

Abstract
Pseudomonas aeruginosa keratitis is characterized by severe corneal ulceration and may lead to blindness if not treated properly in a timely manner. While the roles of the interleukin 1 (IL-1) subfamily of cytokines are well-established, IL-36 cytokines, as a newly discovered subfamily, their regulation, immunological relevance, and relation with IL-1 cytokines in host defense remain largely unknown. Here we showed that Pa-infection induces the expression of IL-36 α and γ, as well as IL-1β and secreted IL-1Ra (sIL-1Ra), but not IL-36Ra. Downregulation of IL-1Ra increases, whereas downregulation of IL-36Ra decreases the severity of Pa keratitis. IL-1R and IL-36Ra downregulation have opposing effects on the expression of IL-1β, sIL-1Ra, IL-36γ, S100A8, and CXCL10, and on the infiltration of innate immune cells. Administration of recombinant IL-1Ra improved, while IL-36Ra worsened the outcome of Pa keratitis. Local application of IL-36γ stimulated the expression of innate defense molecules S100A9, mouse β-defensin 3, but suppressed IL-1β expression in B6 mouse corneas. IL-36γ diminished the severity of Pa keratitis and its protective effects were abolished in the presence of S100A9-neutralizing antibody and partially affected by CXCL10 and CXCR3 neutralizations. Thus, our data reveal that IL-1Ra and IL-36Ra have opposing effects on the outcome of Pa keratitis and suggest that Il-36 agonists may be used as an alternative therapeutic to IL-1β-neutralizing reagents in controlling microbial keratitis and other mucosal infections.
Keywords: Il-36Ra, Il-1Ra, Pseudomonas aeruginosa keratitis, IL-36γ, antimicrobial peptide
Topic: interleukin 1, interleukin 1Ra, interleukin 36, interleukin 36, host defense, inflammation
Introduction
The interleukin (IL)-1 family of cytokines are fundamental regulators of the innate immune response and known to be involved in multiple inflammatory diseases (1, 2). The family contains 7 ligands with agonistic (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ), 3 with antagonistic (IL-1Ra, IL-36Ra, and IL-38), and 1 with anti-inflammatory activity (IL-37) (3, 4). The two large subfamilies, IL-1 and IL-36, are located within a gene cluster: IL1A-IL1B-IL137-IL36G-IL36A-IL36B-IL36RA-IL38-IL1RN, suggesting a close evolutionary relationship. IL-1β is considered to be a key orchestrator of innate immune responses (3). IL-1β activation is regulated at the transcriptional, post-translational processing, and secretion levels (5). However, regardless of how it is released or how the precursor is cleaved, the final activity of IL-1β is determined by the ratio of active IL-1β and its natural inhibitor, soluble IL-1Ra (sIL-1Ra) (3, 6, 7). Disturbance in the IL-1β/sIL-1Ra balance usually results in inflammation diseases (8-11). More importantly, human recombinant IL-1Ra (Anakinra), a biologic designed to treat rheumatoid arthritis, has been found to be effective in treating a broadening list of inflammatory diseases, cardiovascular diseases (12, 13), and type 1 and 2 diabetes (14, 15).
IL-36 cytokines have attracted much attention ever since IL-36RN mutations were found to cause generalized pustular psoriasis, the most severe form involving not only skin inflammation but also systemic symptoms such as fever and malaise (16-19). Binding of IL-36 agonists to IL-36R recruits IL-1RAcP, a shared accessory protein with IL-1R and IL-33R. This, ultimately results in the activation of NF-κB and MAPKs through MyD88 pathway (3, 20). The fact that IL-36R has three agonists, IL-36α,β,γ, and two antagonists, IL-36Ra and IL-38 (21-24) underpins the importance of fine-tuning the IL-36R signaling pathway. In host defense, while the roles of the IL-1β and IL-1R pathways are well established, the regulation of IL-36 cytokines and their immunological relevance remain largely unknown. Recently, IL-36 expression and signaling have been linked to viral (25), mycobacterial (26, 27), Gram-negative and -positive (28) bacterial infections of the lung. The IL-36/IL-36R axis has been shown to play contradicting roles in host response to mycobacterial infection (26, 27). It is also shown to play a protective role in Gram positive Streptococcus pneumoniae or Gram-negative Klebsiella pneumoniae pneumonia by driving protective type-1 responses and classical macrophage activation in the lung (28). Interestingly, the same group, using IL-36γ and IL-36R knockout mice, showed that the IL-36 signaling pathway impaired host immunity and exuberated lung injury in cytotoxic Pseudomonas aeruginosa pulmonary infection (29). The involvement of IL-36/IL-36R in mediating tissue/cell responses to infection in other mucosal tissues, including the cornea, have not been reported to date.
Bacterial keratitis is a sight-threatening disease, commonly associated with extended contact lens wear (30). Pseudomonas aeruginosa (Pa) is a common causative agent of keratitis (31). To cause keratitis, microbes must traverse the epithelial layer and cross the BM to reach the stroma (32, 33), a process that usually takes ~18-24 h to accomplish since the basement membrane serves as a physiological barrier (34, 35). PA-keratitis usually presents as a rapidly progressing condition, with distinct inflammatory epithelial edema and stromal ulceration, which can lead to significant stromal tissue destruction and loss of vision (36). These consequences are largely caused by the host’s inflammatory responses, although bacterial toxins and exoproducts may contribute to Pa keratitis (36). The innate immune system is critical for host defense against infectious challenges. Recognition of pathogens by TLRs results in the initiation of innate immune responses, including the production/secretion of inflammatory mediators, especially IL-1β and its natural inhibitor IL-1Ra (37). In the cornea, epithelial cell–produced IL-1 was suggested to act as a master regulator of corneal wound healing; topical application of IL-1Ra was shown to inhibit inflammatory cell infiltration of the cornea (7). Because they were only discovered recently, the role of IL-36 cytokines in Pa-keratitis and microbial infection of the corneas remains elusive.
In this study, we assessed the expression, contribution, and role of IL-36R signaling pathway, in comparison with that of IL-1R signaling, in the control of Pa-infection by using siRNA knockdown and recombinant receptor antagonists. We found that the expression of IL-36α and γ was induced upon Pa-infection and augmenting IL-36R signaling, either by downregulating IL-36Ra or by exogenous IL-36γ, greatly reduced the severity of Pa-keratitis, in sharp contrast to that reported in Pa-infection of the lung (29). The data support our hypothesis that IL-36 cytokines initiate innate defense and tissue repair in response to microbial infection by inducing epithelial cells to produce AMPs and other cytokines/chemokine and by antagonize the IL-1 subfamily by mediating the balanced expression of IL-1β and IL-1Ra.
Methods
Animals
Wild-type C57BL6 (B6) mice (eight weeks of age; female) were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal procedures were performed in compliance with the ARVO Statement for the use of animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of Wayne State University.
Cell culture
Primary human corneal epithelial cells (HCECs) were obtained from diseased human corneal samples. Corneal epithelial cells were dislodged from the underlying basement membrane by dispase and then digested by trypsin. Cells were pelleted, re-suspended and grown in defined keratinocyte serum-free medium (DK-SFM) (Gibco, Waltham, MA, USA) with supplements and antibiotics.
P3 or P4 of the primary HCECs were used in the experiments. Before treatment, cells were starved overnight in growth factor-free and antibiotic-free keratinocyte basic medium (KBM-2, Lonza, Basel, Switzerland). Subsequently, cells were challenged with heat-killed bacterial (1:50 multiplicity of infection [MOI]) for 1, 2, 4, 8 or 16 hours. At the end of the incubation period, cells were harvested for detecting gene expression.
Mouse model of P. aeruginosa Keratitis
Mice were anesthetized with an intraperitoneal injection of Ketamine (90 mg/kg) and Xylazine (10 mg/kg) before surgical procedures. Mouse corneas were scratched gently with a sterile 26-gauge needle to create three 1-mm incisions to break the epithelial barrier and inoculated with 1.0×104 CFUs of ATCC 19660 in 5μl PBS.
The application of siRNA, recombinant protein, or neutralizing antibody
All the siRNAs used for this study were SMARTpool (a mixture of 4 siRNAs) ON-TARGETplus siRNAs designed by GE Dharmacon Company (Lafayette, CO, USA). Mice were subconjunctivally injected twice with siRNA targeting to a specific gene (50 picomoles in 5μl RNase-free water) over two days. Six hours after the second siRNA injection, mouse corneas were inoculated with PA. To apply recombinant proteins or neutralizing antibody, mice were subconjuntivally injected with 150 ng recombinant protein (IL36γ, IL36Ra) (R&D systems, Minneapolis, MN, USA), 150 ng anakinra, 100 ng anti-S100A9 ((kindly provided by Dr. Philippe Tessier, Laval University Hospital Center, 2, Quebec, Canada), CXCL10 or CXCR3 neutralizing antibodies (R&D systems) four hours before the inoculation with Pa on the corneas.
Clinical examination
Eyes were examined daily to monitor the disease progression with a dissection microscope equipped with a digital camera. For the assessment of clinical scores, mice were color coded and examined in a blinded fashion by two independent observers at 1, and 3 days post infection (dpi) to visually grade the disease severity. Ocular disease was graded in clinical scores ranging from 0 to 12. A total score of 5 or less indicated mild eye disease, 6 to 9 signaled moderate disease, and 9 showed severe disease (35, 38, 39).
Bacteria Load Determination, Cytokine ELISA, and MPO Measurement
We used our previously modified methods that allowed all three assays (bacteria load, MPO determination, and cytokine ELISA measurement) to be performed with a single mouse cornea. Briefly, the corneas were excised, minced, and homogenized in 100 μl PBS with Dounce Micro Tissue Grinder. The homogenates were divided into two. The first part was subjected to plate bacterium counting. Aliquots (50 μl) of serial dilutions were plated onto Pseudomonas Isolation agar plates in triplicates, and colonies were counted next day. The results were expressed as the mean number of CFU/cornea ± standard error. The second part of the homogenates was mixed with 5 μl of 1% SDS and 10% Triton-X 100. For MPO assay, 30 μl homogenates was mixed with 270 μl of hexadecyltrimethylammonium bromide (HTAB) buffer (0.5% HTAB in 50 mM phosphate buffer, pH 6.0). The samples were then subjected to three freeze-thaw cycles, followed by centrifugation at 16,000 × g for 20 min. Twenty μl of the supernatant was mixed with 180 μl of 50 mM phosphate buffer (pH 6.0) containing 16.7 mg/ml O,O-dianisidine hydrochloride and 0.0005% hydrogen peroxide at a 1:30 ratio in a well of 96-well plate. The change in absorbance at 460 nm was monitored continuously for 5 min in a Synergy2 Microplate reader (BioTek). The results were expressed in units of MPO activity/cornea. One unit of MPO activity corresponded to approximately 2.0 × 105 PMN. For ELISA, protein concentration was first determined and using Micro BCA™ protein assay kit (Pierce) and 1 μg of total protein was used for ELISA assay for CXCL2, IL1β, CXCL10 and calprotectin (R & D Systems).
RNA extraction and Real-time PCR
For RT- PCR, mouse corneas or CECs scraped off the cornea were extracted for RNA using RNeasy Mini Kit (QIAGEN). cDNA was generated with an oligo(dT) primer (Invitrogen) followed by analysis using real-time PCR or RT-PCR. For quantitative PCR, cDNA was amplified using StepOnePlus Real-Time PCR system (Applied Biosystems, University Park, IL, USA) with the SYBR® Green PCR Master Mix (Applied Biosystems). Data were analyzed by using ΔΔCT method with β-actin as the internal control. For RT-PCR, PCR products were subjected to electrophoresis on 1% agarose gels containing ethidium bromide. Stained gels were captured by using a digital camera. The following primer pairs were used (Table 1).
Table 1.
Primer Sequences Used for PCR
| Primers | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| mβ –actin | GACGGCCAGGTCATCACTATTG | AGGAAGGCTGGAAAAGAGCC |
| mIL-1β | AAGGAGAACCAAGCAACGACAAAA | TGGGGAACTCTGCAGACTCAAACT |
| mIL1RA | GGGACCCTACAGTCACCTAA | GGTCCTTGTAAGTACCCAGAC |
| mCXCL10 | ATGAACCCAAGTGCTGCCGTC | TTAAGGAGCCCTTTTAGACCTTT |
| mS100a8 | TGCGATGGTGATAAAAGTGG | GGCCAGAAGCTCTGCTACTC |
| mIL36α | CCAAGAACTGGGGGAAATCT | GGAGGGCTCAGCTTTCTTTT |
| mIL36γ | CCCATGCAAGTACCCAGAGT | GGGAAAGCCACTGATTCAAA |
| mIL36Ra | GGATGCTCCCATCACAGACT | CAGAAGCGGGAGTCCAGTAG |
| mIL24 | GCTTTCACCAAAGCGACTTC | GCCCAGTAAGGACAATTCCA |
| mIL6 | GTGGCTAAGGACCAAGACCA | TAACGCACTAGGTTTGCCGA |
| hIL36α | GAAAGCCACAGACTCGAAGG | AAGACAGAGGGAACCCCATC |
| hIL36β | GCACCTGAATTCCCACAGAG | AGCGTTTCACTCCTCATGCT |
| hIL36γ | AATTGTGGGCTGTCCAAC | GGTCAGAACCTTGTGGCAGT |
| hIL36Ra | AGGTTTCAGCAGAGCCGTTA | AAGGTCATGCTGGATGAAGG |
| hIL1β | ACCAAACCTCTTCGAGGCAC | AGCCATCATTTCACTGGCGA |
| hIL1Ra | CTGGGGGTTCTTTCTTCCTC | TAGGGAACTTTGCACCCAAC |
| hβ –actin | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
| mBD3 | GCTAGGGAGCACTTCTTTGCATTTAA | GCTTCAGTCATGAGGATCCATTACCT |
| mMMP13 | TGATGAAACCTGGACAAGC | CTGGACCATAAAGAAACTGAA |
Western Blot
Mouse corneal samples were lysed with RIPA buffer. The lysates were centrifuged to obtain the supernatant. Protein concentration was determined by BCA assay. The protein samples were separated by SDS-PAGE and electrically transferred onto nitrocellulose membranes (Bio-Rad; Hercules, CA, USA). The membranes were blocked with 3% BSA and subsequently incubated with primary and secondary antibodies. Signals were visualized using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific, Pittsburgh, PA, USA) using a Kodak Imaging Station 4000R. β-Actin was used as the loading control. Antibodies: anti-human IL36 α, γ and RA (α, 1 μg/ml; γ, 0.5 μg/ml; RA (0.1 μg/ml, R&DR&D) anti-mouse IL36Ra IL1Ra (1.0 μg/ml) or IL1Ra (0.1 μg/ml, Thermo Fisher Scientific), rabbit anti-mouse IL36γ was generated in Dr. Standiford lab, University of Michigan (29); and anti-β-actin (A1978; Sigma, 1:10000).
Immunohistochemistry
At the indicative time points, the corneas were excised from sacrificed mice and were frozen in OCT compound; posterior parts along with the lens were removed shortly after the global parts frozen. The corneas were cut into 6μm thick sections by cryostat sectioning and the sections were mounted to polylysine-coated glass slides. After a 10-min fixation in 4% paraformaldehyde, the sections were blocked with 10 mM PBS, containing 2% BSA, for 1 hour at room temperature. Sections were then incubated with primary antibodies: rat anti-mouse NIMP-1 (BD; 1:1000) or F4/80 (Ebioscience 1:50), followed by the secondary antibody, FITC conjugated goat anti-rabbit IgG (Jackson Immunoresearch. INC). Slides were mounted with mounting media containing DAPI. Controls were treated similarly, but the primary antibody was replaced with nonspecific rabbit IgG.
In vitro bactericidal activity assay
Corneas, 3 each group were treated with BSA (Control) or IL-36γ were homogenized and sonicated 4 hpi. Each cornea in 0.1 ml homogenate were incubated with 1000 CFU freshly prepared Pa for 1 h at 37°C; 3 20 μl homogenate were spread onto TB Agarose plates; the colonies formed overnight at 37°C were counted and averaged. The average numbers of colonies of 3 corneas were used to calculate the mean of CFU for each condition.
Statistical analyses
Data were presented as means ± SD. Statistical differences among three or more groups were identified using one way analysis of variance (ANOVA). Differences were considered statistically significant at p < 0.05.
Results
Human corneal epithelial cells express IL-36α and γ in response to heat-kill Pa challenge
We first assessed the induction of the IL-I family of cytokines in cultured HCECs challenged with heat-killed (H-K) Pa (strain ATCC 19660). Among IL-36 cytokines, IL-36α and β were not detectable, while IL-36γ and IL-36Ra had a basal-level of expression by RT-PCR in unchallenged cells. Pa-Challenge resulted in the induction of IL-36α and the upregulation of IL-36γ. As for IL-1 cytokines, Pa stimulated robust expression of IL-1β and sIL-1Ra (V2, secreted form) (7) as early response genes in cultured HCECs (Fig. 1A). The expression of IL-36 cytokines was also assessed on the protein level by Western blotting (Fig. 1B). While IL-36γ and IL-36Ra were readily detectable in unstimulated cells, the presence of H-K Pa upregulated the expression of IL-36α, IL-36γ, at 4 hpi. There was a detectable elevation of IL-36Ra at 16 hpi. The image acquisition for IL-36α and RA required a much longer exposure time than that of IL-36γ. Importantly, the immunoreactivity of IL-36γ, but not IL-36α or RA, was also detected in the culture media of HCECs at 8 and 16 h post stimulation, suggesting the cytokine was secreted in response to Pa-challenge.
Figure 1. IL-1 family cytokine expression in human corneal epithelial cells challenged with Heat-killed Pa (ATCC19660).
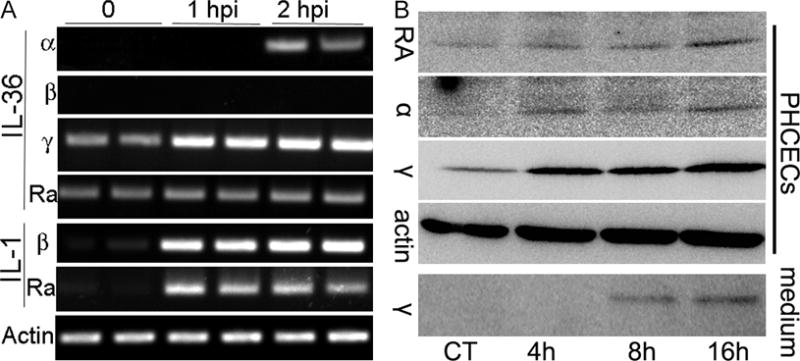
Primary human corneal epithelial cells were cultured in 6-well plates and challenged with heat-killed Pa (1:50) for the indicated times with 1 ml culture media. At the indicated times, cells were lysed and subjected to RT-PCR or Western blotting analyses. (A) Two samples at each time point were assessed for the expression of IL-1β, 1Ra, IL-36α, β, γ and Ra using RT-PCR with β-actin as internal control. (B) Western blot analysis of IL-36α, γ and Ra expression in HCEC extracts (PHCECs) and conditioned media (medium). For cell lysates, 30 μg cellular proteins were used. To assess IL-36 secretion, culture media was collected and 0.5 ml was subjected to TCA precipitation; total TCA precipitates were loaded in each lane for Western blotting. Chemiluminescence images were acquired using a Kodak Imaging Station 4000mm Pro with multiple exposure times for each blot. The figure is representative of 3 independent experiments.
IL-36α and γ, like IL-β and sIL-1Ra, are early response genes in Pa-infected B6 mouse corneas
To assess IL-36 expression in vivo, we inoculated B6 mouse corneas with 104 colony forming units (CFUs) of Pa (strain ATCC19660). Corneas were collected at 3, 6, 9 and 18 hpi and subjected to PCR analysis. Realtime PCR analysis revealed a gradual and robust upregulation of IL-1β and IL-1Ra (Fig. 2A) from 3 to 18 hpi in the whole cornea. IL-36α and IL-36γ was rapidly elevated with no significant differences from 3 to 9 hpi (Fig. 2A). The increase for IL-36α and IL-36γ were relatively moderate compared to IL-1β and IL-1Ra. This may be related to the low level basal expression of IL-36 subfamily of cytokines in naïve corneas, as shown by RT-PCR (Fig. 2B). The induction of IL-36γ at the protein level can be detected in the infected corneas at 1 dpi (Fig. 2C). As in human CECs, IL-36Ra remained detectable but unchanged from 0 to 18 hpi.
Figure 2. IL-1 cytokines are early responsive genes after Pa infection in B6 mouse cornea.
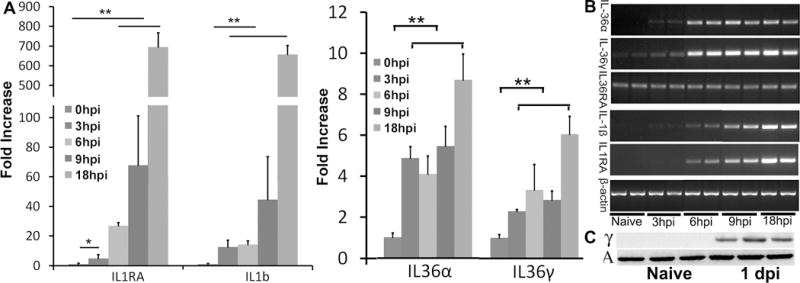
Mouse corneas were scratched with a needle and inoculated with Pa (ATCC 19660, CFUs: 1.0×104). Whole corneas were collected at 3 hpi, 6 hpi, 9 hpi, and 18 hpi for RNA preparation or at 24 hpi for protein extraction and Western blotting with the naïve corneas as the control. (A) qPCR analysis of IL-1Ra, IL-1β, IL36Ra, IL-36α, and IL-36γ. Results are presented as fold increase relative to those of naïve corneas, set as value 1. Stars on top of columns are P value results comparing with the control. *P < 0.05 and **P < 0.01 (ANOVA). Data are representative of three independent experiments (AD: mean + s.e.m.). (B) RT-PCR analysis to show basal expression of IL-1 cytokines. Whole corneas collected as described for A were also subjected to RT-PCR to determine basal (Naïve) and infection induced expression with β-actin as the internal control. (C) Immunoblot analysis of IL-36γ, in cell lysates of whole corneas infected with Pa for 1 day. β-actin serves as the loading control.
Downregulation of IL-1Ra and IL-36Ra had opposing effects on the outcome of Pa keratitis
Having shown the expression pattern of IL-1 cytokines in human and mouse CECs, we next used an siRNA approach to investigate the role of IL-36 signaling in mediating corneal response to infection, in comparison with that of IL-1 (Fig. 3). We first assessed whether antagonist-specific siRNAs can effectively down-regulate targeted genes in the cornea. In the control, naïve corneas low levels of Il-1Ra and IL-36Ra were barely detectable. At 1 dpi, infection induced the upregulation of these receptor antagonists. Presence of specific siRNA suppressed infection-induced expression of these two factors to a level similar to the naïve corneas, indicating effective knockdown of the target genes (Fig. 3A). Since our time course study showed no detectable elevation of IL-36Ra mRNA up to 18 hpi (Fig. 2), we compared mRNA levels of IL-36Ra at 18 and 24 hpi; the results were shown in the insert of Figure 3A. There was an increase in IL-36Ra mRNA from 18 to 24 hpi 1 dpi.
Figure 3. The opposing effects of IL-36RA and IL-1Ra down-regulation on the outcome of Pa keratitis.
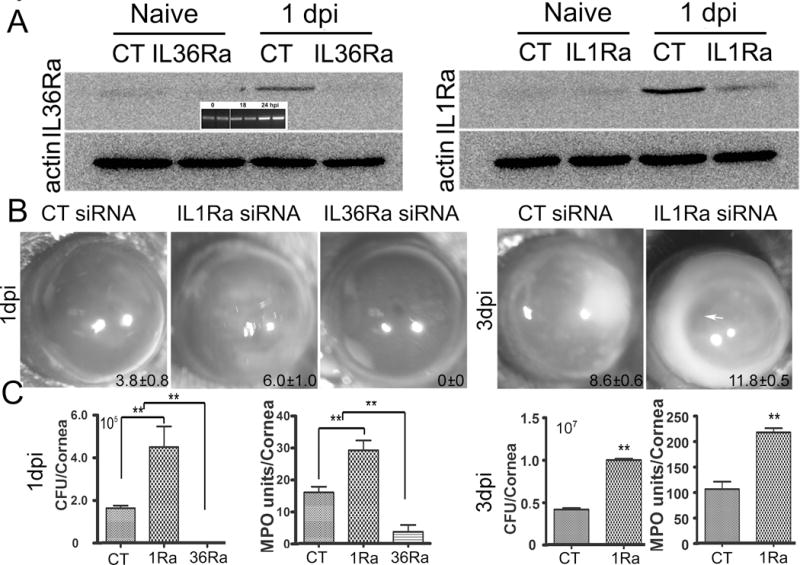
Mice were subconjunctivally injected with IL-1Ra, IL-36Ra specific, or the control, non-specific siRNA (50 picomoles in 5ul RNase free water) twice in two days. Corneas were inoculated with Pa (ATCC 19660, 1.0×104 CFU), 6h after the second siRNA injection. (A) Western blotting assessing the efficacy of siRNA mediated knockdown of IL-1RA and IL-36RA in the cornea in response to Pa infection at 1 dpi with non-inoculated corneas (Naive) as the controls; β-actin serves as the loading control. CT: control, non-specific siRNA. Insert shows RT-PCR showing an increase in IL-36Ra levels at 1 dpi, compared with 0 and 18 hpi. (B) Mice were monitored and photographed daily up to 3 dpi, showing IL-36Ra- and IL-1Ra for 1 dpi and IL-1Ra for 3 dpi in-siRNA treated corneas. The numbers within each eye micrographs are the clinical scores assigned and presented as median + interquartile range. (C) At 1 dpi or 3 dpi, the corneas were excised and subjected to bacterial plate counting and MPO assay. The data were presented as total number of bacteria or MPO units per cornea and were representative of three independent experiments (N=6 each) and indicated p values were generated using unpaired student’s t test. (**p<0.01).
We next examined the pathology of Pa-infected corneas treated with different siRNAs. At 1 dpi, IL-1Ra knockdown resulted in more severe keratitis with significantly higher clinical score (CS, 3.8±0.8 vs 6.0±1.0), bacterial burden (1.63×105 vs 4.50 ×105 CFU), and MPO levels (16.0 vs 29.17 units) than that of control siRNA treated corneas. Knockdown of IL-36Ra, on the other hand, protected the corneal from infection with no sign of infection and inflammation (CS 0), no recoverable bacteria, and a greatly reduced PMN infiltration detected by MPO assay (3.68 units) in the cornea (Fig. 3B and 3C). As the invading pathogens in IL-36Ra siRNA-treated corneas were eradicated with minimal signs of inflammation at 1 dpi, no further observation was made with the group (Fig. 3B). At 3 dpi, knockdown of IL-1Ra resulted in an increase in the severity of keratitis with much higher clinical score (8.6±0.6 vs 11.8±0.5), ~10 fold increases in bacterial burden (4.13×106 vs 9.97×106 CFU), and in MPO activity (106 vs 217 units) (Fig. 3 B and 3C). There was heavy infiltration in the aqueous humor and clear signs of corneal melting in IL-1Ra siRNA-treated corneas (arrow). Since the IL-1Ra siRNA treated corneas were about to perforate, the experiments were terminated at 3 dpi.
IL36 signaling differentially regulates the expression of IL-1β, -1Ra, IL-36γ, and antimicrobial genes in CECs at 6 hpi
We next investigated the expression of IL-1 cytokines and innate defense molecules in IL-1Ra and IL-36Ra downregulated corneas. Our previous study revealed that at 6 hpi, while invading bacteria were mostly within the epithelium with low numbers of infiltrates, the expression of cytokines/chemokines and antimicrobial peptides (AMPs) in epithelial cells had a profound impact of the outcome of infection (39-41). Figure 4A shows the expression of IL-1 cytokines and two known AMPs in the isolated CECs detected with qPCR. Except IL-36Ra, the expression of all 5 genes in control (CT) siRNA-treated, infected corneas were significantly increased to a different extent than the value seen in non-infected CECs, which was set as 1 (NL). Downregulation of IL-36Ra further significantly augmented infection-induced expression of IL-1Ra (36.3 vs 70.3 fold), IL-36γ (3.17 vs 7.87 fold), S100A8 (5.25 vs 19.66 fold), CXCL10 (2.66 vs 7.37 fold), but not IL-1β in infected CECs. Downregulation of IL-1Ra vastly increased IL-1β expression (9.69 vs 22.52 fold), while exhibited no effects on other genes, compared to the control siRNA treated CECs.
Figure 4. Effects of IL36Ra or IL1Ra knockdowns on the gene expression in B6 mouse corneal epithelia at an early stage of Pa infection, 6 hpi.
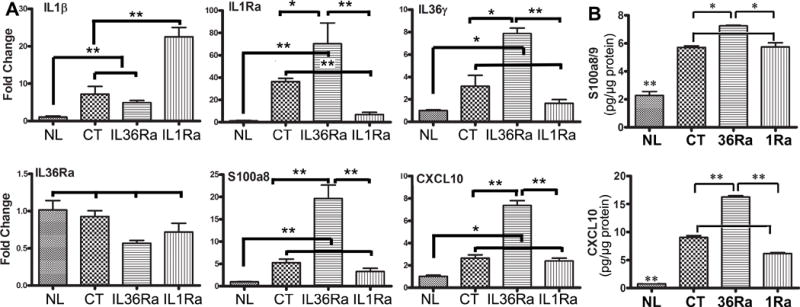
Mouse corneas were treated with siRNAs and inoculated with Pa as in Figure 3. Corneal epithelial samples were collected at 6 hpi for qPCR analysis for the expression of IL-1β, IL-1Ra, IL-36γ, IL-36Ra, S100A8 (A) and CXCL10 or ELISA for calprotectin and CXCL10 (B). The results of qPCR are presented relative increase (fold) to those of control siRNA treated, uninfected corneas, set as 1 after normalization to the level of β-actin as the internal control. The results of ELISA are presented as pg/μg total protein. NL, naïve corneas; CT control siRNA *P < 0.05 and P < 0.01 (ANOVA for qPCR; unpaired T test for ELISA). Data are representative of three independent experiments with 3 corneas per group (mean + s.e.m.).
To determine whether the aforementioned factors were also expressed at the protein level, we selected CXCL10, a multifaceted function protein, and calprotectin (S100A8/A9), for ELISA analyses of mouse CECs (Fig. 4B). At 6 hpi, there was marked upregulation, compared to the naïve corneas, of both CXCL10 (7.52 vs 90.52 pg/μg total proteins) and calprotectin (22.75 vs 57.02). IL-36Ra downregulation augmented CXCL10 markedly (90.52 vs 160.62) and calprotectin moderately (57.02 vs 72.43), whereas IL-1Ra downregulation suppressed CXCL10 (90.52 vs 61.44), but not calprotectin expression in Pa-infected corneas.
Downregulation of IL-1Ra and IL-36Ra had opposing effects on the expression of IL-1 cytokines and antimicrobial peptides in B6 mouse corneas at 1 dpi
The expression of the aforementioned 6 genes were also investigated at 1 dpi in the corneas where heavy infiltrates, including neutrophils, macrophages, dendritic cells, NK cells, and γδT cells, can be detected in the control corneas. At this time point, all 6 genes, including IL-36Ra, were up-regulated in control siRNA-treated corneas at 1 dpi. Consistent with the minimal signs of infection and inflammation observed, the levels of IL-1β, IL-1Ra, IL-36γ, IL-36Ra, S100A8 mRNA were similar to that of naive corneas (NL) with CXCL10 slightly elevated, but significantly lower than infected and control siRNA treated corneas (35.4 vs 15.4 fold increase over the naïve corneas). IL-1Ra downregulation, on the other hand, greatly upregulated IL-1β (77.3 vs 316.6 fold increase), IL-36γ (5.88 vs 8.86), S100A8 (38.74 vs 81.18 fold), CXCL10 (35.4 vs 142.72 fold) in Pa-infected corneas. This elevation may contribute to the severe inflammation in IL1Ra-downregulated corneas (Fig. 5).
Figure 5. Effects of IL36Ra or IL1Ra knockdowns on the gene expression in B6 mouse cornea at 1 dpi.
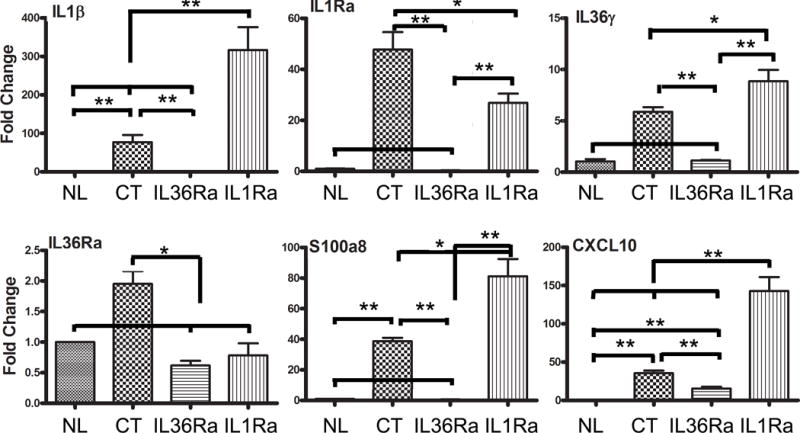
Mouse corneas were treated with siRNAs and inoculated with Pa as in Figure 3. Corneas were excised and processed for real-time PCR analysis at 1dpi. The results are presented as a relative increase (fold) to those of control siRNA-treated, uninfected corneas, set as 1 after normalization to the level of β-actin as the internal control. NL, naïve corneas; CT control siRNA *P < 0.05 and **P < 0.01, and (ANOVA). Data are representative of three independent experiments with 3 corneas per group (mean + s.e.m.).
IL-36Ra and IL-1RA exhibits opposing effects on neutrophil and macrophage infiltrations in infected corneas
Having shown differential control of IL-36Ra and IL-1Ra on the expression of cytokines, chemokines, and AMP, we next assessed their effects on the infiltration of innate immune cells in Pa-infected corneas (Fig. 6). While immune cells can be detected in the naïve cornea, infiltration and an even distribution of neutrophils and macrophages within the stroma were observed in Pa-infected corneas at 1 dpi. The downregulation of IL-1Ra greatly increased the numbers of infiltrated neutrophils and macrophages. Similar to the cytokine expressions, the large numbers of infiltrated cells in IL-1Ra siRNA-treated corneas may be related to strong host responses to the presence of the increased bacterial burden. No infiltrated neutrophils and macrophages were detected in the corneas treated with IL-36Ra siRNA, consistent with the lack of signs of keratitis in Pa-infected corneas at 1 dpi.
Figure 6. Effects of IL36Ra or IL1Ra knockdowns on PMN and macrophage infiltration in B6 mouse cornea at 1 dpi.
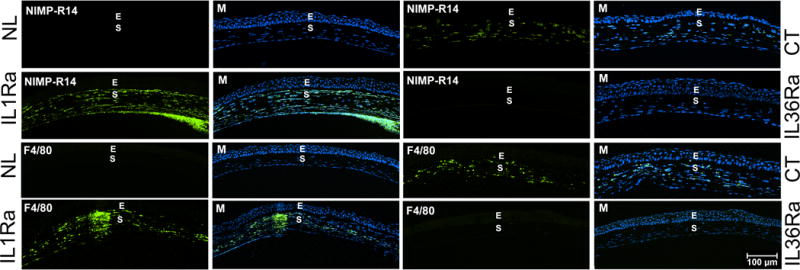
Mouse corneas were treated with control (CT, marked at right side), IL-1Ra (marked at left side), or IL-36Ra (marked at right side) siRNAs and inoculated with Pa as in Figure 3. Naïve corneas (NL, marked at left side) were used as negative control. The corneas were excised and processed for immunohistochemistry analysis at 1dpi. The 6 μ cryostat sections of the corneas were stained with antibody NIMP-R14 for neutrophils and F4/80 for macrophages. The images of infiltrated cells (Green) were merged with DAPI (blue nuclei) staining (M). E epithelium; S stroma. Two independent experiments were performed, 1 representative image for each condition is presented. Scale bar: 100 μm.
Exogenously added IL36Ra and IL-1Ra had opposing effects on the pathogenesis of Pa keratitis in B6 mouse corneas
To further explore the function of IL-36Ra and IL-1Ra, we investigated the effects of exogenous receptor antagonists on the pathogenesis of Pa-keratitis, using 150 μg/ml Anakinra (human recombinant protein), IL-36Ra, or BSA as the control. Figure 7A and 7B showed that at 1 dpi, while mild keratitis can be seen in the control, BSA-treated corneas (CS 4.0), recombinant IL-36Ra increased the severity of keratitis (CS 8.88), including greatly augmented opacification of the cornea, 3 fold surge of bacterial burden, and significantly elevated MPO activity (21.7 vs 38.6 units). Presence of IL-1Ra, on the other hand, inhibited the development of keratitis, resulting in little visible opacification, a very low number of bacteria (176.4±167.2), and low, but measurable, MPO activity (5.19 units), indicating residual levels of PMNs. The same cell lysates used for CFU and MPO determination were also subjected to ELISA measurements of IL-1β and CXCL2, also termed MIP-2. Infection induced IL-1β and CXCL2 expressions (160.1 and 34.96 pg/μg total protein) were markedly augmented by IL-36Ra-treatment (720.7 and 111.5 ng), but were greatly suppressed by IL-1Ra-treatment (5.17 and 4.51 ng) (Fig. C).
Figure 7. Effects of Recombinant IL36Ra or IL1Ra (anakinra) on the severity of Pa keratitis and on the gene expression in B6 mouse corneas.
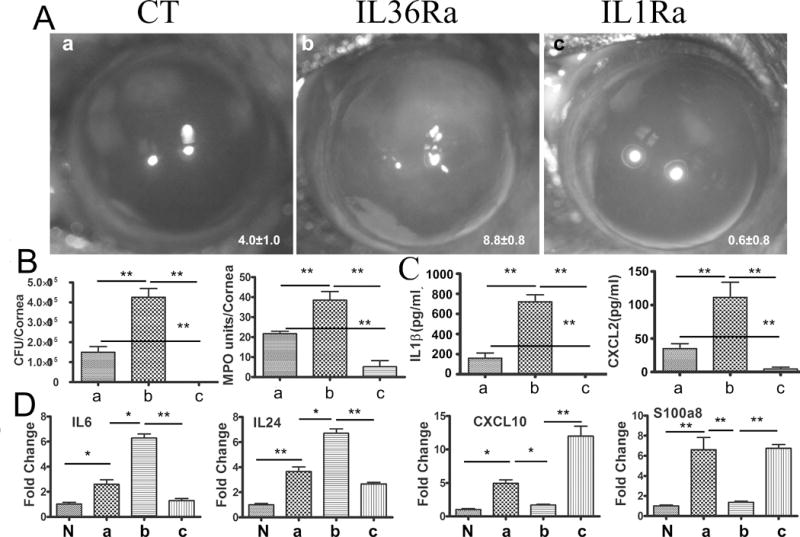
Mice were subconjunctivally injected with either recombinant IL36Ra, anakinra (150 ng) or with 0.1% BSA as the control. Mouse corneas were inoculated with Pa 4 h after the protein injection. (A) Pa-infected corneas were photographed at 1 dpi. The numbers within each eye micrographs are the clinical scores assigned and presented as median + interquartile range. (B) The corneas at 1 dpi were excised and subjected to bacteria load, MPO unit determination, and (C) ELISA assay analysis for CXCL-2 and IL-1β. The letters a, b, and C correspond to the corneas shown in Panel A. (D) To determine the effects of receptor antagonists on corneal gene expression, recombinant IL36Ra or anakinra injected corneas with or without (N, naïve) Pa infection were collected and subjected qPCR analysis. The letters a, b, and C correspond to the corneas shown in Panel A. P values were generated by one-way ANOVA. *P < 0.05 and **P < 0.01. Data are representative of three independent experiments with five mice per group.
To understand the mechanisms underlying IL-36Ra-augmented severity of Pa-keratitis, the expression of cytokines (IL-6, IL-24), and the antimicrobial chemokines CXCL10 and S100A8 in response to exogenous receptor antagonists in CECs at 6 hpi were assessed using qPCR (Fig. 6D). Presence of IL-36Ra augmented the expression of both IL-6 (2.6 vs 6.28 fold increase over the naïve CECs) and IL-24 (3.65 vs 6.70 fold) but suppressed CXCL10 (4.94 vs 1.71 fold) and S100A8 (6.61 vs 1.37 fold) expression. IL-1Ra exhibited opposing effects on the expression of these factors except for S100A8, (2.6 vs 1.2f fold for IL-6; 3.65 vs 2.67 fold for IL-24; and 4.94 vs 11.98 fold for CXCL10).
Exogenous IL-36γ enhances corneal innate immunity and ameliorates Pa keratitis via induction of innate defense genes in B6 mouse corneas
The aforementioned effects of IL-36Ra on the pathogenesis of Pa-keratitis suggest that modulating IL-36 signaling may be used to enhance innate immunity and/or to ease infection-associated inflammation. To test this hypothesis, we subconjunctivally injected murine IL-36γ and assessed the expression innate protective (S100A9, beta-defensin 3, and CXCL10), and IL-1 cytokines (IL-1β and IL-1Ra)(35) in the treated corneas prior to Pa infection (Fig. 8A). IL-36γ treatment without infection stimulated the expression of S100A9 (12.21 fold increase over the naïve corneas), and BD3 (1.83 fold), and CXCL10 (10.56 fold) whereas it exhibited no effects on IL-1Ra and yet downregulated IL-1β expression (1.0 vs 0.26) in the whole corneas (Fig. 8A). The IL-36γ-stimulated expression of S100A9, BD3 and CXCL10 in the corneas were likely participating in pathogen killing at the early stages of infection. To test this hypothesis, we assessed in vitro bactericidal activity by mixing 2000 CFU Pa with whole corneal homogenates. The homogenates derived from IL-36γ treated corneas killed and/or inhibited the growth of Pa (1687 CFU remaining) while in the BSA-treated corneal homogenates Pa numbers increased to 2750 CFU (Fig. 8B).
Figure 8. Recombinant IL-36γ induces the expression of innate immune defense molecules in B6 mouse corneas. (A).
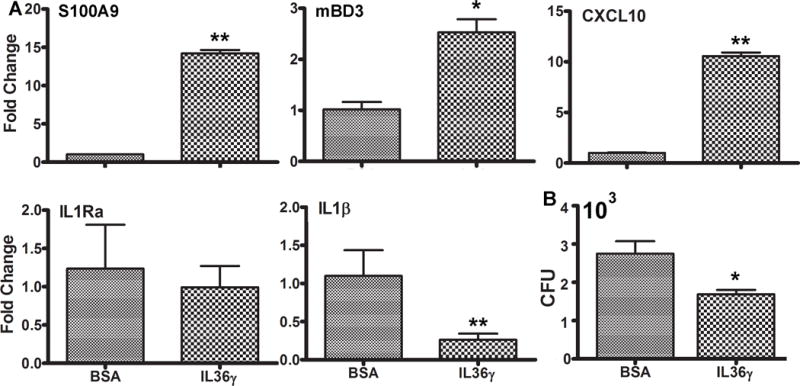
B6 mice were either treated with 0.1%BSA as the control or IL-36γ for 4 hours. The treated corneas were excised and processed for real-time PCR analysis. The results are presented as relative increase (fold) to those of BSA treated, uninfected corneas, set as 1 after normalization to the level of β-actin as the internal control. *P < 0.05 and **P < 0.01, (T test). Data are representative of three independent experiments with 3 corneas per group (mean + s.e.m.). (B) 0.1%BSA or IL-36γ treated corneal homogenates were incubated with 1000 CFU Pa for 1 h at 37°C. The homogenates were then subjected to plate bacterial counting. The results were presented as total CFU per cornea, N=3, *P < 0.05.
The aforementioned IL-36α treated corneas were then inoculated with Pa. At 1 dpi, in the presence of exogenous IL-36γ, the corneas had almost no detectable signs of infection (only 1 of 5 corneas had slight opacification, giving an average clinical score 0.2), no bacteria recoverable by culture, and very low levels of MPO, consistent with corneal photographs showing no sign of opacification and inflammation (Fig. 9). Our previous study revealed that both S100A8 and A9 were the most highly induced AMPs and that neutralizing either subunit resulted in much more severe keratitis in Pa-infected corneas (40). We therefore selected S100A9 neutralizing antibody to be applied concomitantly with IL-36γ (Fig. 9, panel 3). Depletion of S100A9 diminished IL-36γ-stimulated protection, resulting in similar clinical scores, partially decreased bacterial burdens and MPO activity compared to the BSA-treated, control corneas.
Figure 9. Recombinant IL36γ prevents B6 mouse corneas from Pa infection in a S100A9-dependnet manner.
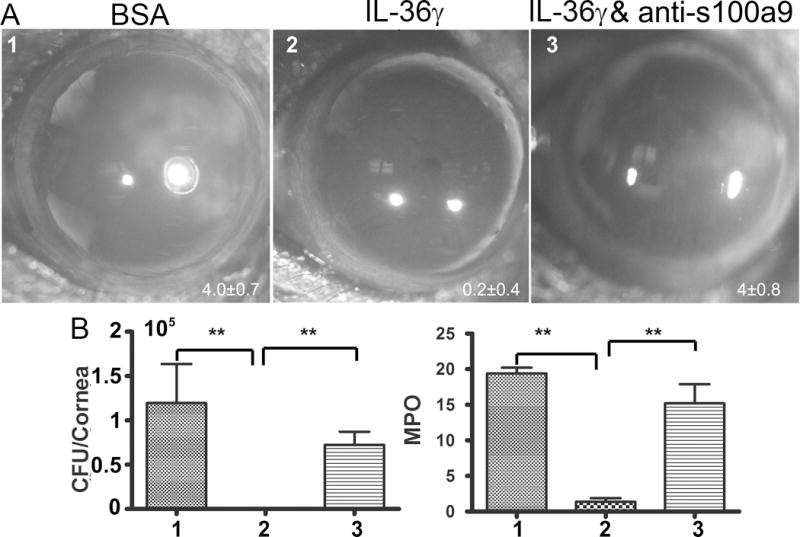
Mice were subconjunctivally injected with BSA as the control, 150ng IL36γ+100 ng IgG, or 150ng IL36γ+100 ng S100A9 neutralizing antibody in 5 μi PBS. Mouse corneas were inoculated with Pa 4 h after the injection. (A) Pa-infected corneas were photographed 1 dpi. The numbers within each eye micrographs are the clinical scores assigned and presented as median + interquartile range. At 1 dpi, the corneas were excised and subjected to bacteria load (B) and MPO unit determination. Data are representative of three independent experiments with five mice per group. (**p<0.01).
Finally, the involvement of CXCL10 as well as its chemokine receptor CXCR3 in IL-36 stimulated innate protection were also assessed using neutralizing antibodies. Neutralizations of CXCL10 and CXCR3 only had limited effects on bacterial burden (229200 CFU for the control infected, 0 for IL-36γ pretreated, 26760 for CXCL10, and 89320 for CXCR3 neutralized corneas) and PMN infiltration assessed by MPO assay (23,17 units/cornea for the control infected, 1.4 for IL-36γ pretreated, 5.4 for CXCL10, and 11.75 for CXCR3 neutralized corneas). Neutralization of CXCR3 had more profound effects on the positive effects of IL-36 than that of CXCL10.
Discussion
In this study, we compared the expression and function of two large IL-1 cytokine subfamilies by manipulating their receptor antagonists in a mouse model of Pa-keratitis. We observed that Pa triggers the expression of IL-1β, IL-Ra, IL-36α, IL-36γ but IL-36Ra in human and mouse CECs and in whole B6 mouse corneas. We also detected IL-36γ secretion in cultured human CECs challenged with H-K Pa. Surprisingly, IL-36Ra and IL-IRa knockdown resulted in opposing effects on the severity of Pa-keratitis: IL-36Ra downregulation prevented corneal infection while IL-1Ra greatly increased the severity of Pa-keratitis. During the early stages of infection (6 hpi), IL-36Ra downregulation decreased the ratio of IL-1β/IL-1Ra, increased the expression of IL-36γ, S100A8, and CXCL10 while IL-1Ra siRNA had opposing effects in mouse CECs. At 1 dpi, IL-1Ra siRNA altered the ratio of IL-1β/IL-Ra and increased expression of IL-36γ, S100A8 and CXCL10; these factors were minimally detected in IL-36-siRNA-treated corneas. Furthermore, exogenously-added IL-1Ra and IL-36γ prevented, while IL-36Ra augmented the pathogenesis of Pa-keratitis. IL-36γ induced or augmented the expression of innate defense molecules S100A9, mouse β-defensin 3, and ISG15 in naïve cornea and enhanced bactericidal activity of corneal homogenates. IL-36β-induced protection was abolished in the presence of S100A9 neutralizing antibody. Thus, IL-36/IL-36R and IL-1/IL-1R signaling pathways had opposing effects on the expression of immunoregulatory and effector genes such as AMPs and, as a consequence, on the outcome of Pa-keratitis.
The expression, secretion, and/or activation of the IL-1 subfamily of cytokines have been studied extensively. In this study, we compared the expression patterns of IL-1 and IL-36 cytokines in CECs and in the corneas in response to infection. Our study revealed that unlike IL-1β and sIL-1RN, IL-36γ and IL-36RN mRNA were present in these naïve cells, suggesting a potential role of IL-36/IL-36R signaling in corneal homeostasis. Infection induced the expression of IL-1β, sIL-1Ra, IL-36α, 36γ as early as 2 hpi, and IL-36Ra at 1 dpi. Moreover, our study for the first time showed the secretion of IL-36γ by Pa-challenged human CECs. The mechanisms underlying IL-36 cytokine secretion, including IL-36Ra remain largely undetermined. The expression pattern demonstrated in the current study suggests that IL-36 cytokines are a part of the network of cytokines/chemokines involved in tissue innate immune response to microbial infection, as that shown in the lung (25, 28, 29).
Taking advantage of the subconjunctival injection procedure and siRNA technology, we observed striking differences in the pathogenesis of Pa keratitis: IL-1Ra downregulation greatly exacerbated keratitis while IL-36Ra siRNA protected B6 mouse corneas from P. aeruginosa. The opposing effects of IL-1Ra and IL-36Ra on the outcomes of P. aeruginosa keratitis were further corroborated by the use of recombinant human IL-1Ra (Anakinra) and mouse IL-36Ra. These antagonists exhibited opposing effects on the pathogenesis of Pa-keratitis: IL-1Ra (Anakinra) supported while IL-36Ra suppressed corneal innate defense against Pa. In these treated corneas, the severities of keratitis were assessed by opacification, quantitated as clinical scores, and by pathogen burdens. Zhou and Pearlman have shown that recruitment of PMN and activation of IL-1R signaling were associated with stromal thickness and haze in S. marcescens-infected corneas (42). Our data also showed a strong association of elevated IL-1β expression and/or altered balance of IL-1β/sIL-1Ra with neutrophil infiltration which in turn caused corneal opacification in an IL-36 related manner. To our knowledge, this is the first study that directly compared the effects of two receptor antagonists on bacterial infection. The striking differences in the outcomes of keratitis between the manipulations of IL-1R and IL-36R mediated signaling pathways indicate that while the activation of IL-1 signaling may be detrimental, IL-36/IL-36R axis play a protective role in controlling P. aeruginosa keratitis in B6 mouse corneas.
Like IL-1 cytokines, IL-36 agonists are generally considered as pro-inflammatory (43, 44). The IL-1 and IL-36 subfamilies share IL-1 receptor accessory protein (IL-1RAcP) as a co-receptor and activate NF-κB and p38 signaling pathways through MyD88 (3, 43). It is now evident that IL-36 cytokines play an important role in inflammatory disorders in different organs such as the gastrointestinal tract (45) and the lung (25, 46). In the skin, all three IL-36R agonists were found to be highly expressed and were involved in the pathogenesis of psoriasis, a chronic inflammatory, multi-system disease (47-49). Mutations of IL-36RN, which encodes IL-36Ra, causes pustular dermatosis (GPP) (19), providing support for the pro-inflammatory nature of IL-36/IL-36R signaling. Interestingly, GPP carrying IL36Ra mutations can be successfully treated with Anakinra, a human recombinant IL-1Ra, suggesting that the culprit for the diseases was excessive activation of IL-1 signaling pathway. Hence, how does Anakinra abrogate the detrimental effects of IL-36Ra insufficiency? One possibility is that IL-1 cytokines are the downstream targets of IL-36. Indeed, our study revealed that downregulating IL-36Ra, exogenous IL-1Ra, in contrast to IL-1Ra knockdown or exogenous IL-1Ra, attenuated IL-1β, but augmented the expression of sIL-1Ra at an early stage (6 hpi) of infection. At this time point, most invading bacteria are restricted within the epithelial layers (35). Hence, IL-36 signaling may alter the IL-1β/IL-1Ra balance in favor of anti-inflammatory effects and IL-36/IL-36R signaling is anti-inflammatory in Pa keratitis.
How might IL-36R activation by siRNA knockdown of IL-36Ra or exogenous IL-36γ mediate the innate killing of invading pathogens, resulting in total eradication of inoculated P. aeruginosa in B6 mouse corneas? Our data shown that the downregulation of IL-36Ra, in contrast to that of IL-1RA, augmented the infection-induced expression of S100A8 and CXCL10 at both the mRNA and protein levels, whereas exogenous IL-36Ra stimulation greatly downregulated their expression in CECs at 6 hpi. Hence, we propose that IL-36R activation-stimulated CXCL10 and/or calprotectin are the effectors that play an important role in innate defense during early time of Pa infection of the cornea. However, at 1 dpi,, the expression of aforementioned factors, except CXCL10, were either undetectable or similar to the naïve controls in IL-36Ra siRNA treated corneas, consistent with the eradication of invading pathogens and the resolution of inflammation, as evidenced by the lack of infiltrated neutrophils and macrophages in the corneas at this time point. In IL-1Ra siRNA treated corneas, while the balance of IL-1β/IL-1Ra remained in favor of pro-inflammatory, the infection induced expressions of IL-36γ, S100A8, and CXCL10 were all elevated, in contrast to that observed in CECS at 6 dpi. This, however, may be related to the strong response of the host to the increased pathogen burden in the treated corneas. Furthermore, in Il-36γ treated corneas, three innate defense molecules, S100A9, mouse β-defensin 3 (homolog of human β-defensin 2), and CXCL10 were induced in naïve corneas while IL-1β was suppressed. Hence, IL-36 cytokines may control the outcome of bacterial keratitis by suppressing proinflammatory IL-1R signaling and by stimulating the expression of AMPs in the epithelium at the initial stage of infection, resulting in the eradication of invading pathogens and rapid resolution of inflammation associated with infection. Our in vitro Pa killing assay results support a role of IL-36R signaling in promoting innate immune killing of invading pathogens in the cornea. Moreover, neutralizing antibody of S100A9 abolished IL-36γ-induced protection, resulting in Pa keratitis with smaller pathogen burden and fewer infiltrated PMNs when comparing to the control, Pa-infected corneas. On the other hand, neutralizing CXCL10 had significant but limited effects on bacterial burden and on PMN infiltration while the effects of neutralizing its receptor CXCR3 were significantly higher. Our previous studies revealed that CXCL10 neutralization had no effect on the outcome of Pa keratitis (50) whereas the neutralizations of CXCL10 and CXCR3 resulted in much severe keratitis in Candida albicans infected corneas (38, 41). Cells expressing CXCR3 at early stage of infection are NK cells that are necessary for innate defense against both Pa and Candida albicans infection of the cornea (38, 41, 50). Thus, CXCL10, likely functioning through CXCR3 as a chemokine, plays a minor role in IL-36γ stimulated innate protection against Pa infection and that other CXCR3 ligands, CXCL9 and/or CXCL11, might also be involved in B6 mouse corneas. We proposed that upregulation of AMPs such as calprotectin and/or β-defensins are an underlying mechanism for IL-36-IL-36R axis to enhance cornea innate immunity against infectious keratitis. We further propose a novel concept that the IL-36 subfamily or IL-36R signaling may antagonize that of the IL-1 subfamily in mediating innate immunity early during P. aeruginosa infection of B6 mouse corneas.
Finally, our study suggests that Anakinra may be used as an adjuvant treatment for bacterial keratitis and that IL-36 agonist may be used as an alternative therapeutic of IL-1 neutralization medications in treating a host of inflammation and infection diseases. This seems paradoxical, given that IL-1 is a master cytokine of local and/or systemic inflammation, which is required for immune cell infiltration and for the effective eradication of invading pathogens. For example, mice deficient in IL-1R had higher Pa burden in the lungs (51, 52) and suffered lethal necrotic pneumonia following M. tuberculosis infection (53). This paradox can perhaps be resolved by the findings regarding long-term use of Anakinra; Anakinra has been used to treat rheumatoid arthritis and related diseases in over 100,000 patients, some over 15 years without significant increases in the rate of infection for the patients (54, 55). In patients with chronic granulomatous disease, an inherited condition with multiple bouts of infections with Gram-positive and Gram-negative bacteria as well as fungi, Anakinra treatment reduces the severity of inflammatory bowel disease and disease-associated granulomas without increased infection (56-58). Hence, local application of Anakinra, IL-36 agonist, alone or in combination, may be used as an adjuvant treatment with antibiotics for infectious keratitis.
Figure 10. CXC10/CXCR3 signaling plays a minor role in IL-36γ stimulated innate protection against Pa infection.
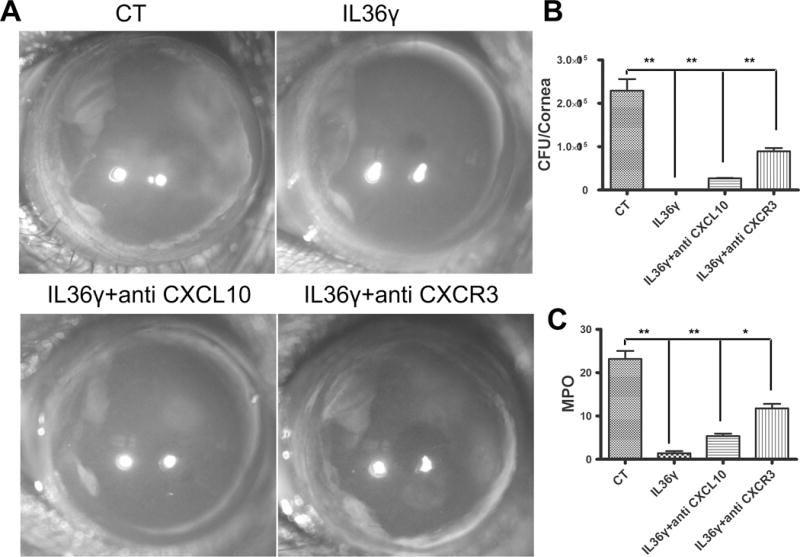
Mice were subconjunctivally injected with 0.1% BSA as the control (CT), 150 ng IL36γ+100 ng IgG, or 150ng IL36γ + 100 ng CXCL10 or CXCR3 neutralizing antibody in 5 μi PBS. Mouse corneas were inoculated with Pa 4 h after the injection. (A) Pa-infected corneas were photographed 1 dpi. The numbers within each eye micrographs are the clinical scores assigned and presented as median + interquartile range. At 1 dpi, the corneas were excised and subjected to bacteria load (B) and MPO unit determination. Data are representative of three independent experiments with five mice per group. (*p<0.05, **p<0.01).
Acknowledgments
The authors would like to thank all the members of the Yu laboratories for assistance and comments on the work. We thank Patrick Lee for critical reading of the manuscript.
We acknowledge support from NIH/NEI R01EY10869, EY17960 (to FSY), p30 EY04078 (NEI core to WSU), Research to Prevent Blindness (to Kresge Eye Institute).
Funding:
This work was supported by the National Eye Institute at the National Institutes of Health [R01 EY017960, R01 EY010869 to FSY] and Research to Prevent Blindness (to Mark Juzych, Chair, Kresge Eye Institute).
Footnotes
The authors declare that there is no conflict of interest associated with this manuscript.
Conflict of Interests: None
The information has previously been presented at the Annual Meeting of the Association for Research in Vision and Ophthalmology (May 2013, #1728)
References
- 1.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 2.Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- 3.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hahn M, Frey S, Hueber AJ. The novel interleukin-1 cytokine family members in inflammatory diseases. Curr Opin Rheumatol. 2017;29:208–213. doi: 10.1097/BOR.0000000000000361. [DOI] [PubMed] [Google Scholar]
- 5.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 7.Yan C, Gao N, Sun H, Yin J, Lee P, Zhou L, Fan X, Yu FS. Targeting Imbalance between IL-1beta and IL-1 Receptor Antagonist Ameliorates Delayed Epithelium Wound Healing in Diabetic Mouse Corneas. Am J Pathol. 2016;186:1466–1480. doi: 10.1016/j.ajpath.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family–Balance between agonists and antagonists in inflammatory diseases. Cytokine. 2015;76:25–37. doi: 10.1016/j.cyto.2015.06.017. [DOI] [PubMed] [Google Scholar]
- 9.Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002;13:323–340. doi: 10.1016/s1359-6101(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 10.Mattii M, Ayala F, Balato N, Filotico R, Lembo S, Schiattarella M, Patruno C, Marone G, Balato A. The balance between pro- and anti-inflammatory cytokines is crucial in human allergic contact dermatitis pathogenesis: the role of IL-1 family members. Exp Dermatol. 2013;22:813–819. doi: 10.1111/exd.12272. [DOI] [PubMed] [Google Scholar]
- 11.Arend WP. Cytokine imbalance in the pathogenesis of rheumatoid arthritis: the role of interleukin-1 receptor antagonist. Semin Arthritis Rheum. 2001;30:1–6. doi: 10.1053/sarh.2001.23693. [DOI] [PubMed] [Google Scholar]
- 12.Cavalli G, Dinarello CA. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology (Oxford) 2015 doi: 10.1093/rheumatology/kev269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Killu AM, Liang JJ, Jaffe AS. Erdheim-Chester disease with cardiac involvement successfully treated with anakinra. Int J Cardiol. 2013;167:e115–117. doi: 10.1016/j.ijcard.2013.04.057. [DOI] [PubMed] [Google Scholar]
- 14.van Asseldonk EJ, van Poppel PC, Ballak DB, Stienstra R, Netea MG, Tack CJ. One week treatment with the IL-1 receptor antagonist anakinra leads to a sustained improvement in insulin sensitivity in insulin resistant patients with type 1 diabetes mellitus. Clin Immunol. 2015 doi: 10.1016/j.clim.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 15.van Poppel PC, van Asseldonk EJ, Holst JJ, Vilsboll T, Netea MG, Tack CJ. The interleukin-1 receptor antagonist anakinra improves first-phase insulin secretion and insulinogenic index in subjects with impaired glucose tolerance. Diabetes Obes Metab. 2014;16:1269–1273. doi: 10.1111/dom.12357. [DOI] [PubMed] [Google Scholar]
- 16.Rajan N, Sinclair N, Nakai H, Shimomura Y, Natarajan S. A tale of two sisters: identical IL36RN mutations and discordant phenotypes. Br J Dermatol. 2015 doi: 10.1111/bjd.14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farooq M, Nakai H, Fujimoto A, Fujikawa H, Matsuyama A, Kariya N, Aizawa A, Fujiwara H, Ito M, Shimomura Y. Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Hum Mutat. 2013;34:176–183. doi: 10.1002/humu.22203. [DOI] [PubMed] [Google Scholar]
- 18.Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO, Burden AD, Capon F, Trembath RC, Barker JN. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89:432–437. doi: 10.1016/j.ajhg.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, Towne JE, Douangpanya J, Pons C, Mansour S, Serre V, Makni H, Mahfoudh N, Fakhfakh F, Bodemer C, Feingold J, Hadj-Rabia S, Favre M, Genin E, Sahbatou M, Munnich A, Casanova JL, Sims JE, Turki H, Bachelez H, Smahi A. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–628. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]
- 20.Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol. 2015;97:645–652. doi: 10.1189/jlb.3RI1014-495R. [DOI] [PubMed] [Google Scholar]
- 21.Tripodi D, Conti F, Rosati M, Maccauro G, Saggini A, Cianchetti E, Angelucci D, Fulcheri M, Tete S, Salini V, Caraffa A, Antinolfi P, Toniato E, Castellani ML, Conti P, Theoharides TC. IL-36 a new member of the IL-1 family cytokines. J Biol Regul Homeost Agents. 2012;26:7–14. [PubMed] [Google Scholar]
- 22.Gresnigt MS, van de Veerdonk FL. Biology of IL-36 cytokines and their role in disease. Semin Immunol. 2013;25:458–465. doi: 10.1016/j.smim.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279:13677–13688. doi: 10.1074/jbc.M400117200. [DOI] [PubMed] [Google Scholar]
- 24.van de Veerdonk FL, Stoeckman AK, Wu G, Boeckermann AN, Azam T, Netea MG, Joosten LA, van der Meer JW, Hao R, Kalabokis V, Dinarello CA. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc Natl Acad Sci U S A. 2012;109:3001–3005. doi: 10.1073/pnas.1121534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoyagi T, Newstead MW, Zeng X, Kunkel SL, Kaku M, Standiford TJ. IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol. 2017;10:1043–1055. doi: 10.1038/mi.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Segueni N, Vigne S, Palmer G, Bourigault ML, Olleros ML, Vesin D, Garcia I, Ryffel B, Quesniaux VF, Gabay C. Limited Contribution of IL-36 versus IL-1 and TNF Pathways in Host Response to Mycobacterial Infection. PLoS One. 2015;10:e0126058. doi: 10.1371/journal.pone.0126058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahsan F, Moura-Alves P, Guhlich-Bornhof U, Klemm M, Kaufmann SH, Maertzdorf J. Role of Interleukin 36gamma in Host Defense Against Tuberculosis. J Infect Dis. 2016;214:464–474. doi: 10.1093/infdis/jiw152. [DOI] [PubMed] [Google Scholar]
- 28.Kovach MA, Singer B, Martinez-Colon G, Newstead MW, Zeng X, Mancuso P, Moore TA, Kunkel SL, Peters-Golden M, Moore BB, Standiford TJ. IL-36gamma is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal Immunol. 2017 doi: 10.1038/mi.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoyagi T, Newstead MW, Zeng X, Nanjo Y, Peters-Golden M, Kaku M, Standiford TJ. Interleukin-36gamma and IL-36 receptor signaling mediate impaired host immunity and lung injury in cytotoxic Pseudomonas aeruginosa pulmonary infection: Role of prostaglandin E2. PLoS Pathog. 2017;13:e1006737. doi: 10.1371/journal.ppat.1006737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleiszig SM, Evans DJ. The pathogenesis of bacterial keratitis: studies with Pseudomonas aeruginosa. Clin Exp Optom. 2002;85:271–278. doi: 10.1111/j.1444-0938.2002.tb03082.x. [DOI] [PubMed] [Google Scholar]
- 31.Hazlett LD. Bacterial infections of the cornea (Pseudomonas aeruginosa) Chemical immunology and allergy. 2007;92:185–194. doi: 10.1159/000099269. [DOI] [PubMed] [Google Scholar]
- 32.Alarcon I, Kwan L, Yu C, Evans DJ, Fleiszig SM. Role of the corneal epithelial basement membrane in ocular defense against Pseudomonas aeruginosa. Infect Immun. 2009;77:3264–3271. doi: 10.1128/IAI.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alarcon I, Tam C, Mun JJ, LeDue J, Evans DJ, Fleiszig SM. Factors impacting corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Invest Ophthalmol Vis Sci. 2011;52:1368–1377. doi: 10.1167/iovs.10-6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans DJ, Fleiszig SM. Why does the healthy cornea resist Pseudomonas aeruginosa infection? Am J Ophthalmol. 2013;155:961–970 e962. doi: 10.1016/j.ajo.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao N, Kumar A, Yu FS. Matrix Metalloproteinase-13 as a Target for Suppressing Corneal Ulceration Caused by Pseudomonas aeruginosa Infection. J Infect Dis. 2015;212:116–127. doi: 10.1093/infdis/jiv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazlett LD. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res. 2004;23:1–30. doi: 10.1016/j.preteyeres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Carvalho FA, Aitken JD, Gewirtz AT, Vijay-Kumar M. TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol. 2011;4:102–111. doi: 10.1038/mi.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, Gao N, Dong C, Zhou L, Mi QS, Standiford TJ, Yu FS. Flagellin-induced expression of CXCL10 mediates direct fungal killing and recruitment of NK cells to the cornea in response to Candida albicans infection. Eur J Immunol. 2014;44:2667–2679. doi: 10.1002/eji.201444490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross BX, Gao N, Cui X, Standiford TJ, Xu J, Yu FX. IL-24 Promotes Pseudomonas aeruginosa Keratitis in C57BL/6 Mouse Corneas. J Immunol. 2017;198:3536–3547. doi: 10.4049/jimmunol.1602087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao N, Sang Yoon G, Liu X, Mi X, Chen W, Standiford TJ, Yu FS. Genome-wide transcriptional analysis of differentially expressed genes in flagellin-pretreated mouse corneal epithelial cells in response to Pseudomonas aeruginosa: involvement of S100A8/A9. Mucosal immunology. 2013;6:993–1005. doi: 10.1038/mi.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao N, Liu X, Wu J, Li J, Dong C, Wu X, Xiao X, Yu FX. CXCL10 suppression of hem- and lymph-angiogenesis in inflamed corneas through MMP13. Angiogenesis. 2017;20:505–518. doi: 10.1007/s10456-017-9561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou R, Zhang R, Sun Y, Platt S, Szczotka-Flynn L, Pearlman E. Innate immune regulation of Serratia marcescens-induced corneal inflammation and infection. Investigative ophthalmology & visual science. 2012;53:7382–7388. doi: 10.1167/iovs.12-10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bassoy EY, Towne JE, Gabay C. Regulation and function of interleukin-36 cytokines. Immunol Rev. 2018;281:169–178. doi: 10.1111/imr.12610. [DOI] [PubMed] [Google Scholar]
- 44.Jensen LE. Interleukin-36 cytokines may overcome microbial immune evasion strategies that inhibit interleukin-1 family signaling. Sci Signal. 2017;10 doi: 10.1126/scisignal.aan3589. [DOI] [PubMed] [Google Scholar]
- 45.Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, Statovci D, Moran T, Fallon PG, Shanahan F, Brint EK, Melgar S, Hussey S, Walsh PT. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol. 2016 doi: 10.1038/mi.2015.134. [DOI] [PubMed] [Google Scholar]
- 46.Ramadas RA, Ewart SL, Iwakura Y, Medoff BD, LeVine AM. IL-36alpha exerts pro-inflammatory effects in the lungs of mice. PLoS One. 2012;7:e45784. doi: 10.1371/journal.pone.0045784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol. 2012;12:486–490. doi: 10.1016/j.coph.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, Young DA, Fouser LA, Nickerson-Nutter C, Collins M, Dunussi-Joannopoulos K, Medley QG. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131:2428–2437. doi: 10.1038/jid.2011.234. [DOI] [PubMed] [Google Scholar]
- 49.Dietrich D, Gabay C. Inflammation: IL-36 has proinflammatory effects in skin but not in joints. Nat Rev Rheumatol. 2014;10:639–640. doi: 10.1038/nrrheum.2014.156. [DOI] [PubMed] [Google Scholar]
- 50.Yoon GS, Dong C, Gao N, Kumar A, Standiford TJ, Yu FS. Interferon Regulatory Factor-1 in Flagellin-Induced Reprogramming: Potential Protective Role of CXCL10 in Cornea Innate Defense Against Pseudomonas aeruginosa Infection. Invest Ophthalmol Vis Sci. 2013;54:7510–7521. doi: 10.1167/iovs.13-12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veliz Rodriguez T, Moalli F, Polentarutti N, Paroni M, Bonavita E, Anselmo A, Nebuloni M, Mantero S, Jaillon S, Bragonzi A, Mantovani A, Riva F, Garlanda C. Role of Toll interleukin-1 receptor (IL-1R) 8, a negative regulator of IL-1R/Toll-like receptor signaling, in resistance to acute Pseudomonas aeruginosa lung infection. Infection and immunity. 2012;80:100–109. doi: 10.1128/IAI.05695-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reiniger N, Lee MM, Coleman FT, Ray C, Golan DE, Pier GB. Resistance to Pseudomonas aeruginosa chronic lung infection requires cystic fibrosis transmembrane conductance regulator-modulated interleukin-1 (IL-1) release and signaling through the IL-1 receptor. Infect Immun. 2007;75:1598–1608. doi: 10.1128/IAI.01980-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, Ryffel B, Quesniaux VF. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–1189. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 54.Dinarello CA. Overview of the interleukin-1 family of ligands and receptors. Semin Immunol. 2013;25:389–393. doi: 10.1016/j.smim.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 55.Fleischmann R, Stern R, Iqbal I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2004;4:1333–1344. doi: 10.1517/14712598.4.8.1333. [DOI] [PubMed] [Google Scholar]
- 56.de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, Begun J, Plantinga TS, Joosten LA, van der Meer JW, Chamilos G, Netea MG, Xavier RJ, Dinarello CA, Romani L, van de Veerdonk FL. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A. 2014;111:3526–3531. doi: 10.1073/pnas.1322831111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van de Veerdonk FL, Netea MG, Dinarello CA, van der Meer JW. Anakinra for the inflammatory complications of chronic granulomatous disease. Neth J Med. 2011;69:95. [PubMed] [Google Scholar]
- 58.Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25:469–484. doi: 10.1016/j.smim.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]