

Abstract
Systemic Lupus Erythematosus (SLE) is an autoimmune disease characterized by increased type I interferons (IFNs), autoantibodies, and inflammatory-mediated multi-organ damage. TLR7 activation is an important contributor to SLE pathogenesis, but the mechanisms by which type I IFNs participate in TLR7-driven pathology remain uncertain. In this study, we examined the requirement for type I IFNs in TLR-7 stimulated lupus nephritis. Lupus-prone NZM2328, INZM (which lack a functional type I IFN receptor), and NZM2328 IL-1β−/− mice were treated at 10 weeks of age on the right ear with R848 (TLR7 agonist) or control (DMSO). Autoantibody production and proteinuria were assessed throughout treatment. Multi-organ inflammation was assessed at the time of decline in health. Renal infiltrates and mRNA expression were also examined after 14 days of treatment. Both NZM2328 and INZM mice exhibited a decline in survival after 3–4 weeks of R848 but not vehicle treatment. Development of splenomegaly and liver inflammation were dependent on type I IFN. Interestingly, autoantibody production, early renal infiltration of dendritic cells, upregulation of IL-1β, and lupus nephritis occurred independent of type I IFN signaling. Development of TLR7-driven lupus nephritis was not abolished by the deletion of IL-1β. Thus, while IFNα is sufficient to induce nephritis acceleration, our data emphasize a critical role for IFN-independent signaling in TLR7-mediated lupus nephritis. Further, despite upregulation of IL-1β after TLR7 stimulation, deletion of IL-1β is not sufficient to reduce lupus nephritis development in this model.
Introduction
Systemic lupus erythematosus (SLE) is a devastating autoimmune disease characterized by cycles of disease flare that lead to permanent organ damage(1). Lupus nephritis is a feared complication of SLE; 25% of lupus nephritis patients go on to develop end-stage renal disease within 10 years after renal compromise(2) despite current treatment regimens(3). Triggers that lead to renal disease flares still remain unclear; thus, there is a critical need to identify factors which promote lupus nephritis in order to develop novel targets and therapies.
Polymorphisms in toll-like receptor 7 (TLR7) are linked with the development of lupus(4, 5). Murine models of lupus have also provided genetic and experimental evidence to support a role for TLR7 activation in the pathogenesis of lupus nephritis(6, 7). Male BXSB mice, which contain an additional copy of TLR7 on the Y chromosome, develop lupus nephritis, whereas female mice are protected(8). Transgenic mice overexpressing TLR 7 also develop lupus (9). Inducible murine lupus models, such as the pristane model, are dependent on TLR7 signaling for lupus pathogenesis (10). Interestingly, even repeated TLR7 agonist epicutaneous application to wild-type mice leads to development of lupus-like characteristics including mild lupus nephritis(11). Despite these supportive data, the mechanisms by which TLR7 signaling leads to nephritis remain unknown.
TLR7 activation can result in both type I IFN production and NFκB activation in various cell populations such as dendritic cells, monocytes, macrophages, and B cells. Type I IFNs, including IFNα, can promote lupus development(12–14) and are sufficient to induce acceleration of nephritis in lupus prone mice(15). In addition, various genetic and inducible models of lupus are protected by deletion of type I IFN signaling(12, 16). Given the importance of type I IFN in lupus, we hypothesized that TLR7-induction of type I IFNs was responsible for its effects. To test this, we examined an inducible model of lupus flare via epicutaneous stimulation of TLR7 in lupus-prone mice. The NZM2328 mouse strain was selected for this study because they have spontaneous development of lupus characteristics including autoantibody production around 14 weeks of age, splenomegaly, and lupus nephritis around 35 weeks of age(17). Surprisingly, we found that TLR7 treatment of young mice accelerated lupus nephritis development, including renal infiltration of innate immune populations, immune complex deposition, and IL-1β upregulation, independently of type I IFNs. Other manifestations, such as liver inflammation and splenomegaly required type I IFNs to occur. Further, we demonstrated that a novel IL-1β−/− mouse on the NZM2328 background was also susceptible to TLR7-accelerated nephritis. These data support IFN-independent immune activation in the presence of robust TLR7 stimulation as sufficient for acceleration of lupus nephritis.
Methods and Materials
Mice
All mice were bred and housed at the University of Michigan in specific pathogen-free housing. All mice were treated according to our University of Michigan IACUC-approved protocol. NZM2328 mice and INZM (lack the α-chain of the IFNα/β receptor) mice were a kind gift of Dr. Chaim Jacob, University of Southern California(12). NZM2328 IL-1β −/− mice were generated through the University of Michigan Transgenic Animal Model Core via CRISPR-Cas9 technology using IL-1β CRISPR/Cas9 KO Plasmid sc-421097 from Santa Cruz Biotechnology (Santa Cruz, CA). An 8-bp deletion resulting in a frame shift mutation was confirmed via Sanger sequencing through the University of Michigan DNA sequencing core (Supplemental Figure 1). NZM2328 IL-1β−/− mice were backcrossed onto the NZM2328 background for three generations to eliminate off-target effects of using the CRISPR system followed by heterozygote crossing to develop mice homozygous for the IL-1β deletion. Female 10-week old mice were utilized for all experiments. The NZM mouse model has minimal autoantibody production and no nephritis at this age(12). Males were not used as they do not achieve a lupus phenotype in this model, and lupus is predominantly a female disease(18).
TLR7 cutaneous stimulation
Female 10-week old mice were treated via epicutaneous application of 100μg of the TLR7 agonist R848 (Enzo Life Science) dissolved in 8 μL DMSO, or DMSO alone as a control, to the right ear three times weekly until euthanasia. DMSO and R848 treated mice were housed in separate cages to avoid cross-contamination. For some studies, mice were treated for only 2 weeks before euthanasia (for evaluation pre-proteinuria); others were treated until they developed proteinuria or became moribund (for survival studies) followed by euthanasia. Control mice were harvested with their paired R848-treated littermates.
Analysis of Anti-dsDNA and IgG serum levels
Serum was collected every 2 weeks. Anti-dsDNA and IgG levels were analyzed with the use of ELISA kits (Alpha Diagnostic International, San Antonio, TX and Innovative Research Inc., Novi, MI).
Analysis of proteinuria
Urine was collected weekly and protein was screened via dipstick followed by albumin measurements via Albuwell kits (Exocell, Philadelphia, PA) and total creatinine (Cr) via commercial kit (Bioassay Systems, Hayward, CA). Urinary protein excretion was represented by the albumin:Cr ratio.
Renal histopathology and immune complex deposition scoring
Glomerular inflammation (activity index) and scarring (chronicity index) of murine kidneys were quantified in a blinded fashion (by JBH) on perfused kidneys fixed in 10% formalin followed by 3μm sectioning and Periodic Acid Schiff (PAS) staining as previously described by us and others(19, 20). In brief, a semi-quantitative scoring system (0, no involvement; 0.5, minimal involvement of 10%; 1, mild involvement (10–30% section); 2, moderate involvement (31–60% of section); and 3, severe (60% of section)) was used to assess 13 different parameters of activity and chronicity (mesangial hypercellularity, mesangial deposits, mesangial sclerosis, endocapillary cellular infiltrate, subepithelial and subendothelial deposits, capillary thrombi, capillary sclerosis, cellular or organized crescents, synechiae, tubular atrophy, and interstitial fibrosis). The chronicity and activity index was generated by compiling the scores from groups of related parameters (for activity: mesangial hypercellularity, mesangial deposits, and endocapillary cellular infiltrate; for chronicity: interstitial fibrosis, tubular atrophy, synechiae, organized crescents, and capillary sclerosis). Glomerular immune complexes were quantified by immunofluorescence microscopy as previously described(19). Briefly, 6μm frozen kidney sections were stained for 1hr at 4°C with Texas-red-conjugated anti-mouse IgG (Sigma) and FITC-conjugated anti-C3 (ICL, Portland, OR) followed by Hoechst (Invitrogen, Eugene, OR) counterstain to stain DNA. Quantification of immune complex staining in 8 glomeruli per mouse was performed at the Center for Live Cell Imaging at the University of Michigan using Metamorph v7.0.6 to calculate the mean fluorescence in a defined area for each stain. Glomeruli were identified based on Dapi staining and outlined to define the area for analysis. Both FITC and Texas Red staining were calculated and shown as staining per glomerulus.
Liver scoring
Inflammation of murine livers were quantified blindly by JBH on livers fixed in 10% formalin followed by 3μm sectioning and Periodic Acid Schiff (PAS) staining as described by others (21). In brief, a scoring system (0: <0.5 inflammatory foci/field; 1, 0: 5–1.0 foci/field; 2: 1.0–2.0 foci/field; 3: >2.0 foci/field) was used.
Microscopy
Images of H&E stained skin sections and PAS-stained kidney and liver sections were captured using an Olympus BX41 microscope with a 100x objective (total magnification=1000x). Images of kidney immune complex staining were captured at the Center for Live Cell Imaging (CLCI) at the University of Michigan Medical School using an Olympus IX70 inverted microscope (Olympus; Center Valley PA) with a 40x objective.
Flow cytometry
Following euthanasia, the lymph nodes (draining lymph node (dLN) from the cervical chain on the treated side and non-draining lymph node from the inguinal chain) and spleen were removed, teased apart and passed through a 70μm filter to generate single cell suspensions. One of the kidneys was also removed, minced and digested as previously described(19) with 0.1 mg/mL Liberase (Roche), 200 U/mL DNAse (Roche) and 2.4 mM CaCl2 in DMEM (Invitrogen) at 37 °C in a humidified incubator for 1 h. The tissue was then passed through a 70 μm cell strainer and RBCs were lysed with multi-species RBC lysis buffer (eBioscience). Live cells were counted via trypan blue exclusion. The cells were incubated in flow block (1% horse serum and 1% bovine serum albumin in PBS) for 1hr, then stained for 1 h on ice using the following antibodies: CD3-APC, F4/80-PE, CD11b-APC, CD11c-PE, CD4 APC/Cy7, CD8 APC/Cy7, B220 APC (BioLegend, San Diego, CA), SA-Qdot 605 (Invitrogen) followed in some cases by permeabilization and intracellular staining for IgH+L A488 (Southern Biotech, Birmingham, AL). The flow data was collected using a BD LSR II flow cytometer and analyzed via FlowJo VX.0.7 (Tree Star). The following gating strategy was used: live cells were gated for: T cells: CD3+, Macrophages: CDllcintCDllb+F480+; Dendritic Cells: CDllc+CDllb+F480−; B cells: CD4−CD8−IgH+L+B220+; and Plasma cells: CD4−CD8−IgH+LhiB220int-low.
Bone Marrow Derived Macrophages
Bone marrow derived macrophages (BMDM) were generated as previously described(22). Bone Marrow was flushed from the tibiae and femurs of 10 weeks old NZM2328 mice and NZM IL1β −/− mice and plated in macrophage differentiation media (59% IMDM, 10% FBS, 30% L-cell supernatant, and 1% penicillin/streptomycin) for 7 days at 37°C in 5% CO2. BMDM were then plated 2×106 cells per 6 well for detection of IL-1β activation.
IL-1β quantification
BMDM were incubated with or without 1ug LPS for 4 hours followed by 1-hour incubation with or without 5mM ATP to activate the inflammasome. Secreted IL-1β was measured via ELISA (DuoSet ELISA kit, R&D, Abingdon, UK). Cells were lysed with RIPA +PI for 10mins on ice. 10μg of each lysate was then run on a 10%SDS-polyacrylamide gel and blotted on nitrocellulose membranes (GE Healthcare). The membranes were blocked in 4% milk/TBST, followed by probing with anti-murine IL-1β Ab 1:500(Cell Signaling Technology) overnight. Then the membrane was incubated with anti-rabbit IgG-HRP 1:1,000 (Abcam). The pro-IL-1β band was detected using Western Bright Quantum (Advansta) and imaged using Omega Lum C system (Aplegen).
RTqPCR analysis
Kidney tissue was homogenized in TriPur (Roche) and RNA was purified via Direct-zol mini RNA prep (Zymo). RNA (100 ng) was reverse-transcribed into cDNA and quantitative real-time PCR analysis was completed on an ABI PRISM 7900HT (Applied Biosystems) by the DNA sequencing core at the University of Michigan. The primers used were as follows (all listed 5′→3′): C-C Motif Chemokine Ligand 2 (ccl2) AGGTCCCTGTCATGCTTCTG (forward), GGATCATCTTGCTGGTGAAT (reverse); Myxovirus (influenza virus) resistance 1 (mx1) GATCCGACTTCACTTCCAGATGG (forward), CATCTCAGTGGTAGTCAACCC(reverse); C-X-C Motif Chemokine Ligand 10(cxcl10) ATCATCCCTGCGAGCCTAT (forward), ATTCTTGCTTCGGCAGTTAC (reverse) chemokine (C–C motif) ligand 5 (ccl5) CAATCTTGCAGTCGTGTTTG (forward), GGAGTGGGAGTAGGGGATTA (reverse); chemokine (C–C motif) ligand 4 (ccl4) AGCAACACCATGAAGCTCTG (forward), CTGTCTGCCTCTTTTGGTCA (reverse); C-X-C motif chemokine 13 (cxcl13) AGAGGTTTGCGAGATGGACT (forward), GAGCCTGGACCTTTAAGCTG (reverse); β-Actin TGGAATCCTGTGGCATCCTGAAAC (forward), TAAAACGCAGCTCAGTAACAGTCCG (reverse); Tumor necrosis factor alpha (tnf) CCCACTCTGACCCCTTTACT (forward), TTTGAGTCCTTGATGGTGGT (reverse); il1b CCCTGCAGCTGGAGAGTGTGGA (forward), CTGAGCGACCTGT-CTTGGCCG (reverse); il6 TAGTCCTTCCTACCCCAATTTCC (forward), TTGGTCCTTAGCCACTCCTTC (reverse). Gene expression was calculated relative to B actin (2^βactin/2^gene).
Statistical analysis
All statistics were completed using GraphPad Prism 6.0. For figures where >2 comparisons were made, ANOVA testing was used. Tukey’s multiple comparison test was used for normally distributed data and a Kruskal-Wallace/Dunn’s multiple comparison test was used for non-parametric data. For figures where comparisons between two groups was completed, a two-tailed student’s t-test for normally distributed data was used. Welch’s correction was applied when required for significant differences in variances. For non-normally distributed data, a Mann–Whitney was used. Survival analysis following R848 application was completed via Log-rank test. Pearson correlation was used for comparison of renal activity scores and albumin/Cr values.
Results
TLR7 Epicutaneous stimulation leads to an interferon-independent decline in survival
TLR7 stimulation in the skin may serve an important role in lupus flare induction (11, 23); thus, we first examined the effects of cutaneous TLR7 stimulation on young, pre-autoimmune NZM2328 lupus-prone mice. These mice typically develop lupus nephritis around 35 weeks of age followed by a decline in survival (24). Intriguingly, R848 topical treatment of 10-week old NZM2328 mice led to a rapid decline in survival when compared to DMSO-treated controls (Figure 1A). This decline was marked by a nephrotic appearance including ascites, edema, and lethargy. Given that TLR7 stimulation is associated with increased IFN signaling, we hypothesized that this accelerated death was driven by type- I IFNs. Thus, we next examined the effects of R848 on NZM2328 mice that are deficient in the type I IFN receptor (INZM). These mice are protected from naturally-occurring development of lupus for up to 2 years of life (12). To our surprise, treatment of these mice with R848 led to edema, ascites and a rapid decline in survival in a time frame similar to NZM2328 mice (Figure 1). No difference in the survival curves between NZM and INZM mice were noted via Log-rank test (p=0.723). Median survival was 30 days for both strains. These data suggest that despite absent type I IFN signaling, both INZM and NZM2328 mice exhibit a rapid decline in survival following epidermal TLR7 activation.
Figure 1. Epicutaneous TLR 7 stimulation leads to type I IFN-independent accelerated decline in survival in lupus-prone mice.
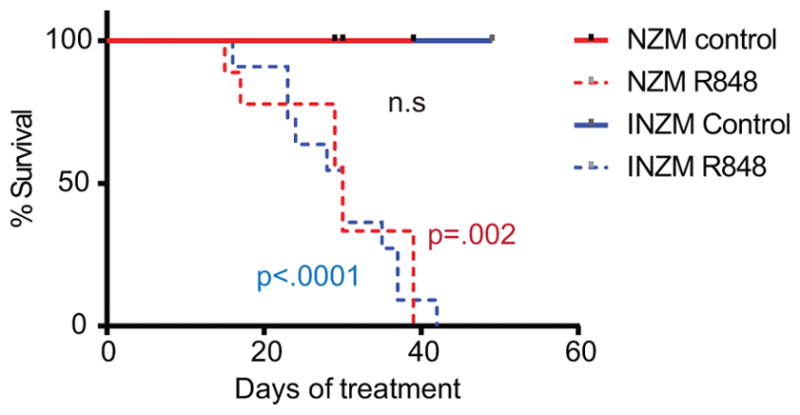
10-week-old NZM2328 and INZM mice were treated with 100μg of the TLR7 agonist R848 or control (DMSO) three times weekly. Survival curve for NZM2328 and INZM mice is shown. n=12 each for DMSO and R848 treated NZM mice. n=11 for DMSO and n=12 for R848 treated INZM mice. The p value in blue shows the difference in the survival curves between INZM DMSO vs. INZM R848 treated mice. The p value in red shows the difference in the survival curves between NZM DMSO vs. NZM R848 treated mice. The p value in black shows the difference in the survival curves between NZM R848 and INZM R848 treated mice
TLR7 epicutaneous stimulation leads to accelerated development of murine lupus
We next examined the development of systemic lupus characteristics following R848 stimulation. Using our assay, dsDNA antibodies typically rise to about 100,000U/ml during nephritis onset in NZM 2328 mice (19). Importantly, while only NZM2328 treated with R848 demonstrated significant acceleration of total IgG production, both NZM2328 and INZM mice demonstrated increased dsDNA antibodies in the serum (Figure 2A and B); however, the rise dsDNA antibodies in the serum was greater in NZM vs. INZM mice (p=.0167 NZM vs. INZM at 2 weeks). This suggests that autoantibody production following TLR7 epicutaneous stimulation is enhanced by but not dependent on type I IFN signaling Consistent with previous literature (12, 25), splenomegaly was detected following R848 stimulation in NZM2328 but not INZM mice, supporting a role for IFNs in TLR7-driven splenomegaly (Figure 2C). An increase in total splenic cells following R848 stimulation was detected in only NZM2328 mice; analysis of cell subsets identified no significant changes in T cells, dendritic cells, or macrophages (Figure 2D and E). Further, NZM2328 mice treated with R848 demonstrated accelerated development of liver inflammation as indicated by necrotic hepatocytes and focal portal inflammation. INZM mice treated with R848 did not develop liver inflammation, indicating that type I IFNs are required for this manifestation following TLR7 activation (Figure 2F and G).
Figure 2. TLR 7 stimulation leads to IFN-independent elevated autoantibody production and IFN dependent splenomegaly and liver inflammation.

10 week-old NZM2328 and INZM mice treated with R848 or DMSO control were analyzed. A. Total IgG in serum of NZM2328 and INZM mice after 2 weeks of treatment. Each dot represents an individual mouse. B. dsDNA IgG in serum at 0 weeks, 2 weeks, and 4 weeks of treatment. n=13 NZM R848; n=14 NZM DMSO; n= 9 INZM DMSO; n=11 INZM R848 C. Spleen weight of NZM2328 and INZM mice from survival studies. Spleens were harvested when mice were moribund (around 20–40 days of treatment). Littermate DMSO controls were harvested at the same times as the moribund mice. Each dot represents an individual mouse. Representative photographs of DMSO and R848 NZM2328 spleens shown in inset. D. Immune cell populations in the spleen were evaluated by flow cytometry after 2 weeks of R848 or DMSO treatment. n=13 NZM R848; n=11 NZM control; n=8 INZM control; n=9 INZM R848. (E and F) 10 week- old NZM2328 and INZM mice treated with R848 or DMSO control were treated until moribund and analyzed for development of liver inflammation. E. Representative photo of the portal vein. F. Graph represents liver inflammation scoring of NZM2328 and INZM mice. Each symbol represents one mouse.
We next examined changes in B cells and antibody (Ab)-secreting cells in the spleen and lymph nodes. As shown in Figure 3, both total B cells (B220+) and Ab-secreting cells (IgH+Lhigh) increased in the spleen of NZM2328 mice but only Ab-secreting cells increased in INZM mice post R848 stimulation (Figure 3A–C), suggesting that amplification of splenic antibody production is independent of type I IFNs. We then examined the lymph node populations to determine if IFN-independent B cell activation was also occurring there. As shown in Figure 3D, stimulation with R848 resulted in increased total number of cells in the cervical (draining) lymph nodes in both NZM2328 and INZM mice. Contrarily, increased cell numbers were noted in inguinal (non-draining) lymph nodes in only NZM mice after R848 treatment. The total number of B cells (B220+) in both NZM2328 and INZM were increased in draining but not the non-draining lymph nodes following R848 stimulation (Figure 3E). NZM2328 showed an increase in the secreting cell population in the draining and non-draining lymph node, but INZM only demonstrated an increase in the draining lymph node (Figure 3F). Of note, the overall numbers of B cells and Ab-secreting cells were significantly fewer in the INZM mice. These data indicate that TLR7 stimulation amplifies local B cell and Ab production responses in the absence of type I IFN signaling, but the responses may be diminished. However, systemic amplification of Ab-secreting cells in the non-draining lymph nodes required the presence of type I IFN signaling. Together, this suggests that the accelerated autoantibody production seen post TLR7 cutaneous stimulation is partially type I IFN-independent and that the Ab-secreting cells in the dLN and spleen may serve as a site for autoantibody production in this model.
Figure 3. TLR 7 stimulation leads to IFN-independent increases in secreting cells in the draining lymph nodes and the spleen.

Immune cell populations in the spleen were evaluated by flow cytometry after 2 weeks of R848 or DMSO treatment. n= 5 NZM DMSO; n= 5 NZM R848; n=5 INZM DMSO; n=5 INZM R848. A. Gating strategy for Ab-secreting cells. B, C. Graphs displaying changes in B cells: CD4−CD8−IgH+IgL+B220+ (shown in B) and Ab-secreting cells: CD4−CD8− IgH+IgLhigh (shown in C). D. Total number of cells isolated from indicated lymph nodes for DMSO or R848 treated mice. E, F. Graphs displaying changes in B cells: CD4−CD8−IgH+IgL+B220+ (shown in E) and Ab-secreting cells: CD4−CD8− IgH+IgLhigh (shown in F) for draining (cervical) and non-draining (inguinal) LN. Data is displayed as mean ± SD. Each symbol represents one mouse. LN=lymph node.
TLR7 epicutaneous stimulation leads to accelerated development of lupus nephritis in a type I IFN-independent manner
Past work has shown type I IFNs are sufficient to induce renal flares in lupus-prone mice and are required for lupus nephritis development in several murine models (12, 15, 26, 27). We next examined whether TLR7 epicutaneous stimulation led to accelerated lupus nephritis development and whether it was dependent on type I IFNs. Surprisingly, both NZM2328 and INZM mice treated with R848 demonstrated a rise in urinary albumin:Cr ratio supportive of glomerular damage (Figure 4A and B). Histopathologic scoring demonstrated a significant increase in renal activity score when mice were treated with R848 until moribund (Figure 4C and D). A strong positive correlation was detected between renal activity score and urinary alb/cr ratio for both NZM2328 and INZM mice (Figure 4E), which supports renal inflammation in R848 treated mice. No significant increase in renal chronicity index score for prolonged exposure in NZM2328 and INZM mice was detected (Figure 4F). As immune complex deposition is a hallmark of lupus nephritis, this was also assessed. R848 treatment of the NZM2328 and INZM mice led to a significant increase in both IgG and C3 deposition within the kidney (Figure 5A–C). In order to examine whether TLR7-induced lupus created similar transcriptional changes to naturally occurring lupus nephritis found in older, untreated NZM 2328 mice, we examined transcriptional signatures of the kidneys of both. As shown in Supplemental Figure 2, identical upregulation of various inflammatory and type-I IFN associated genes were noted in both R848-induced and naturally occurring nephritis. Together, these parameters support development of accelerated lupus nephritis following cutaneous stimulation with a TLR7 agonist in a type I interferon independent manner.
Figure 4. TLR 7- mediated lupus nephritis occurs in an interferon-independent manner.

10 week- old NZM2328 and INZM mice treated with R848 or DMSO control were analyzed for development of lupus nephritis. A. Urine alb/cr ratio was measured serially in NZM2328 treated mice n=12 NZM R848; n=15 NZM DMSO B. Urine alb/cr ratio was measured serially in INZM treated mice. n=5 INZM DMSO; n=6 INZM R848 C. Representative photo of the glomeruli in the kidney of NZM2328 and INZM mice following treatment until moribund. Littermate control mice were harvested at the time of illness in R848 treated mice. Scale bar equals 20um. D. Renal activity score for NZM2328 and INZM mice after 2 weeks of treatment or when moribund from R848 treatment (long-term treatment). E. The moribund renal activity score for NZM2328 and INZM mice treated with R848 and DMSO was plotted versus the alb/cr ratio at euthanasia and analyzed via Pearson correlation. F. Renal Chronicity index for NZM2328 and INZM mice after 2 weeks of treatment and when moribund (long-term treatment).
Figure 5. Immune complex deposition in the kidney is interferon-independent.

10 week- old NZM2328 and INZM mice treated with R848 or DMSO until moribund and were analyzed for immune complex deposition. A. Representative immunofluorescence microscopy of glomeruli (outlined by white dashed line). Texas Red- IgG, Green- C3, Blue- DAPI. (B–C). Quantification of immune complex staining/area was completed. Littermate DMSO controls were harvested when littermates were ill. B. Quantification of IgG/area. C. Quantification of C3/area. Each dot represents the average fluorescence of 8 glomeruli from a single mouse.
TLR7 epicutaneous stimulation leads to upregulation of IL-1β, but nephritis is not dependent on IL-1β
As TLR7-mediated lupus nephritis developed in the absence of type I IFN signaling, we next assessed what other pathways may play a role in TLR7-mediated lupus nephritis. We chose to examine NFκB-regulated cytokines as this transcription factor is also activated downstream of TLR7 with the hypothesis that cytokines upregulated in the kidney in both NZM and INZM mice may be important in TLR7-mediated nephritis. We examined mRNA changes in the kidney of INZM and NZM mice treated for 2 weeks with R848 or DMSO control. Following stimulation, there were significant changes in the expression of NFκB-regulated cytokines tnf, and il1b, (Fig. 6A–C) in the NZM mice treated with R848 vs. control, but there was only a significant upregulation of il1b in INZM mice treated with R848 vs. DMSO (Fig. 6B). These data support a potential common role for IL-1β in TLR7 mediated lupus nephritis flare in NZM and INZM mice.
Figure 6. TLR 7-mediated upregulation of IL-1β is not required for lupus nephritis.

A–C. RNA was isolated from the kidney of NZM or INZM mice treated for two weeks with DMSO control or R848. Real-time PCR was completed using primers for the genes listed. Graphs display the mean±SD for each gene as compared to the average of b-actin. Each dot represents an individual mouse. D.10 week- old NZM IL-1β KO mice were treated with R848 or DMSO control and survival was plotted. n=9 NZM IL-1β −/− DMSO; n= 8 NZM IL-1β −/− R848 E. Anti-dsDNA IgG in serum at 2 weeks of treatment. n=8 NZM IL-1β −/− R848; n=8 NZM IL-1β −/− DMSO. F. Representative photomicrograph of the glomeruli in the kidney of moribund NZM IL-1β −/− mice treated with R848 or DMSO. Littermate controls were harvested when R848 treated mice were moribund. Scale bar equals 20um. G. Renal activity score for NZM IL-1β−/− mice when moribund. H. Renal chronicity index for mice in G.
To examine the role of IL-1β in TLR7-mediated nephritis, we generated NZM2328 mice that lack IL-1β (Supplemental Figure 1) and treated them epicutaneously with R848 or DMSO. These mice also exhibited a decline in survival (Figure 6D), increase in dsDNA IgG antibodies (Figure 6E) and development of lupus nephritis (Figure 6F–H). These data indicate that like type I IFNs, IL-1β is not required for TLR7-mediated nephritis.
TLR7 epicutaneous stimulation leads to early infiltration of dendritic cells and upregulation of ccl2
Previous work has identified important roles for macrophage and dendritic cell infiltration into the kidney prior to nephritis onset (19, 26). We thus examined inflammatory cell recruitment into the kidney prior to lupus nephritis onset in order to determine the earliest cellular contributors to TLR7-induced nephritis. Two weeks after R848 or DMSO treatment, prior to proteinuria onset, kidneys were removed, digested and pre/early-nephritic renal immune cell populations were examined by flow cytometry. (T cell, macrophage, and dendritic cell population gating strategies are shown in Fig. 7A). Consistent with previous data (19), we did not see a change in the T cell (CD3+) and B cell (CD19+) population prior to proteinuria onset (Fig. 7B and C). We also did not see changes in the macrophage (CDllb+CDllcint F4/80+) population at this time point (Fig. 7D). However, we detected a significant infiltration of the dendritic cell population (CDllb+ CDllc+ F4/80−) in both the NZM and INZM mice treated with R848 but not DMSO (Fig. 7E). The number of infiltrating cells was overall fewer in INZM mice, but this was not statistically different between NZM and INZM mice (p=0.2391 by unpaired student’s t-test). This suggests that TLR7 activation promotes early infiltration of dendritic cells into the kidney, similar to what has been observed in other genetic models of murine lupus (26), but in an interferon-independent fashion.
Figure 7. Early dendritic cell infiltration is a common feature of TLR7 driven nephritis in NZM and INZM mice.

Immune cell population changes in the kidney of NZM and INZM mice following 2 weeks of R848 or DMSO treatment. A. Gating strategy for immune cell populations. B–E. Graphs displaying changes in T cells (B): CD3+, B cells (C): CD19+, Macrophages (D): CDllcintCDllb+F480+, and Dendritic Cells (E): CDllc+CDllb+F480−. n=12 NZM DMSO; n=13 NZM R848; n=8 INZM DMSO; n=8 INZM R848. Data is displayed as mean ± SD.
To identify potential mechanisms of chemotaxis into the kidney, we examined chemokine expression in the kidney following DMSO or R848 treatment with the hypothesis that relevant chemokines should rise in both NZM and INZM mice with R848 treatment. As shown in Figure 8, there were no changes in ccl4 (MIP-2) or cxcl13 expression in either strain following R848 treatment (Figure 8A, B). ccl5 expression increased in NZM2328 but not INZM mice after R848 exposure (Figure 8C). IP-10 (cxcl10) expression also increased only NZM2328 mice after R848 exposure (Figure 8D). Importantly, upregulation of ccl2 (MCP-1), a chemokine that rises during lupus nephritis(28) and whose receptor (CCR2) is expressed on monocytes, macrophages, and dendritic cells(29, 30), was demonstrated to increase in both NZM and INZM mice after R848 treatment (Figure 8E). These data suggest TLR7-mediated upregulation of ccl2 may serve as a potential trigger for induction of inflammatory cell infiltration in an interferon-dependent manner.
Figure 8. CCL2 is upregulated in NZM and INZM mice after TLR7 stimulation.

NZM and INZM mice were treated with R848 or DMSO for two weeks followed by harvest of RNA from the kidney. Real-time PCR was completed for the genes listed. Graphs display the mean ± SD for each gene as compared to the average of β-actin. Each dot represents a single mouse.
Discussion
In this paper, we examine a novel model of lupus flare in which lupus nephritis is induced in genetically-prone mice after three-four weeks of epicutaneous application of a TLR7 agonist, R848. R848 treatment led to accelerated development of autoantibodies, splenomegaly, liver inflammation, and lupus nephritis. With the use of NZM mice lacking the type I IFN receptor, we demonstrated that TLR7-mediated splenomegaly and liver inflammation were dependent on type I IFN signaling. Surprisingly, however, renal injury was independent of type I IFNs. Indeed, INZM mice demonstrated proteinuria, increased renal immune complex formation, upregulation of NFκB regulated cytokines, and infiltration of CDllb+CDllc+F4/80low DCs in the kidney similar to NZM mice.
Human genetics support a role for TLR7 in lupus (4, 5), and murine data has further supported this9, 10,(11). BXSB male mice develop lupus secondary to the Yaa locus that contains a duplication of the TLR7 gene (23). The role of TLR 7 has also been demonstrated in transgenic mice overexpressing TLR 7(9) and in lupus inducible models such as the pristine model (10). A recent paper also demonstrated that TLR7 epicutaneous stimulation of wild type mice led to development of mild lupus characteristics after long-term (13 to 15 weeks) treatment(11). Our model furthers these observations and demonstrates that TLR7 stimulation rapidly (in 3–4 weeks) accelerates lupus development in young, lupus-prone mice. Further, we show that mice that are otherwise protected from lupus development in the absence of functional IFN signaling (12) are similarly susceptible to the nephritis-inducing effects of TLR7. Others have shown that overexpression of IFNα is sufficient to stimulate lupus nephritis in a similar time frame to TLR7 treatment (15). However, in the presence of TLR7 agonist, type I IFN signaling is not required. This observation may reflect duplicative roles of other inflammatory cytokines induced by TLR7.
Recently, the role of type I IFN has been explored in other murine models of lupus. Treatment of mice with HgCl2 is able to induce autoantibody production in a lysosomal TLR-dependent but type I IFN independent fashion(31), similar to the data we see in the R848 NZM model. Development of renal disease and direct contribution of TLR7 in this xenobiotic model were not assessed(31). In a different model, exacerbation of autoimmunity via deletion of TLR9 in the MRL/lpr mouse was found to be type I IFN dependent(32). In this model, IFNs are required for development of renal disease but not anti-nucleosome antibody production. HepG-2 positive ANA antibodies and anti-RNA antibodies were dependent on type I IFN signaling(32). Combined with our findings, these data suggest that the role for IFNs in lupus development may vary depending on the stimuli. Autoantibody production may be independent of IFNs when the trigger is strong environmental exposure (such as R848 or HgCl2). Conversely, lupus that develops based on genetic factors without the need for external triggers (such as TLR9-deficient MRL/lpr or NZM2328 mice) may require IFN signaling for autoantibody and nephritis development.
NFκB activation occurs downstream of TLR7 stimulation and activation of this pathway is important for stimulation of lupus nephritis (33). Thus, TLR7 activation of NFκB pathways may be a critical step in driving lupus flares. Mutations in A20, which drive NFκB activation, in WT mice led to lupus nephritis (34, 35) possibly through decreased regulation of inflammasome activity (36). Though roles for TNF alpha and IL-6 in lupus development have been suggested (37), and IL-6 can exacerbate TLR7-driven lupus(38), we do not see these cytokines significantly upregulated in the kidney in a, interferon-independent manner in our TLR 7 stimulation model. Intriguingly, we see an upregulation of IL-1β in the kidney prior to the onset of lupus nephritis in both NZM2328 and INZM mice. This finding coincides with data that support a potential role for IL-1β and inflammasome activation in lupus development (reviewed in (39)). However, deletion of IL-1β in NZM2328 mice did not reduce the development of lupus nephritis after TLR7 exposure. Given that TLR7 activates pleotropic pathways, generation of double or triple knockouts may be required to hinder lupus nephritis development in the presence of such a strong inflammatory activator.
Similar to others(12), we found that splenomegaly following TLR7 stimulation was type I IFN dependent. Interestingly, the massive R848-induced splenomegaly was out of proportion to the small increases in B and T cell populations in the spleen that were identified by flow. This supports a possible role for extra medullary hematopoiesis resulting in R848-mediated splenomegaly, consistent with findings by other groups (40, 41).
TLR7 epicutaneous stimulation can lead to development of autoantibody production in the absence of type I IFN signaling. We were able to see an increase in dsDNA antibodies in NZM2328 and INZM mice following R848 stimulation, although the increase in anti-dsDNA levels was less robust in INZM mice. Increases in B (B220+) cell numbers in the spleen were noted in only the NZM mice, which suggests that expansion of B cell populations in the spleen may require type I IFNs following TLR7 exposure. This would be consistent with a recently described role for splenic follicular dendritic cell production of type I IFN in promotion of autoreactive B cell populations (42, 43). Type I IFNs also enhance the TLR7 signaling response, allowing for autoantibody production(44), and they support survival of transitional stage B cells in the spleen(45). Thus, the more extensive systemic immune activation seen in NZM mice may be secondary to these effects of type I IFN. Overexpression of TLR7 stimulation has also been shown to lead to expansion of transitional stage B cell populations in a type I IFN independent manner(46). Given that we see a rise in Ab-secreting cells in the spleen and dLN but not in the non-draining LN of INZM mice, this may indicate that a certain threshold of TLR7 stimulation, and possibly other cytokines, is needed to generate secreting cell expansion in a type I IFN independent manner.
TLR7 stimulation results in the recruitment of CD11b+CD11c+F4/80− DCs in the kidney prior to proteinuria onset without significant changes in the T cell or B cell population. Interestingly, this recruitment occurs in a type I IFN-independent manner. The DC chemoattractant MCP-1/CCL2 was increased in the kidney in both NZM and INZM mice, which supports its potential role for instigating nephritis downstream of TLR7 activation. Intriguingly, CCL2 has been shown to rise during human lupus nephritis development(28) and is proposed as a urinary biomarker for disease(47). Blockade of CCL2 demonstrates some efficacy in murine lupus models(48). Macrophages, which also are recruited by CCL2, were not increased in the kidney after R848 treatment, which may reflect differential upregulation of CCL2’s receptor, CCR2, on DCs and macrophages in this model. Overall, consideration of the role of CCL2 downstream of TLR7-driven lupus should be made in future studies.
In summary, we have demonstrated a novel role for TLR7 epicutaneous stimulation in mediating lupus flare in lupus-prone mice. Following stimulation, splenomegaly and liver inflammation occur in a type I IFN-dependent manner. Importantly, autoantibody production and lupus nephritis occur independent of IFN signaling. We demonstrate that IL-1β is not required for development of TLR7-activated lupus nephritis. Future studies should address the role of CCL2 in TLR7-mediated lupus nephritis. In addition, our data lend a note of caution to ongoing trials utilizing type I IFN blockade in lupus nephritis: consideration of the upstream drivers of nephritis (which may vary in individual patients) may be important for identifying effective treatment modalities. In particular, our data would suggest that blockade of type I IFN signaling my not be effective if TLR7 is driving the phenotype.
Supplementary Material
Acknowledgments
Funding of this project was provided by the Stone Family Foundation and the National Institutes of Health via the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) under Award Numbers K08AR063668 and R01AR071384 (to JMK) and the National Institute of Allergy and Infectious Diseases Research Training in Experimental Immunology Training Grant T32AI007413.
ABBREVIATIONS
- dLN
draining LN
- IFN
interferon
- LN
lymph node
- SLE
systemic lupus erythematosus
- TLR
toll-like receptor
References
- 1.Bagavant H, Fu SM. Pathogenesis of kidney disease in systemic lupus erythematosus. Current opinion in rheumatology. 2009;21:489–494. doi: 10.1097/BOR.0b013e32832efff1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Zubiria Salgado A, Herrera-Diaz C. Lupus nephritis: an overview of recent findings. Autoimmune diseases. 2012;2012:849684. doi: 10.1155/2012/849684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tektonidou MG, Dasgupta A, Ward MM. Risk of End-Stage Renal Disease in Patients With Lupus Nephritis, 1971–2015: A Systematic Review and Bayesian Meta-Analysis. Arthritis & rheumatology. 2016;68:1432–1441. doi: 10.1002/art.39594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tian J, Ma Y, Li J, Cen H, Wang DG, Feng CC, Li RJ, Leng RX, Pan HF, Ye DQ. The TLR7 7926A>G polymorphism is associated with susceptibility to systemic lupus erythematosus. Molecular medicine reports. 2012;6:105–110. doi: 10.3892/mmr.2012.865. [DOI] [PubMed] [Google Scholar]
- 5.Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen JY, Yang W, Wong M, Kawasaki A, Tsuchiya N, Sumida T, Kawaguchi Y, Howe HS, Mok MY, Bang SY, Liu FL, Chang DM, Takasaki Y, Hashimoto H, Harley JB, Guthridge JM, Grossman JM, Cantor RM, Song YW, Bae SC, Chen S, Hahn BH, Lau YL, Tsao BP. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15838–15843. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horton CG, Farris AD. Toll-like receptors in systemic lupus erythematosus: potential targets for therapeutic intervention. Current allergy and asthma reports. 2012;12:1–7. doi: 10.1007/s11882-011-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton CG, Pan ZJ, Farris AD. Targeting Toll-like receptors for treatment of SLE. Mediators of inflammation. 2010 doi: 10.1155/2010/498980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis and rheumatism. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- 9.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, van Rooijen N, Kumar H, Kawai T, Satoh M, Akira S, Reeves WH. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. The Journal of experimental medicine. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Fujimoto C, Kataoka S, Terada Y, Sano S. Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: a new model of systemic Lupus erythematosus. Arthritis & rheumatology. 2014;66:694–706. doi: 10.1002/art.38298. [DOI] [PubMed] [Google Scholar]
- 12.Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, Kovats S, Jacob CO. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol. 2009;183:6021–6029. doi: 10.4049/jimmunol.0803872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crow MK. Type I interferon in the pathogenesis of lupus. Journal of immunology. 2014;192:5459–5468. doi: 10.4049/jimmunol.1002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairhurst AM, Xie C, Fu Y, Wang A, Boudreaux C, Zhou XJ, Cibotti R, Coyle A, Connolly JE, Wakeland EK, Mohan C. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J Immunol. 2009;183:6831–6838. doi: 10.4049/jimmunol.0900742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, Davidson A. Interferon-alpha accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis and rheumatism. 2011;63:219–229. doi: 10.1002/art.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nacionales D, Kelly-Scumpia K, Lee P, Weinstein J, Lyons R, Sobel E, Satoh M, Reeves W. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis and rheumatism. 2007;56:3770–3783. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudofsky UH, Lawrence DA. New Zealand mixed mice: a genetic systemic lupus erythematosus model for assessing environmental effects. Environmental health perspectives. 1999;107(Suppl 5):713–721. doi: 10.1289/ehp.99107s5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petri M. Epidemiology of systemic lupus erythematosus. Best practice & research Clinical rheumatology. 2002;16:847–858. doi: 10.1053/berh.2002.0259. [DOI] [PubMed] [Google Scholar]
- 19.Clark KL, Reed TJ, Wolf SJ, Lowe L, Hodgin JB, Kahlenberg JM. Epidermal injury promotes nephritis flare in lupus-prone mice. Journal of autoimmunity. 2015;65:38–48. doi: 10.1016/j.jaut.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, Kaplan MJ. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis & rheumatology. 2014;66:152–162. doi: 10.1002/art.38225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, Havekes LM, Kleemann R, van den Hoek AM. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PloS one. 2014;9:e115922. doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trouplin V, Boucherit N, Gorvel L, Conti F, Mottola G, Ghigo E. Bone marrow-derived macrophage production. Journal of visualized experiments: JoVE. 2013:e50966. doi: 10.3791/50966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ansel JC, Mountz J, Steinberg AD, DeFabo E, Green I. Effects of UV radiation on autoimmune strains of mice: increased mortality and accelerated autoimmunity in BXSB male mice. The Journal of investigative dermatology. 1985;85:181–186. doi: 10.1111/1523-1747.ep12276652. [DOI] [PubMed] [Google Scholar]
- 24.Jacob CO, Zang S, Li L, Ciobanu V, Quismorio F, Mizutani A, Satoh M, Koss M. Pivotal role of Stat4 and Stat6 in the pathogenesis of the lupus-like disease in the New Zealand mixed 2328 mice. Journal of immunology. 2003;171:1564–1571. doi: 10.4049/jimmunol.171.3.1564. [DOI] [PubMed] [Google Scholar]
- 25.Sun X, Wiedeman A, Agrawal N, Teal TH, Tanaka L, Hudkins KL, Alpers CE, Bolland S, Buechler MB, Hamerman JA, Ledbetter JA, Liggitt D, Elkon KB. Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. Journal of immunology. 2013;190:2536–2543. doi: 10.4049/jimmunol.1202689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai C, Wang H, Sung SS, Sharma R, Kannapell C, Han W, Wang Q, Davidson A, Gaskin F, Fu SM. Interferon alpha on NZM2328.Lc1R27: enhancing autoimmunity and immune complex-mediated glomerulonephritis without end stage renal failure. Clinical immunology. 2014;154:66–71. doi: 10.1016/j.clim.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, Satoh M, Reeves WH. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis and rheumatism. 2007;56:3770–3783. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee YH, Song GG. Urinary MCP-1 as a biomarker for lupus nephritis: a meta-analysis. Zeitschrift fur Rheumatologie. 2017;76:357–363. doi: 10.1007/s00393-016-0109-z. [DOI] [PubMed] [Google Scholar]
- 29.Sahu R, Bethunaickan R, Singh S, Davidson A. Structure and function of renal macrophages and dendritic cells from lupus-prone mice. Arthritis & rheumatology. 2014;66:1596–1607. doi: 10.1002/art.38410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao X, Pirapakaran T, Luo XM. Chemokines and Chemokine Receptors in the Development of Lupus Nephritis. Mediators of inflammation. 2016;2016:6012715. doi: 10.1155/2016/6012715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollard KM, Escalante GM, Huang H, Haraldsson KM, Hultman P, Christy JM, Pawar RD, Mayeux JM, Gonzalez-Quintial R, Baccala R, Beutler B, Theofilopoulos AN, Kono DH. Induction of Systemic Autoimmunity by a Xenobiotic Requires Endosomal TLR Trafficking and Signaling from the Late Endosome and Endolysosome but Not Type I IFN. J Immunol. 2017;199:3739–3747. doi: 10.4049/jimmunol.1700332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nickerson KM, Cullen JL, Kashgarian M, Shlomchik MJ. Exacerbated Autoimmunity in the Absence of TLR9 in MRL. Faslpr Mice Depends on Ifnar1. The Journal of Immunology. 2013;190:3889–3894. doi: 10.4049/jimmunol.1203525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanda SK, Lopez-Pelaez M, Arthur JS, Marchesi F, Cohen P. Suppression of IRAK1 or IRAK4 Catalytic Activity, but Not Type 1 IFN Signaling, Prevents Lupus Nephritis in Mice Expressing a Ubiquitin Binding-Defective Mutant of ABIN1. Journal of immunology. 2016;197:4266–4273. doi: 10.4049/jimmunol.1600788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, Arthur JS, Cohen P. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. The Journal of experimental medicine. 2011;208:1215–1228. doi: 10.1084/jem.20102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caster DJ, Korte EA, Nanda SK, McLeish KR, Oliver RK, G’Sell T, Sheehan RRM, Freeman DW, Coventry SC, Kelly JA, Guthridge JM, James JA, Sivils KL, Alarcon-Riquelme ME, Scofield RH, Adrianto I, Gaffney PM, Stevens AM, Freedman BI, Langefeld CD, Tsao BP, Pons-Estel BA, Jacob CO, Kamen DL, Gilkeson GS, Brown EE, Alarcon GS, Edberg JC, Kimberly RP, Martin J, Merrill JT, Harley JB, Kaufman KM, Reveille JD, Anaya JM, Criswell LA, Vila LM, Petri M, Ramsey-Goldman R, Bae SC, Boackle SA, Vyse TJ, Niewold TB, Cohen P, Powell DW. ABIN1 dysfunction as a genetic basis for lupus nephritis. Journal of the American Society of Nephrology: JASN. 2013;24:1743–1754. doi: 10.1681/ASN.2013020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M, Shi X, Qian T, Li J, Tian Z, Ni B, Hao F. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-kappaB and NLRP3 inflammasome activation in macrophages of mice. International journal of clinical and experimental medicine. 2015;8:17430–17440. [PMC free article] [PubMed] [Google Scholar]
- 37.Jacob N, Yang H, Pricop L, Liu Y, Gao X, Zheng SG, Wang J, Gao HX, Putterman C, Koss MN, Stohl W, Jacob CO. Accelerated pathological and clinical nephritis in systemic lupus erythematosus-prone New Zealand Mixed 2328 mice doubly deficient in TNF receptor 1 and TNF receptor 2 via a Th17-associated pathway. Journal of immunology. 2009;182:2532–2541. doi: 10.4049/jimmunol.0802948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain S, Park G, Sproule TJ, Christianson GJ, Leeth CM, Wang H, Roopenian DC, Morse HC., 3rd Interleukin 6 Accelerates Mortality by Promoting the Progression of the Systemic Lupus Erythematosus-Like Disease of BXSB. Yaa Mice. PloS one. 2016;11:e0153059. doi: 10.1371/journal.pone.0153059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Current opinion in rheumatology. 2014;26:475–481. doi: 10.1097/BOR.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weindel CG, Richey LJ, Bolland S, Mehta AJ, Kearney JF, Huber BT. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy. 2015;11:1010–1024. doi: 10.1080/15548627.2015.1052206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuang H, Han S, Xu Y, Li Y, Wang H, Yang LJ, Reeves WH. Toll-like receptor 7-stimulated tumor necrosis factor alpha causes bone marrow damage in systemic lupus erythematosus. Arthritis & rheumatology. 2014;66:140–151. doi: 10.1002/art.38189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, Carroll MC. Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity. 2017;46:106–119. doi: 10.1016/j.immuni.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang NH, Li TT, Kim JJ, Landolt-Marticorena C, Fortin PR, Gladman DD, Urowitz MB, Wither JE. Interferon-alpha induces altered transitional B cell signaling and function in Systemic Lupus Erythematosus. Journal of autoimmunity. 2015;58:100–110. doi: 10.1016/j.jaut.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Green NM, Moody KS, Debatis M, Marshak-Rothstein A. Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. The Journal of biological chemistry. 2012;287:39789–39799. doi: 10.1074/jbc.M112.383000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Hong H, Li J, Walter MR, Fish EN, Hsu HC, Mountz JD. Cutting Edge: Endogenous IFN-beta Regulates Survival and Development of Transitional B Cells. J Immunol. 2017;199:2618–2623. doi: 10.4049/jimmunol.1700888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, Kim J, Tanaka L, Buechler MB, Hamerman JA, Imanishi-Kari T, Clark EA, Elkon KB. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. The Journal of experimental medicine. 2013;210:2773–2789. doi: 10.1084/jem.20122798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta R, Yadav A, Aggarwal A. Longitudinal assessment of monocyte chemoattractant protein-1 in lupus nephritis as a biomarker of disease activity. Clin Rheumatol. 2016;35:2707–2714. doi: 10.1007/s10067-016-3404-9. [DOI] [PubMed] [Google Scholar]
- 48.Devarapu SK, Kumar Vr S, Rupanagudi KV, Kulkarni OP, Eulberg D, Klussmann S, Anders HJ. Reprint of “Dual blockade of the pro-inflammatory chemokine CCL2 and the homeostatic chemokine CXCL12 is as effective as high dose cyclophosphamide in murine proliferative lupus nephritis”. Clin Immunol. 2017 doi: 10.1016/j.clim.2017.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.