serum resulted in greater p110PI3K•Irs1 complex formation that approached the normal range (Fig. 5a).
Figure 5. Fst315 promotes WAT insulin resistance, HGP, and glucose intolerance in WT mice.
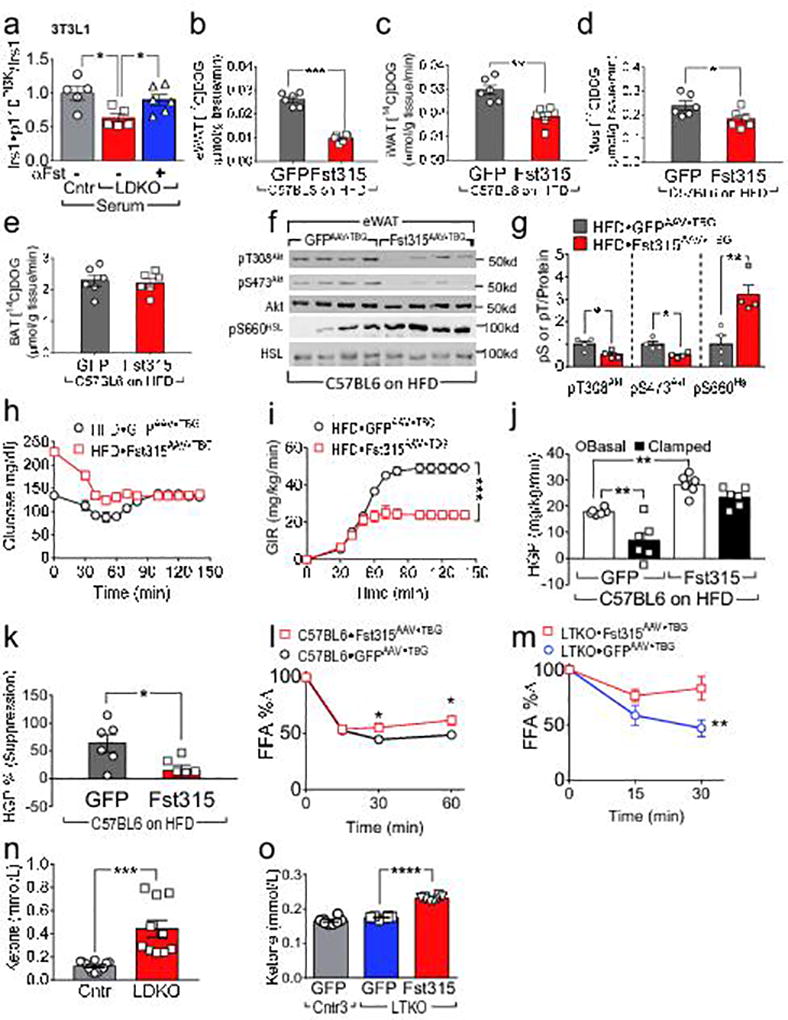
(a) Insulin-stimulated Irs1•p110PI3K complex formation in 3T3-L1 adipocytes treated with serum from overnight-fasted four-month old LDKO (vs Cntr) mice, with or without the addition of Fst antibody (R&D, AF669, 10 µg/ml) (n = 5–6). Panels b–k: Five-week old C57BL6 mice were challenged with high fat diet for four months before infection with Fst315AAV•TBG or GFPAAV•TBG, and hyperinsulinemic-euglycemic clamps were performed four weeks later. (b–e) Uptake of 2DOG ([1-14C] 2-deoxy-D-glucose) tracer into eWAT (b), iWAT (c), skeletal muscle (d), and BAT (e), measured at the end of the clamp (n = 6). (f,g) Western blot analysis (f) and densitometric quantitation (g) of insulin-regulated Akt and HSL phosphorylation in eWAT upon completion of the clamp. (h,i) Blood glucose concentrations (h) and glucose infusion rates (i) before and during hyperinsulinemic-euglycemic clamp (n = 6). (j,k) Basal and clamped hepatic glucose production (j), and calculated suppression of HGP by insulin during the clamp (k) (n = 6). (l) Change in serum FFAs (% of overnight-fasted baseline) following insulin treatment (1 U/kg, i.p.) of 14-week old C57BL6 mice (maintained 10 weeks on high fat diet) two weeks after infection with Fst315AAV•TBG or GFPAAV•TBG (n = 10). (m) Change in serum FFAs (% of overnight-fasted baseline) following insulin treatment (1 U/kg, i.p.) of four-month old LTKO mice two weeks after infection with Fst315AAV•TBG or GFPAAV•TBG (n = 6–7). (n) Urinary ketones in four-month old LDKO and control mice after five hour fast (n = 10–12). (o) Urinary ketones (5 hours fasting) in four-month old LTKO and control mice two weeks after infection with GFPAAV•TBG or Fst315AAV•TBG (n = 8). Data in (a) were analyzed by ANOVA and compared by the FDR method of Benjamini and Hochberg (*q < 0.05); other data were analyzed by unpaired Student’s t-test (b–e,g,k,n), one-way ANOVA (j and o) and two-way ANOVA (i,l,m). Data are reported as the mean±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
To test whether Fst315 induced WAT insulin resistance in WT mice, C57BL6 mice maintained on a HFD for four months were infected with control GFPAAV•TBG or Fst315AAV•TBG. C57BL6•Fst315AAV•TBG-mice displayed less 2-DOG ([1-14C] 2-deoxy-D-glucose) uptake (as a measure of insulin action) into eWAT, iWAT and skeletal muscle upon completion of the hyperinsulinemic-euglycemic clamp (Fig. 5b–d); however, uptake by BAT was not affected (Fig. 5e). Both pT308Akt and pS473Akt were significantly higher while pS660Hsl was significantly lower in eWAT of C57Bl/6•Fst315AAV•TBG mice (Fig. 5f,g). Unlike Fst315, Fst288 has a functional heparin binding domain that can anchor it to the cell membrane upon secretion, where it might compete with receptors for binding TGFβ-superfamily ligands27. We placed C57BL6 mice on a HFD for two months and then infected them with Fst288AAV•TBG. Four weeks later, hepatic Fst288 mRNA and protein were 128-fold and 4-fold higher, respectively, than in control mice (Supplemental Fig. 7a,b). Unexpectedly, circulating Fst was elevated 12-fold one week after infection indicating that hepatic Fst288 entered the circulation (Supplemental Fig. 7c). Compared to control C57BL6•GFPAAV•TBG-mice on the HFD, fasting plasma insulin and blood glucose concentrations increased significantly in C57BL6•Fst288AAV•TBG-mice (Supplemental Fig. 7d,e), which also developed glucose intolerance (Supplemental Fig. 7f). Fst288AAV•TBG infection resulted in a 2-fold lowering of 2-DOG uptake into eWAT and iWAT upon completion of hyperinsulinemic-euglycemic clamp (Supplemental Fig. 7g,h).
We next conducted hyperinsulinemic-euglycemic clamps to measure HGP in C57BL6 mice infected with GFPAAV•TBG, Fst315AAV•TBG or Fst288AAV•TBG. On a HFD, the GIR required to maintain euglycemia during the clamp in the GFPAAV•TBG-infected mice was significantly higher than in either Fst315AAV•TBG- or Fst288AAV•TBG-mice (Fig. 5h,i and Supplemental Fig. 7i,j). Moreover, hepatic expression of either Fst315 or Fst288 increased HGP during basal and clamp conditions, and strongly reduced the ability of insulin to suppress HGP (Fig. 5j,k and Supplemental Fig. 7k,l).
Compared to fasted Cntr-mice on the HFD, insulin failed to suppress circulating FFA in fasted C57BL6•Fst315AAV•TBG-mice maintained on HFD (Fig. 5l). To confirm the inhibitory effect of Fst upon the suppression of lipolysis, metabolically normal LTKO-mice were infected with Fst315AAV•TBG or control GFPAAV•TBG. Two weeks later, FFA levels after insulin treatment (1U/kg) decreased normally in the Cntr-mice, but only weakly in the LTKO-Fst315AAV•TBG mice (Fig. 5m). As in LDKO-mice, urinary ketones were greater in the LTKO•Fst315AAV•TBG mice than in Cntr-mice (Fig 5n,o). These results are consistent with unsuppressed lipolysis owing to Fst-induced WAT insulin resistance.
Regulation of hepatic gene expression by Fst depends partially on hepatic FoxO1
To investigate how Fst affects hepatocytes per se, we used Affymetrix microarrays to quantitate liver mRNAs from fasted mice infected with Fst288AAV•TBG or GFPAAV•TBG (Supplemental Fig. 8a). Fst288AAV•TBG infection modestly up- or down-regulated the expression of many genes, including several related to hepatic glucose metabolism (Pck1, G6pc and Gck) (Supplemental Fig. 8a), while qPCR confirmed that Fst288AAV•TBG affected the expression of these and other genes during both fasting (Pck1, G6pc and Gck) and feeding (Foxo1, Igfbp1, Pck1, and Ppargc1a) (Fig. 6a,b). We also found that Fst288 expression was associated with a significant increase in glucose-6-phosphatase enzyme activity (encoded by G6pc) during fasting, which is essential for increased HGP (Fig. 6c). Microarray analysis revealed that Fst288 modestly down-regulated several lipid and cholesterol synthetic genes (Supplemental Fig. 8a). We used qPCR to confirm that Fst288 reduced the expression of hepatic Srebf1, Fasn, and Hmgcs1 (Supplemental Fig. 8b); moreover, immunoblotting showed that Fst288AAV•TBG increased Hsl and decreased Srebp1 protein in C57BL6 mouse liver (Supplemental Fig. 8c).
Figure 6. Regulation of hepatic gene expression by Fst depends partially on hepatic FoxO1.

Panels a–d: C57BL6 mice challenged with high fat diet for 2 months were infected with Fst288AAV•TBG or GFPAAV•TBG. (a,b) Hepatic mRNA expression was measured by qPCR after a 16 h overnight fast without (a) or with 4 hours of refeeding (b) one week after AAV infection. (c) Glucose-6-phosphatase (G6Pase) activity after a 16 h overnight fast in liver one week after infection with Fst288AAV•TBG or GFPAAV•TBG (n = 5). (d) Western blot analysis and densitometric quantitation of FoxO1 protein in nuclear and cytoplasmic fractions, or total liver lysate of C57BL6 mice, with or without 4 hours refeeding following a 16 h overnight fast, and one week after infection with Fst288AAV•TBG or GFPAAV•TBG (n = 5). Panels e–i: Two-month old LTKO mice were infected with GFPAAV•TBG or Fst288AAV•TBG to reconstitute hepatic Fst. GTTs (e) were performed two weeks after AAV infection (n = 7). (f,g) Hyperinsulinemic-euglycemic clamps were conducted four weeks after infection; shown are the blood glucose concentrations (f) and glucose infusion rates before and at steady state (g) during hyperinsulinemic-euglycemic clamp (n = 5). (h,i) Basal and clamped hepatic glucose production (h) and calculated suppression of HGP by insulin during the clamp (i) (n = 5). Data were analyzed by unpaired Student’s t-test (a–e,g,i) and one-way ANOVA (h). Data are reported as the mean ± SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Hepatic FoxO1 mediates many effects of insulin upon glucose and lipid metabolism13. To determine whether Fst288 modulates FoxO1 cellular distribution, we compared the concentration of FoxO1 in nuclear and cytoplasmic fractions of liver lysates from fasting and fed C57BL6•Fst288-mice. Compared to control C57BL6•GFP-mice, total FoxO1 protein was ~2-fold greater in the liver of C57BL6•Fst288-mice (Fig. 6d). Both nuclear and cytoplasmic FoxO1 were greater during fasting; however, during feeding nuclear FoxO1 was greater while cytoplasmic FoxO1 was lower, opposite to the regulation of FoxO1 by insulin (Fig. 6d).
To determine whether Fst288-regulated hepatic gene expression depends upon FoxO1, we infected LTKO-mice with Fst288AAV•TBG. RNA expression profiles showed that Fst288 regulated the expression of genes involved in gluconeogenesis and glycolysis, decreasing Gck and increasing Pck1 and G6pc, even without hepatic FoxO1; however, Fst288-mediated down-regulation of lipogenic and cholesterogenic genes such as Srebf1 was lost in the LTKO-mice (Supplemental Fig. 8a). Next, to determine whether Fst288 required hepatic FoxO1 to promote HGP, GTTs and hyperinsulinemic-euglycemic clamp were performed. Compared to control GFPAAV•TBG•LTKO-mice, glucose tolerance was impaired mildly in Fst288AAV•TBG•LTKO-mice (Fig. 6e). The GIR required to maintain euglycemia during clamp was also much less in Fst288AAV•TBG•LTKO-mice than in GFPAAV•TBG•LTKO-mice (Fig. 6f,g). As in LDKO-mice, insulin failed to suppress HGP in the Fst288AAV•TBG•LTKO-mice (Fig. 6h,i); thus, the detrimental effect of Fst288 upon HGP developed without hepatic FoxO1.
Fst–mediated WAT insulin resistance is independent of hepatic FoxO1
Finally, we investigated whether reduced Fst in LTKO-mice contributed to their improved glucose tolerance and eWAT insulin sensitivity. We infected LTKO-mice with Fst315AAV•TBG to increase circulating Fst315 about 9-fold (Supplemental Fig. 9a). Compared to control GFPAAV•TBG•LTKO-mice, the LTKO•Fst315AAV•TBG-mice developed profound glucose intolerance that phenocopied LDKO-mice (Supplemental Fig. 9b). During insulin stimulation, both pT308Akt and pS473Akt decreased significantly—and pS660Hsl increased significantly—in eWAT of LTKO•Fst315AAV•TBG-mice (Supplemental Fig. 9c,d). These results suggested that Fst315 promoted WAT insulin resistance independently of hepatic FoxO1. Moreover, the GIR required to achieve euglycemia decreased in clamped LTKO•Fst315AAV•TBG-mice (Supplemental Fig. 9e,f); and insulin failed to suppress HGP in LTKO•Fst315AAV•TBG-mice compared against LTKO•GFPAAV•TBG-mice (Supplemental Fig. 9g,h). Thus, FoxO1-dependent glucose dysregulation in LDKO-mice owes, at least in part, to FoxO1→Fst regulation rather than hepatic FoxO1 alone.
Discussion
Our results identify the hepatokine Fst as a mediator of systemic metabolic dysregulation driven by hepatic FoxO1 activity. Prior investigations have highlighted the FoxO1-dependent nature of excessive HGP and/or glucose intolerance in mice with hepatic insulin resistance12,15,18. We previously showed that insulin fails to suppress HGP in our LDKO-mice14; herein, we confirm that HGP suppression is restored in LDKO-mice by disruption of hepatic FoxO1 (LTKO-mice). Correlating with HGP, expression and secretion of Fst is strongly upregulated in LDKO-liver, but nearly normalized in LTKO-liver. Within primary hepatocytes, knockdown of FoxO1 decreases—and its overexpression increases—the expression and secretion of Fst. Compatible with direct regulation by FoxO1, we observe enriched recovery of Fst promoter regions containing FoxO1 binding motifs. Importantly, reducing hepatically-produced Fst in LDKO-mice using CRISPR/Cas9- or Fst shRNA mimics the benefits of FoxO1 deletion in LTKO-mice. Conversely, viral expression of Fst in the liver of high fat-fed WT mice or chow-fed LTKO-mice reproduces an LDKO-like phenotype, including impaired WAT insulin sensitivity, unsuppressed HGP, and severe glucose intolerance. Thus, reduced expression and secretion of Fst may largely explain the restoration of metabolic homeostasis in LTKO- versus LDKO-mice12. Nonetheless, we do not rule out that other liver-intrinsic factors might contribute—particularly as viral expression of Fst alters RNA expression profiles in livers of both WT and LTKO-mice.
Although HGP and glucose tolerance are normalized in LTKO-mice, the IRS→PI3K→Akt cascade in liver of LTKO-mice remains insensitive to insulin12. Our data support that normalization of HGP in LTKO-mice (and similar models) owes substantially to the restoration of WAT insulin sensitivity, which reduces lipolysis and the availability of FFA and glycerol that potentiate hepatic gluconeogenesis1,16,17. Demonstrating the negative influence of hepatic FoxO1 upon WAT insulin sensitivity, we observe significantly impaired IRK→IRS→Akt signaling and Akt→PDE3b─┤HSL signaling in LDKO-WAT, as well as impaired insulin suppression of serum FFA—each of which is normalized in LTKO mice.
Within the insulin-resistant liver, excess FFA are oxidized to acetyl-CoA and further metabolized to produce ketone bodies28. Our observation of elevated urinary ketones in LDKO-mice—but not LTKO-mice—validates the oxidation of excess FFA in the LDKO liver to acetyl-CoA, which allosterically upregulates pyruvate carboxylase-dependent hepatic gluconeogenesis and HGP1. Forced expression of Fst in LTKO-liver likewise increases ketone production, in conjunction with impaired suppression of HGP and glucose intolerance. Together, these data support that WAT insulin resistance mediated by Fst contributes significantly to glucose intolerance in LDKO-mice12,29–31; however, additional secreted or non-secreted hepatic gene products regulated by FoxO1 might combine with Fst to produce the systemic effects of severe hepatic insulin resistance (e.g., Patatin Like Phospholipase Domain Containing 2 (Pnpla2) and G0/G1 Switch 2 (G0S2)32.
Two Fst isoforms are produced by alternative mRNA splicing: membrane-bound (autocrine/paracrine) Fst288 that contains a functional heparin binding site; and the longer circulating Fst315 that does not21,27,33. Though our assays distinguish the hepatic mRNAs encoding these isoforms, available immunoassays cannot distinguish the protein isoforms. Both Fst288 and Fst315 mRNAs are upregulated in LDKO versus control or LTKO mice. Although its heparin binding domain reportedly anchors Fst288 to cell membranes wherein it is produced27, infection with either Fst288AAV•TBG or Fst315AAV•TBG increased circulating Fst in LTKO-mice that weakly express endogenous Fst; moreover, the two isoforms equivalently dysregulated WAT insulin sensitivity, HGP and glucose tolerance. Notwithstanding these results, mice engineered to produce only Fst288—without circulating Fst315—were shown earlier to have mildly decreased fasting glucose34. We conclude that its over-expression in Fst288AAV•TBG-infected mice confers hepatokine/endocrine function upon normally autocrine/paracrine Fst288.
Fst was identified originally as a soluble protein in follicular fluid that suppresses the biosynthesis and release from the pituitary of follicle stimulating hormone35. However, Fst binds to and neutralizes diverse TGFβ-superfamily ligands. TGFβ-superfamily signaling is initiated by heterotetrameric receptor serine kinases, composed of ‘type II’ and ‘type I’ receptors that establish the binding specificity and selective phosphorylation of R-Smads—Smad2 and Smad3 downstream of TGFβ/Activin/Nodal, or Smad1, Smad5 and Smad8 in the BMP/GDF pathway; these phosphorylated Smads assemble with Smad4 to enter the nucleus and modulate gene expression20,36,37. Fst isoforms bind primarily activin A and B, myostatin (GDF8), and with lower affinity BMP4 and BMP11 (refs. 27,34,38,39). Circulating follistatin like-3 (Fstl3) binds the same ligands as Fst40 and deletion of Fstl3 improves glucose tolerance in mice41. Neutralization of activin B might be relevant for production of WAT insulin resistance, as it was shown recently to suppress lipolysis in mouse embryo fibroblast-derived adipocytes and human adipocytes42. However, as Fst is an inhibitor of TGFβ superfamily ligands its activity upon cultured adipocytes might depend upon the mix of TGFβ ligands present in the medium. Moreover, BMP4 was shown to suppress lipolysis in pre-adipocytes by inhibiting HSL43. Neutralization of other ligands of TGFβ-superfamily receptor Acvr2b could also be involved, since a soluble form of this receptor enhances gluconeogenesis in insulin-insufficient mice44.
Future assessment of changes in WAT mRNA transcription caused by ectopic Fst and/or TGFβ-superfamily ligands would aid in validating the mechanism by which Fst impairs WAT insulin sensitivity. Along these lines, we observe that virus-mediated Fst288 expression significantly alters the pattern of hepatic gene expression in high fat-fed WT mice. Since Fst expression produces a different pattern of hepatic gene expression in the LTKO-liver, we suggest, provisionally, that excess circulating Fst may amplify, or alter, FoxO1-dependent hepatic gene expression changes. Regardless, as viral expression of Fst does not require hepatic FoxO1 to produce an LDKO-like phenotype (i.e., in LTKO-mice), Fst-induced changes in hepatic gene expression might be less critical for systemic dysregulation than its ability to promote WAT insulin resistance45.
Previously it was reported that insulin resistant individuals and those with T2D exhibit modestly elevated circulating FST, and that this increase in FST is positively correlated with %HbA1c, fasting glucose and glucose excursion during oral GTT26. While we did not measure circulating FST in healthy controls, we found that RYGB surgery significantly reduced circulating Fst in obese individuals with diabetes and that this reduction correlated with reduced %HbA1c, an indicator of improved medium-term glycemic control. Thus, an unknown therapeutic aspect of RYGB might be to reduce the effect of circulating FST upon WAT, thereby facilitating control of HGP and glycaemia. Although other interpretations of these data are possible, our mouse data support the notion that targeting hepatically-produced and secreted FST in diabetic adults might prove useful in restoring systemic metabolic homeostasis. Because of its clear relation to hepatic insulin resistance and possible pleiotropic effects, targeting excess circulating Fst might also prove more fruitful than attempts to upregulate TGFβ superfamily-mediated signals in the insulin-resistant WAT.
Online Methods
Mice
We generated the liver-specific Irs1 and Irs2 double knockout (LDKO), and Irs1, Irs2 and FoxO1 triple knockout (LTKO) mice as previously described12,13. We purchased C57BL6 mice (Stock No. 000664), ob/ob mice (Stock No. 000632), B6.129S2-Il6tm1Kopf/J mice (Stock No. 002650) and B6;FVB-Tg(Adipoq-cre)1Evdr/J mice (Stock No. 010803) from The Jackson Lab. We generated FDKO mice by crossing B6;FVB-Tg(Adipoq-cre)1Evdr/J with Irs1 and Irs2 double floxed mice. We generated LDKO:IL6−/− mice by crossing B6.129S2-Il6tm1Kopf/J with LDKO mice. All mice were housed in plastic cages on a 12:12 h light–dark cycle with free access to water and food. We performed animal experiments according to all relevant ethical regulations and protocols approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee.
Hyperinsulinemic euglycemic clamp in conscious and unrestrained mice
Prior to the clamp experiment, one catheter was inserted into the right jugular vein for infusions. After 5–7 days of recovery, mice that had lost less than 10% of their preoperative weight were subjected to the clamp. On the day of the experiment, mice were deprived of food for 3.5 hours at 8:00am and then infused continuously with D-[3-3H]-glucose (PerkinElmer) (0.05 µCi/min) at a rate of 1ul/min for 1.5 h. After basal sampling from the tail vein, a 140min hyperinsulinemic euglycemic clamp was conducted with a primed-continuous infusion of human regular insulin (4 mU/kg/min, Humulin, Eli Lilly) at a rate of 2µl/min and continuously with D-[3-3H]-glucose (PerkinElmer) (0.1 µCi/min) at a rate of 2ul/min throughout the clamp experiment. The insulin solutions were prepared with 3% BSA in 0.9% saline. 20% glucose was infused at variable rates as needed to maintain plasma glucose at ~130 mg/dl (except in Fig 1D,F(Cntr±SEM: 138±9 mg/dl; LDKO±SEM: 204±21 mg/dl; P < 0.05)). All infusions were done using micro infusion pumps (KD Scientific). Blood glucose concentrations were monitored regularly according to a fixed scheme from tail vein. To estimate insulin-stimulated glucose uptake in WAT, BAT and skeletal muscle, 2-deoxy-D-[1-14C] glucose (10 µCi/mice; PerkinElmer) was administered as a bolus at 95 min after the start of clamp. Blood samples (20ul) were taken at −5, 100, 110, 120, 130, and 140 min of clamp for the measurement of plasma D-[3-3H]-glucose and 2-deoxy-D-[1-14C] glucose concentrations. Steady state was considered achieved during 100–140min, when a fixed glucose-infusion rate maintained the glucose concentration in blood constantly for 40 min (see Supplemental Fig. 12). At the end of the experiment, mice were killed by ketamine/xylazine and WAT, BAT, skeletal muscle and liver were dissected and store at −80°C until further analysis.
The D-[3-3H]-glucose and 2-deoxy-D-[1-14C] glucose concentrations in plasma were measured according to the procedure of “GLUCOSE CLAMPING THE CONSCIOUS MOUSE” from the Vanderbilt-NIDDK Mouse Metabolic Phenotyping Center with some modifications. Briefly, 6 µl of plasma sample mixed with 14 µl saline was treated with 100ul 3N Ba(OH)2 and ZnSO4 (add Ba(OH)2 prior to ZnSO4) and 100 µl of supernatant was pipetted into ascintillation vial and dried in oven overnight; 8 ml of scintillation fluid as added to the dried vial, or to 50 µl non-dry supernatant for measuring radioactivity in a liquid scintillation counter. For measuring 2-deoxy-D-[1-14C] glucose, lysates of adipose tissue and skeletal muscle were processed using a perchloric acid Ba(OH)2/ZnSO4 precipitation46. Glucose uptake into WAT, BAT and skeletal muscle in vivo was calculated based on 2-deoxy-D-[1-14C]-glucose 6-phosphate accumulation and specific activity of 2-deoxy-D-[1-14C]-glucose in serum.
High-fat diet treatment
Wild-type C57BL6 male mice were feed with a high-fat diet (Research Diet: R1245i, 45 kcal% from fat) for 8–16 weeks.
Glucose tolerance tests (GTT)
For GTTs, mice were fasted, but with free access to water, for 5 hours before the procedure. At the start of the procedure, the mice were weighed and a basal glucose levels were measured. The mice were then injected intraperitoneally with glucose (2g/kg body weight) 6 hours after fasting. Glucose levels were measured again at times 15, 30, 60 and 120 minutes after injection.
Insulin tolerance test (ITT)
For ITTs, mice were fasted, but with free access to water, for 4 hours before the procedure. At the start of the procedure, the mice were weighed and a basal glucose levels were measured. The mice were then injected intraperitoneally with human insulin (1U/kg body weight, Humulin, Eli Lilly) 5 hours after fasting. Glucose levels were measured again at times 15, 30 and 60 minutes after injection.
Fasting/refeeding experiment
Mice were fasted, but with free access to water, for 16 hours overnight and then fed with regular chow diet for 4 hours. Mice were sacrificed after they were anaesthetized with ketamine/xylazine and liver and serum were collected for further analysis.
Insulin treatment
In vivo insulin signaling was analyzed 5-min after insulin injection of 1U/kg human insulin via vena cava after mice were fasted for 16 hours overnight and anaesthetized with ketamine/xylazine. Mice were sacrificed and skeletal muscle, eWAT, iWAT, BAT and liver tissues were immediately frozen in liquid nitrogen for further analysis.
Blood Chemistry Analysis
We used commercial ELISA kits to measure circulating insulin and Fst according to the manufacturers’ instructions. Insulin kit (80-INSMSU-E01) was purchased from Alpco Inc. Fst protein levels were measured using the human Follistatin ELISA kit (R&D Systems, DFN00; 22% cross reactivity with mouse Follistatin) following the manufacturer’s protocols with some modification. In brief, we applied 150µl mouse serum (150µl vs 100µl) and increased the incubation time (overnight vs 2 hours). IL6 protein levels were measured using Mouse IL-6 ELISA Kit (Thermo Scientific Pierce, EM2IL6). Blood glucose was measured using a glucose meter (Bayer Contour). Urine ketone bodies were measured using commercial assay kits (Wako USA, 415-73301 and 411-73401). Serum IL1β, IL2, IL4, IL5, KC and MIP1a concentrations were measured using Milliplex MAP mouse magnetic bead panel kit.
Primary hepatocytes isolation
Eight to ten-week-old mice were anesthetized by intraperitoneal injection of ketamine/xylazine (100mg/kg and 10mg/kg body weight). Following anesthesia, the abdominal cavity was opened by incisions with scissors and the vena cava and portal vein were located. A perfusion catheter was placed into the vena cava. Pre-warmed Liver Perfusion Medium (Invitrogen, 17701) (37C) was delivered at 1.6 ml/min for 12 min using a peristaltic pump. An incision was made at the portal vein as an outlet for the perfusion solution. Immediately following the Liver Perfusion Medium, pre-warmed Liver Digest Medium (Invitrogen, 17703) (37°C) was delivered at 1.6 ml/min for 12 min using a peristaltic pump. At the end of the perfusion, liver was dissected and transferred to a Petri dish on ice containing 10 ml of L-15 medium (Invitrogen, 21083) with 10%FBS. After washing three times with Hepatocyte Wash Buffer (Invitrogen, 17704), the primary hepatocytes were re-suspended in William’s E medium (Invitrogen, 12551) containing Percoll beads (Sigma, p4937). After 4 hrs incubation in William’s E medium containing 10%FBS penecillin and streptomycin, the unattached cells were removed, and the dishes were washed with PBS and incubated with maintenance media (William’s E medium containing 100µM Dex, Insulin-Transferrin-Selenium, 2mM glutamine and PenStrep).
3T3-L1 cell culture
3T3-L1 pre-adipocytes obtained from a mycoplasma-free stock were cultured in DMEM/F12 with 10% BCS in 5% CO2. Two days post-confluence, cells were exposed to DMEM/10% FBS with isobutylmethylxanthine (0.5 mM), dexamethasone (1 µM) and insulin (5 µg/ml). After 2 days, cells were maintained in DMEM/10% FBS until ready for treatment at day 7. On day 9, cells were treated with insulin (10nM) for 3min after being maintained in DMEM/5% mouse serum for 24 hours.
Luminex Assays
As described previously47,48, the Irs1 capture antibody (rabbit monoclonal antibody 58-10C-31, Millipore catalog number 05-784R) was coupled to magnetic carboxylated microspheres. The p110 subunit of PI3K was detected with antibodies from Cell Signaling Technology (CST #4249). For Luminex assays, cell lysates (10 µg) or mouse tissue lysates (80 µg) were diluted with Irs1 capture beads (4000 beads/well) into a total volume of 50 µl of phosphoprotein detection wash buffer (Bio-rad) and incubated overnight in 96-well round bottom plates. After washing twice with the same buffer, the beads were incubated with 50 µl of detection antibody for 1 h on a rotary plate shaker (80 rpm). After removal of the biotinylated detection antibody, the beads were incubated with shaking in 25 µl of 1 µg/ml streptavidin-phycoerythrin (Prozyme) for 15 min. All solutions were then removed, and beads were suspended in PBS-BN (Sigma) for analysis in a Luminex FlexMap 3D instrument.
Histological and immunohistochemical analyses
Liver and WAT were fixed in phosphate buffered paraformaldehyde (10%) and embedded in paraffin. H&E staining was conducted by the Pathology Core in Dana-Farber Harvard Cancer Center. Fst immunostaining was conducted as described previously2. To probe Fst in the liver sections, tissues were deparaffinized in xylene and rehydrated in a series of alcohol/water mixtures. Antigen retrieval was performed in 10 mM sodium citrate, pH 6.0 for 10 min. Tissue sections were blocked in 10% normal goat serum, incubated with the primary antibodies (Abcam: 203131) overnight at 4°C, goat anti-rabbit secondary antibody (Molecular Probes, A11012) for 1 hour at room temperature (25°C). Slide mounting was performed using Prolong Antifade Gold with DAPI (Life Technologies, 1652731). Composite images were created from a 10 × 10 array of adjacent non-overlapping 10× magnification images taken using a Zeiss Axiovert LSM 510 microscope.
Nuclear and cytoplasmic protein extraction
The nuclear and cytoplasmic proteins were differentially extracted as directed using a kit from Thermo Scientific (NE-PER Nuclear and Cytoplasmic Extraction Kit, 78835).
G6Pase activity measurement
G6Pase activity assays were performed as previously described49. Briefly, 100 mg liver was homogenized in buffer (Hepes, 5mM; Sucrose, 250 mM) using a dounce homogenizer. Microsomes were prepared the using ultracentrifuge at 100,000×g. The reaction was started by adding microsomes prep into the substrates mix (G6P: 1–5 mM; sodium cacodylate-,183.3 mM; Histone Type-IIA, 10mg/ml).
Bypass surgery samples information
Human serum used in this study was obtained as a part of a larger clinical study approved by both the Boston Children’s Hospital IRB (IRB-P00021478; PI: Nicholas Stylopoulos) and the University of Pittsburgh Medical Center IRB (MOD15090464-02/PRO15090464). Informed and consenting individuals underwent RYGB surgery by the Minimally Invasive Bariatric and General Surgery (MIBGS) group of the University of Pittsburgh (UPMC). Blood was collected by venipuncture at a research visit prior to surgery (baseline) and 6 months after surgery. Individuals taking Metformin or statins were asked to temporarily discontinue these medications starting on the night prior to surgery. Diabetes was defined as either a documented fasting blood glucose level >126 mg/dl or a %HbA1c of 6.5 or higher, or treatment with an anti-diabetic medication. Mean %HbA1c in diabetic individuals was 8.35±1.3.
Adenoviral Vector Preparation
To make FoxO1 and GFP overexpression adenoviruses, mouse FoxO1 and GFP coding sequences were first subcloned into a pShuttle-IRES-hrGFP-2 vector (Agilent) and then transferred to pAdEasy vector (Agilent) as described earlier50. The constructs were digested by enzyme PacI (NEB, R0547) and then transfected into human embryonic kidney (HEK) 293A cells for adenoviral production. To make short hairpin RNA (shRNA) adenovirus to knock down Foxo1, a gene-specific shRNA (top strand: 5’ GAGCGTGCCCTACTTCAAGGA) was designed using an online tool (BLOCK-iT, Life Technologies). The hairpin-encoding oligonucleotides were cloned in vector pENTR/U6 (Life Technologies), which was recombined with pAd/BLOCK-iT vectors. The positive clones were used for transfection of HEK293A cells to make adenoviruses, as previously described51.
AAV virus vector preparation
AAV8.TBG.PI.eGFP (GFPAAV•TBG, Lot: AV-8-PV0146), AAV8.TBG.PI.Cre (CreAAV•TBG, Lot: AV-8-PV1091) and AAV8.TBG.PI.Null.bGH (nullAAV•TBG, Lot: AV-8-PV0148) were purchased from the Vector Core, University of Pennsylvania. To make overexpression AAV viruses, mouse genes coding sequences were cloned into a pAAV2 backbone with TBG promoter vector and then sent to BCH viral core for AAV2/8 virus production and purification. Mouse genes were cloned by PCR using specific primers listed below:
| Cloning primer | |
|---|---|
| Saa4-F | AATTGGTACCATGAGGCTTGCCACCGTCATTGTC |
| Saa4-B | ACAAGTCGACCTCAGAACTTCTCAGGAAG |
| Inhbe-F | AATTGGTACCATGAAGCTTCCAAAAGCCCAGC |
| Inhbe-B | ACAAGTCGACCCTAGCTGCAGCCACAGGCCT |
| Apol7-F | TACTTACGCGTACCATGGACACCCCAGACAGAGAA |
| Apol7-B | ACAAGTCGACCTCACTTAGTCAGGCTGTCATGA |
| Lect1-F | AATTGGTACCATGACAGAGAACTCAGACAAA |
| Lect1-B | ACAAGTCGACCTTACACCATGCCCAAGATG |
| Serpinb1a-F | AATTGGTACCATGGAGCAGCTGAGTTCAGC |
| Serpinb1a-B | ACAAGTCGACCCTATGGGGAACAAACCCTGCC |
| Serpinb6b-F | AATTGGTACCATGGATCCACTGCTGGAAGCAAAT |
| Serpinb6b-B | ACAAGTCGACCTCATGGGGAGGAGAACCGACCA |
| Fst-F | AATTGGTACCATGGTCTGCGCCAGGCACCAGCCC |
| Fst288-B | ACAAGTCGACCTCAGTTGCAAGATCCAGAATGC |
| Fst315-B | ACAAGTCGACCTTACCACTCTAGAATGGAA |
| Lpl-F | TATCGCTAGCATGGAGAGCAAAGCCCTGCT |
| Lpl-B | TATGCTCGAGTCAGCCAGACTTCTTCAGAG |
| Il1rn-F | TATCGCTAGCATGACAGCAGCACAGGCTGAGGC |
| Il1rn-B | TATGCTCGAGCTATTGGTCTTCCTGGAAGT |
| Igf1-F | ATCAGCTAGCATGACCGCACCTGCAATAAAGAT |
| Igf1-B | ATCACTCGAGCTACATTCTGTAGGTCTTGT |
| Ntf3-F | TACTTACGCGTACCATGGTTACTTCTGCCACGATCT |
| Ntf3-B | ACAAGTCGACCTCATGTTCTTCCAATTTTTC |
| Ghr-F | ATCAGCTAGCATGGATCTTTGTCAGGTCTT |
| Ghr-B | ATCACTCGAGCTACTGCATGATTTTGTT |
| Serpinb6a-F | AATTGGTACCATGGATCCTCTACAGGAAGCA |
| Serpinb6a-B | ACAAGTCGACCTCAGGGAGAGGAGAACCGGC |
| Gpx3-F | TATCGCTAGCATGGCCCGGATCCTCCGGGCATCCT |
| Gpx3-B | TATGCTCGAGTTACTTCCCCCTGGCGCTCAGGG |
| Mpzl1-F | AATTGGTACCATGGCAGAGGCCGTCGGAG |
| Mpzl1-B | TAGCGGCCGCTTAGTCTTTCCGGATGTCCG |
sgFST AAV•TBG
SaCas9 (Cas9 from Staphylococcus aureus) is short enough to be delivered with a U6-driven single guide RNA.Plasmid (pX602-AAV-TBG::NLS-SaCas9-NLS-HA-OLLAS-bGHpA;U6::BsaI-sgRNA, #61593) was purchased from Addgene. To make Fst gene knock down AAV viruses, the sgRNAs target to Fst were designed on the Broad Institute’s website (sgFST-1: GGAGTGCACATTCGTTGCGGT; sgFST-2: GGCACACTCGCTGGCGTATG), purchased from Integrated DNA Technologies, then cloned and packaged in BCH viral core.
shFst AAV•TBG
For preparation of pscAAV-TBG-GFP-shFst, the 377bp CMV promoter of pscAAV-GFP (Addgene #32396) was removed (AvrII-BspeI digest) and replaced with a 706 bp PCR-generated fragment containing the TBG (thyroid binding globulin) promoter and paired upstream alpha micro-globulin enhancers. The resulting plasmid was linearized at its unique NotI site (between eGFP and SV40 polyA sequences), into which was placed a 131 bp, EagI-digested synthetic/PCRed cassette containing a 107 bp miRNA-like hairpin targeting Fst. Design of the cassette was based on previously described miRNA stem-loop sequences and design rules52. The primers of anti-sense Fst sequence are as below:
- TBG-ShFst1-TOP:
- CGTACGCAGGTACCAGGTGATAGCAATGTCAGCAGTGCCTTTGAATGAACAAGAAGAATAAAGTGAAGCCACAGATG
- TBG-ShFst1-BOTTOM:
- GCTCGGCCGAGCTCGTAGAGTATGGTCAACCTTACTTTTTATTCTTCCTGTTCATTCACCATCTGTGGCTTCAC
Adenovirus injection and AAV virus injection
Delivery of adenoviral DNA constructs into mouse liver was achieved by intravenous injection via the tail vein. Mice were administered with 1× 108 particles of adenovirus per gram of body weight in 100–200 microliters saline (0.9% NaCl). Delivery of adeno-associated virus DNA constructs into mouse liver was achieved by intravenous injection via tail vein with 2 × 1011 genome copies (GC) of AAV virus per mice in 100–200 microliters saline (0.9% NaCl).
Western blot analysis
Tissue or cells were homogenized in the lysis buffer (50 mm Hepes, pH 7.5, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1.5 mm MgCl2, 1 mm EGTA, 10 mm sodium pyrophosphate, 100 mm sodium fluoride, and freshly added protease inhibitor cocktail and phosphatase inhibitor cocktail). Protein extracts were resolved on an SDS-PAGE gel and transferred to nitrocellulose membrane (Bio-Rad). Detection of proteins was carried out by incubations with HRP-conjugated secondary antibodies followed by ECL detection reagents. Uncropped images can be found in Supplemental Figure. 10,11.
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed as previously described with minor modifications53,54. Briefly, cells were cross-linked with 1% formaldehyde at room temperature for 15 minutes. The cross-linking was stopped with 125 mM glycine for 5 min at room temperature. Cells were washed with PBS and collected into harvest buffer (100 mM Tris-HCl PH 9.4, 10 mM DTT) and incubated on ice for 10 minutes followed by centrifugation at 2000g for 5 minutes. Cells then were sequentially washed with ice cold PBS, buffer I (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5), and buffer II (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5). Pellets were resuspended in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0 supplemented with protease and phosphatase inhibitor cocktail (Sigma)) and sonicated to reduce the DNA length to 0.3 to 1.5 kb followed by centrifugation at 8000g for 1 minute at 4C. The soluble chromatin were diluted 10-fold in dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0), precleared with sheared salmon sperm DNA and protein A/G sepharose (Santa Cruz Biotechnology) at 4oC for one hour. The amounts of chromatin were measured and equal amounts of chromatin were used in subsequent IPs with 3 µg of control IgG or FoxO1 antibody overnight. After immunoprecipitation, 25 µl protein A/G sepharose were added and the incubation was continued for another two hours. Precipitates were washed sequentially for 5 minutes each in TSE I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.0, 150 mM NaCl) for three times, TSE II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), and TE buffer (10 mM Tris PH 8.0, 1 mM EDTA). Precipitates were then eluded with 1% SDS, 0.1 M NaHCO3. Eluates were heated at 65°C for overnight to reverse the formaldehyde cross-linking, treated by proteinase K, and DNA fragments were purified with a DNA purification Kit (Qiagen). Purified DNAs were amplified and quantified using Power SYBR Green PCR Master Mix (Life Technologies) and promoter specific primers in QuantStudio 6 Flex. Promoter specific primers are listed below:
| Chip-PCR primer | |
|---|---|
| mPck1-chip-F | TGCAGCCAGCAACATATGAA |
| mPck1-chip-R | TGATGCAAACTGCAGGCTCT |
| mGapdh-chip-F | GCCCTTGAGCTAGGACTGG |
| mGapdh-chip-R | AGGGCTGCAGTCCGTATTTA |
| Fst-chip-F1 | CAAGTGCCTTACCTCCCATT |
| Fst -chip-R1 | TTTTTGCACAGCTGTTGGTT |
| Fst -chip-F2 | TCTAAGCCTTCGTGGTTTTCA |
| Fst -chip-R2 | GGTGACCCCAAAATGTGAAG |
| Fst -chip-F3 | CAAGGTGCAGCTGAAAAACA |
| Fst -chip-R3 | GTTTGGCCTTGTGAATACGG |
| Fst -chip-F4 | AATTTGTATGGGTAGGCCAAA |
| Fst -chip-R4 | GGGGTTGCAGTGTTTCAGAC |
| Fst -chip-F5 | CCTGAGAGCTTGTGTGCACTT |
| Fst -chip-R5 | CCAGAGAAACACCCACCTTC |
| Fst -chip-F6 | GTGGGGACATAGGAAGGTGA |
| Fst -chip-R6 | GCCACTTAACGGCAGAATTT |
| Fst -chip-F7 | CAAGGGGGACAGTTTGCTAT |
| Fst -chip-R7 | CTCTTCCCTCCAACGCATTA |
| Fst -chip-F8 | AGGGAGGAAAAAGCCTGAAG |
| Fst -chip-R8 | GGGGCTTCACATCCTTCTTA |
| Fst -chip-F9 | TGATTCATGCCCACTTTCAA |
| Fst -chip-R9 | CCGAAATACAGCCGTGCTAA |
| Fst -chip-F10 | TTCGCTGTTTGTTGTCGTTC |
| Fst -chip-R10 | AACGATTACTTCACGGAAAAGG |
| Fst -chip-F11 | GGGGAGAAAAGAAAGGGAGA |
| Fst -chip-R11 | GCTCCACAAGTCAGAAGCAA |
| Fst -chip-F12 | GGGCAGGGGTTCTACTTTTC |
| Fst -chip-R12 | GGTTCCAACATCTTCCGAGA |
| Fst -chip-F13 | TACCAGCCTTGCTGATAGGG |
| Fst -chip-R13 | GAAATTTCCGAGGGCTGCT |
RNA extraction and qPCR analysis
RNA isolation was performed as described previously12. Real time RT-PCR was performed in two steps. First, cDNA was synthesized using a cDNA synthesis kit (Bio-Rad). Second, cDNA was analyzed by real time PCR using the SYBR Green Master Mix (Applied Biosystems). Specific PCR primers are listed below:
| Q-PCR primer | |
|---|---|
| Saa4-F | AGGTTGTCCCGATAGGCTCT |
| Saa4-B | CCACCGTCATTGTCCTCTG |
| Inhbe-F | CTGGCAACCGAGAGAAAGTC |
| Inhbe-B | TACAGGTGGTGGGACCAAAG |
| Apol7-F | GGTAAGTGTGGTGTCTGCTGA |
| Apol7-B | ATAAAGAAAGAAGT |
| Lect1-F | CCTTCACCTGTGCTTTGATG |
| Lect1-B | ACCTTTAAAATGGGAAGCGG |
| Serpinb1a-F | ACTCTGAAACTTGCTAACAGAC |
| Serpinb1a-B | AGGCATGCAGAAAATCCACA |
| Serpinb6b-F | TCACCCAGTGTTTTCAAAAGG |
| Serpinb6b-B | GCTTGCTGACAGCCTGAAC |
| Fst288/315-F | TGCTCTTCTGGCGTGCTTCT |
| Fst288-B | GGAAAGCTGTAGTCCTGGTCTT |
| Fst315-B | AAATTGCAGAAAGTTCTGA |
| Lpl-F | TCAGCTGTGTCTTCAGGGGT |
| Lpl-B | TTTGGCTCCAGAGTTTGACC |
| Il1rn-F | TCCCAGATTCTGAAGGCTTG |
| Il1rn-B | GTGAGACGTTGGAAGGCAGT |
| Igf1-F | CTTCAGTTCGTGTGTGGA |
| Igf1-B | CACAGTACATCTCCAGTC |
| Fst-total-F | TGCTGCAACACTCTTCCTTG |
| Fst-total-B | TGCTGCTACTCTGCCAGTTC |
| Ghr-B | GTACTATGCTAAAAGGGAAA |
| Ghr-B | GTTGAAGTAACAGCTGTTTTT |
| Gpx3-F | GATGGTGAGGGCTCCATACT |
| Gpx3-B | CATCCTGCCTTCTGTCCCT |
| Serpinb6a | TAGCACCCTGAAAGTCCAGC |
| Serpinb6a | TTGCTCAGAACAGCCAACAG |
| Mpzl1-F | ATGTGCAAGTCAGCTTCCCT |
| Mpzl1-B | TGGCTGTGGTCGGTGCTA |
| Ntf3-F | GCCACGGAGATAAGCAAGAA |
| Ntf3-B | ACGGATGCCATGGTTACTTC |
| Foxo1-F | ACGAGTGGATGGTGAAGAGC |
| Foxo1-B | TGCTGTGAAGGGACAGATTG |
| Igfbp1-F | TCCTCTGTCATCTCTGGGCT |
| Igfbp1-B | TGTGTACCAGAACCTGCTGC |
| Pck1-F | AGAAGGAGTACCCATTGAG |
| Pck1-B | CTGAGGGCTTCATAGACA |
| G6pc-F | ATG GTCACTTCTACTCTTGC |
| G6pc-B | CAAGATGACGTTCAAACAC |
| Ppargc1a-F | TGAAGTGGTGTAGCGACCAA |
| Ppargc1a-B | CGCTAGCAAGTTTGCCTCAT |
| Gck-F | TCACTGGCTGACTTGGCTTGCA |
| Gck-B | AAGGACAGGGACCTGGGTTCCA |
| Srebf1-F | GGA GCCATGGATTGCACATT |
| Srebf1-B | CCTGTCTCACCCCCAGCATA |
| Fasn-F | CTGCCACAACTCTGAGGACA |
| Fasn-B | TTCGTACCTCCTTGGCAAAC |
| Acaca-F | GGCCAGTGCTATGCTGAGAT |
| Acaca-B | TATCACACAGCCAGGGTCAA |
| Srebf2-F | ACAGCCGCCCTTCAAGTG |
| Srebf2-B | TCACAGGCATTGTGGTCAGAA |
| Hmgcs1-F | GCATTCAAAGGAAGTGACCC |
| Hmgcs1-B | CGTTCTTCTTCCAGGGTCTG |
| Hmgcr-F | AGAGCGAGTGCATTAGCAAAG |
| Hmgcr-B | TCACAAGGCATTCCACAAGA |
| Pnpla2-F | TCA TCA GGT CCT TTG GTT CC |
| Pnpla2-B | TCC GAG AGA TGT GCA AAC AG |
| Lipe-F | TCT CGT TGC GTT TGT AGT GC |
| Lipe-B | ACG CTA CAC AAA GGC TGC TT |
| Hmgcs2-F | GGT GGA TGG GAA GCT GTC TA |
| Hmgcs2-B | ACA TCA TCG AGG GTG AAA GG |
Affymetrix MoGene-2_0-st arrays
RNA was extracted using TRI reagent (Sigma) and then subjected to a cleanup step using an RNeasy Mini Kit with on-column DNase digestion (Qiagen). Gene expression profiles were obtained using Affymetrix MoGene-2_0-st Chip in Genetics Core facility of Boston Children’s Hospital.
Statistical Analysis
Two-tailed unpaired Student’s t-tests were used to assess statistical significance between two groups. Multiple groups or treatment were compared using one-way ANOVA or two-way ANOVA, or Kruskal-Wallis test as appropriate to the design and/or data distribution. When ANOVA indicated a significant difference among the groups, differences between groups were identified using a stricter criterion for significance according to the Bonferroni rule. Due to non-normal distribution of data in Fig 4b,d,e, data are reported as the median±95%CI. No data were considered outliers by testing or arbitrarily excluded. The sample sizes (indicated throughout) were chosen to equal to three or greater for in vitro studies and five or greater (e.g. mice per group) for in vivo experiments. Data collection for screening of putative hepatokines (Fig. 2) in WT mice was performed in blind fashion and unblinded for treatment (hepatokine). Arrays (Supplemental Fig. 2i and Supplemental Fig. 8a) were performed by service for fee that was blind to genotype. GraphPad Prism 7 software was used for all data analysis.
Data Availability and Accession Code
Array Express accession number for data in Fig. 3b,c is #E-MEXP-1649. The data in Supplemental Fig. 2i and Supplemental Fig. 8a have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE111809.
Supplementary Material
Acknowledgments
We thank Sudha Biddinger’s lab for providing the cDNA from LIRKO liver. We thank Evan Rosen’s lab for providing 3T3-L1 cells. We thank Umut Ozcan’s lab for providing pAAV2.TBG.PI vector. We thank Ronald DePinho’s lab for providing floxed FoxO1 mice. This work was supported by NIH grants DK098655 and project 2 GM021700 (MFW); DK108642 (NS); and DK091592, DK107682 and AA024550 (XCD).
Footnotes
AUTHOR CONTRIBUTIONS
M.F.W and R.T designed the research direction, and R.T. performed the majority of the experiments with specialized assistance from C.W., K.D.C., O.S., W.Q., Y.H., J.M., S.L., X.C.D., L.L., M.S. and N.S. All data were analyzed by R.T. and M.F.W. The manuscript was written by R.T. and M.F.W. with assistance from K.D.C.
COMPETING FINANCIAL INTERESTS
MFW is a scientific consultant for Housey Pharmaceutical Research Laboratories.
References
- 1.Perry RJ, et al. Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell. 2015:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin X, Moskophidis D, Mivechi NF. Heat Shock Transcription Factor 1 Is a Key Determinant of HCC Development by Regulating Hepatic Steatosis and Metabolic Syndrome. Cell Metab. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126:12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017;25:777–796. doi: 10.1016/j.cmet.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Stefan N, Haring HU. The metabolically benign and malignant fatty liver. Diabetes. 2011;60:2011–2017. doi: 10.2337/db11-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Ouaamari A, et al. SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell Metab. 2016;23:194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu.Rev.Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 8.Edgerton DS, et al. Insulin's direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. 2017;2:e91863. doi: 10.1172/jci.insight.91863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer C, et al. Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. American journal of physiology. Endocrinology and metabolism. 2002;282:E428–434. doi: 10.1152/ajpendo.00116.2001. [DOI] [PubMed] [Google Scholar]
- 10.White MF, Copps K. The Mechanisms of Insulin Action. In: Jameson JL, DeGroot LJ, editors. Endocrinology. Vol. 1. Elsevier; Philadelphia: 2016. pp. 556–585. [Google Scholar]
- 11.Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab. 2017;28:497–505. doi: 10.1016/j.tem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong XC, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. doi: 10.1016/j.cmet.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng Z, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat.Med. 2009;15:1307–1311. doi: 10.1038/nm.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo S, et al. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol.Cell Biol. 2009;29:5070–5083. doi: 10.1128/MCB.00138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Sullivan I, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu M, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Titchenell PM, et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefan N, Haring HU. The role of hepatokines in metabolism. Nat Rev Endocrinol. 2013;9:144–152. doi: 10.1038/nrendo.2012.258. [DOI] [PubMed] [Google Scholar]
- 20.Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. The international journal of biochemistry & cell biology. 2013;45:2333–2347. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Hansen JS, Plomgaard P. Circulating follistatin in relation to energy metabolism. Mol Cell Endocrinol. 2016;433:87–93. doi: 10.1016/j.mce.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Bluher M, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev.Cell. 2002;3:25–38. doi: 10.1016/s1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- 23.Shearin AL, Monks BR, Seale P, Birnbaum MJ. Lack of AKT in adipocytes causes severe lipodystrophy. Mol Metab. 2016;5:472–479. doi: 10.1016/j.molmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, et al. Inhibition of TNF-alpha improves the bladder dysfunction that is associated with type 2 diabetes. Diabetes. 2012;61:2134–2145. doi: 10.2337/db11-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen JS, et al. Circulating Follistatin Is Liver-Derived and Regulated by the Glucagon-to-Insulin Ratio. J Clin Endocrinol Metab. 2016;101:550–560. doi: 10.1210/jc.2015-3668. [DOI] [PubMed] [Google Scholar]
- 26.Hansen J, et al. Plasma follistatin is elevated in patients with type 2 diabetes: relationship to hyperglycemia, hyperinsulinemia, and systemic low-grade inflammation. Diabetes Metab Res Rev. 2013;29:463–472. doi: 10.1002/dmrr.2415. [DOI] [PubMed] [Google Scholar]
- 27.Brown ML, Schneyer AL. Emerging roles for the TGFbeta family in pancreatic beta-cell homeostasis. Trends Endocrinol Metab. 2010;21:441–448. doi: 10.1016/j.tem.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kucejova B, et al. Hepatic mTORC1 Opposes Impaired Insulin Action to Control Mitochondrial Metabolism in Obesity. Cell reports. 2016 doi: 10.1016/j.celrep.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 30.Walton RG, et al. Increasing adipocyte lipoprotein lipase improves glucose metabolism in high fat diet-induced obesity. The Journal of biological chemistry. 2015;290:11547–11556. doi: 10.1074/jbc.M114.628487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JK, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A. 2001;98:7522–7527. doi: 10.1073/pnas.121164498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang W, et al. Integrated Regulation of Hepatic Lipid and Glucose Metabolism by Adipose Triacylglycerol Lipase and FoxO Proteins. Cell reports. 2016;15:349–359. doi: 10.1016/j.celrep.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneyer AL, Wang Q, Sidis Y, Sluss PM. Differential distribution of follistatin isoforms: application of a new FS315-specific immunoassay. J Clin Endocrinol Metab. 2004;89:5067–5075. doi: 10.1210/jc.2004-0162. [DOI] [PubMed] [Google Scholar]
- 34.Brown ML, et al. Follistatin and follistatin like-3 differentially regulate adiposity and glucose homeostasis. Obesity. 2011;19:1940–1949. doi: 10.1038/oby.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueno N, et al. Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci U S A. 1987;84:8282–8286. doi: 10.1073/pnas.84.23.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samanta D, Datta PK. Alterations in the Smad pathway in human cancers. Front Biosci (Landmark Ed) 2012;17:1281–1293. doi: 10.2741/3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 38.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidis Y, et al. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology. 2006;147:3586–3597. doi: 10.1210/en.2006-0089. [DOI] [PubMed] [Google Scholar]
- 40.Lambert-Messerlian GM, et al. Inhibins and activins in human fetal abnormalities. Mol Cell Endocrinol. 2004;225:101–108. doi: 10.1016/j.mce.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 41.Mukherjee A, et al. FSTL3 deletion reveals roles for TGF-beta family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci U S A. 2007;104:1348–1353. doi: 10.1073/pnas.0607966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo T, et al. Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. eLife. 2014;3:e03245. doi: 10.7554/eLife.03245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Modica S, et al. Bmp4 Promotes a Brown to White-like Adipocyte Shift. Cell reports. 2016;16:2243–2258. doi: 10.1016/j.celrep.2016.07.048. [DOI] [PubMed] [Google Scholar]
- 44.Wang Q, Guo T, Portas J, McPherron AC. A soluble activin receptor type IIB does not improve blood glucose in streptozotocin-treated mice. International journal of biological sciences. 2015;11:199–208. doi: 10.7150/ijbs.10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hansen JS, et al. Exercise-Induced Secretion of FGF21 and Follistatin Are Blocked by Pancreatic Clamp and Impaired in Type 2 Diabetes. J Clin Endocrinol Metab. 2016;101:2816–2825. doi: 10.1210/jc.2016-1681. [DOI] [PubMed] [Google Scholar]
- 46.Ferre P, Leturque A, Burnol AF, Penicaud L, Girard J. A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem J. 1985;228:103–110. doi: 10.1042/bj2280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hancer NJ, et al. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. Journal of Biological Chemistry. 2014;289:12467–12484. doi: 10.1074/jbc.M114.554162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Copps KD, Hancer NJ, Qiu W, White MF. Serine 302 Phosphorylation of Mouse Insulin Receptor Substrate 1 (Irs1) Is Dispensable For Normal Insulin Signaling and Feedback Regulation by Hepatic S6 Kinase. Journal of Biological Chemistry. 2016 doi: 10.1074/jbc.M116.714915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu A, Romero R, Petty HR. An enzymatic fluorimetric assay for glucose-6-phosphate: application in an in vitro Warburg-like effect. Anal Biochem. 2009;388:97–101. doi: 10.1016/j.ab.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tao R, et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J.Biol.Chem. 2011;286:14681–14690. doi: 10.1074/jbc.M110.201061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiong X, Tao R, DePinho RA, Dong XC. Deletion of hepatic FoxO1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PloS one. 2013;8:e74340–e74340. doi: 10.1371/journal.pone.0074340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Myburgh R, et al. Optimization of Critical Hairpin Features Allows miRNA-based Gene Knockdown Upon Single-copy Transduction. Mol Ther Nucleic Acids. 2014;3:e207. doi: 10.1038/mtna.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miao J, et al. Functional specificities of Brm and Brg-1 Swi/Snf ATPases in the feedback regulation of hepatic bile acid biosynthesis. Mol Cell Biol. 2009;29:6170–6181. doi: 10.1128/MCB.00825-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Array Express accession number for data in Fig. 3b,c is #E-MEXP-1649. The data in Supplemental Fig. 2i and Supplemental Fig. 8a have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE111809.