

Abstract
A palladium(II)-catalyzed β,γ-aminohydroxylation reaction of non-conjugated alkenyl carbonyl compounds has been developed. This reaction utilizes a cleavable bidentate directing group to achieve regioselective aminopalladation. The resulting chelation-stabilized alkylpalladium(II) intermediate is then hydroxylated using oxygen/2,6-dimethylbenzoquinone in HFIP as the mild oxidation system. Under optimized conditions, various nucleophiles and alkene substrates are capable of delivering good yields and high diastereoselectivity of the aminohydroxylated product.
Graphical Abstract
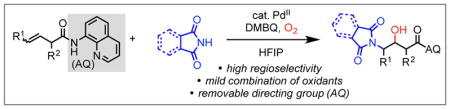
Since its discovery in 1996 by Sharpless and coworkers,1 catalytic aminohydroxylation of alkenes has been recognized as a powerful tool in synthetic chemistry to enable expedient access to 1,2-amino alcohols, including those embedded in bioactive compounds.2 Since then, significant effort has been devoted to expanding the scope of the coupling partners3 and achieving analogous aminooxygenation reactivity using palladium,4,5 copper,6 iron,7 platinum,8 and nonmetal9 catalysts to replace osmium. In particular, the past two decades have witnessed significant advancements in the area of palladium(II)-catalyzed aminooxygenation. Mechanistically, such reactions generally require rapid interception of the reactive aminopalladated alkylpalladium(II) species with a strong oxidant to facilitate C–O reductive elimination from a high-valent intermediate. Intramolecular palladium(II)-catalyzed aminohydroxylations have generally tolerated a broader scope of oxidants than intermolecular variants. Examples of the former category have been reported with hypervalent iodine,4a copper chloride,4b hydrogen peroxide/acetic acid,4c oxygen/acetic acid,4d and nitrogen dioxide from copper(II) nitrate trihydrate4e (Scheme 1A). On the other hand, the latter category, has only been described with hypervalent iodine oxidants to the best of our knowledge (Scheme 1B).5 Though efficacious, these strong oxidants are disadvantageous in terms of functional group compatibility and atom economy. The goal of the present study was to develop an intermolecular alkene 1,2-aminooxygenation reaction using a mild oxidation system by taking advantage of a substrate directivity approach.
Scheme 1.

Background and Project Synopsis
Our group has previously developed a series of intermolecular alkene hydrofunctionalization and 1,2-difunctionalization reactions by utilizing a bidentate directing group to govern regioselectivity in the Wacker-type nucleometallation step and also to stabilize the resulting alkylpalladium(II) species for downstream elementary steps.10,11 Based on this precedent, we questioned whether these intermediates would be sufficiently long-lived to react with weaker electrophilic O-atom sources. We were particularly interested in accessing aminooxygenated products bearing free OH groups directly, as such reactions are historically less precedented.
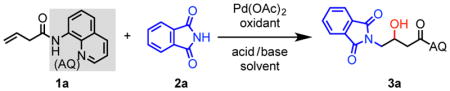
To initiate our investigation, we considered 3-butenoic acid masked as its 8-aminoquinoline (AQ) amide (1a) as the model substrate, phthalimide as nitrogen nucleophile, hexafluoroisopropanol (HFIP) as solvent, and 2,6-dimethylbenzoquinone (DMBQ) as oxidant under air at 100 °C (Table 1). We were excited to find that these conditions yielded 43% of the aminohydroxylated product. Upon screening oxidant (entry 1–3), acid/base (entry 4–7), solvent (entry 8–10), and temperature (entries 1, 11, and 12), we found that our first attempt gave the highest yield. We then performed control experiments in an attempt to identify the oxygen atom source in the system (entries 13 and 14) and found that no product was detected when the reaction was run under nitrogen atmosphere. This led us to speculate that oxygen in the air was the oxygen source and an essential component of the catalytic cycle. Therefore, we continued to optimize the equivalents of DMBQ (entry 15–18) under oxygen atmosphere, and we found that 2 equiv DMBQ yielded 80% product (entry 17).12
Table 1.
Optimization of the Reaction Conditionsa
| |||||
|---|---|---|---|---|---|
| entry | acid / base | oxidant | atmosphere | solvent | yieldb |
| 1 | KHCO3 | DMBQ | air | HFIP | (59) |
| 2 | KHCO3 | benzoquinone | air | HFIP | (34) |
| 3 | KHCO3 | dimethoxybenzoquinone | air | HFIP | (7) |
| 4 | AcOH | DMBQ | air | HFIP | n.d. |
| 5c | KHCO3 | DMBQ | air | HFIP | (40) |
| 6 | K2HPO4 | DMBQ | air | HFIP | (42) |
| 7 | iPr2NEt | DMBQ | air | HFIP | (30) |
| 8 | KHCO3 | DMBQ | air | tAmylOH | (10) |
| 9 | KHCO3 | DMBQ | air | DMF | (5) |
| 10 | KHCO3 | DMBQ | air | dioxane | (10) |
| 11d | KHCO3 | DMBQ | air | HFIP | (12) |
| 12e | KHCO3 | DMBQ | air | HFIP | (14) |
| 13f | KHCO3 | DMBQ | air | HFIP | (23) |
| 14 | KHCO3 | DMBQ | N2 | HFIP | n.d. |
| 15 | KHCO3 | DMBQ | O2 | HFIP | 65 |
| 16 | KHCO3 | DMBQ (0.5 equiv) | O2 | HFIP | 50 |
| 17 | KHCO3 | DMBQ (2.0 equiv) | O2 | HFIP | 80 |
| 18 | KHCO3 | DMBQ (3.0 equiv) | O2 | HFIP | 73 |
Reaction conditions: 1a (0.1 mmol), 2a (1.5 equiv), Pd(OAc)2 (10 mol %), oxidant (1.5 equiv), base (1 equiv), solvent (0.2 mL), 100 °C, 12–16 h.
Isolated yield. Values in parentheses represent yields determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-triisopropylbenzene as internal standard.
KHCO3 (0.5 equiv).
120 °C.
80 °C.
4Å molecular sieves as additive.
Having identified optimal conditions, we next investigated the scope of this Pd(II)-catalyzed alkene aminohydroxylation reaction. First, nitrogen nucleophiles were tested with alkene substrate 1a. Similar to earlier work,5 we found that cyclic imide nucleophiles with structures similar to phthalimide were compatible coupling partners (3a–3e). However, several other nitrogen nucleophiles that had previously been proven to be competent in related reactions,10a,10d,10e including saccharin, carbazole, isatin, glutarimide, and thalidomide, were incompatible under these reaction conditions (see Supporting Information). We also attempted selected 1,3-dicarbonyl carbon nucleophiles, but no carbohydroxylated product was observed in these cases. The nucleophile scope for this transformation is much more restricted than that of our other palladium(II)-catalyzed difunctionalization reactions, and at this stage we are unclear on the underlying origin of this phenomenon. One possibility is that after nucleopalladation, a carbonyl group on the phthalimide coordinates to the palladium as a third ligand,10b,10f thereby tuning the steric and electronic environment around the palladium to enable coordination and reaction with the active O-atom electrophile (e.g., O2).
Subsequently, we examined the scope of non-conjugated alkene substrates with phthalimide 2a as the nucleophile. An array of α-substituted terminal alkene substrates performed well under the reaction conditions, leading to formation of a single diastereomer in all cases (3f–3k).10c The transformation, however, was found to be sensitive to steric hinderance at the α-position. In particular, attempts to react alkenes with a bulky α-isopropyl group or α, α-gem-dimethyl substitution led only to recovered starting material. We were pleased to find that an internal 1,2-dialkyl alkene also worked to some extent in this reaction (3l), though it required longer reaction time, higher catalyst loading, and elevated temperature. More sterically congested internal alkenes did not yield the desired product. Interestingly, this reaction also gave small quantities of product when an alternative non-conjugated alkene substrate class was used, namely homoallyl amines masked by a picolinamide bidentate directing group (3m). Several other bidentate directing groups were also tested, but none were effective in this reaction (see Supplementary Information).
To demonstrate the practical utility and operational simplicity of this palladium(II)-catalyzed alkene aminohydroxylation method, we performed the reaction on 2.0-mmol scale with alkene substrate 1a and 1.0-mmol scale with 1f using phthalimide (2a) as nucleophile. In both cases, reaction performance was consistent with that of smaller scale reactions, providing 75% and 82% yields, respectively (Scheme 2).
Scheme 2.

Aminohydroxylation Scale-Up
Several downstream manipulations of the aminohydroxylated product were next performed (Scheme 3). To cleave the AQ directing group, the free hydroxy group was first protected with a tert-butyldimethylsilyl (TBS) group, and the AQ was then hydrolyzed in two steps, affording the deprotected carboxylic acid product 4 in quantitative yield.10d The AQ directing group could also be transformed in one step to ester derivative 5 via nickel-catalyzed methanolysis.14 Moreover, after phthalimide deprotection the resulting primary amine was found to undergo cyclization to cleave the directing group and form β-hydroxy-γ-lactam derivative 6.10d,10e We discovered, perhaps unsurprisingly, that this β-hydroxy AQ amide motif is quite susceptible to elimination upon activation; for instance, heating the mesyl-protected alcohol in triethylamine gave 96% of protected trans-4-aminocrotonic acid 7,15 which is a potent agonist of GABA(A) and GABA(C) receptors.16
Scheme 3.

Product Deprotection and Diversification
Finally, we sought to elucidate the mechanism of this palldium(II)-catalyzed intermolecular aminohydroxylation reaction. We first considered a mechanism in which alkene hydroamination10a was followed by directed C(sp3)–H oxidation. However, this pathway was ruled out because the putative hydroaminated intermediate was found to be unreactive in C(sp3)–H oxidation under the reaction conditions (Scheme 4A). This indicated that a general reactivity paradigm involving reaction of an aminopalladated intermediate with an O-atom electrophile was operative. We then turned to isotopic labeling experiments to investigate the source of the hydroxy group in the final product. To eliminate the possibility of labeled oxygen sources being exchanged with the oxygen atoms in the inorganic base of KHCO3, reactions were conducted under modified conditions using potassium phthalimate as nucleophile; under these conditions standard product 3a was isolated in 65% yield after 44 hours (Scheme 4B, entry 1). We next ran the reaction under 18O2 atmosphere and observed 26% yield of 70% singly labeled product, and 18% doubly labeled product according to MS (entry 3).17 On the other hand, using 16O2 atmosphere and doping in 1 equiv of H218O led to high yield but only 12% 18O incorporation (entry 4). The high amount of oxygen incorporation with 18O2 atmosphere but not with H218O is consistent with a mechanism in which the OH in the product originates primarily from O2. This finding also agrees with our initial observation that the reaction does not take place under nitrogen atmosphere (Table 1, entry 13). Additional labeling experiments with H218O have established that O-atom exchange between water and the carbonyl groups of the product takes place under the reaction conditions. This pathway is likely responsible for the detection of singly labeled product in the case of entry 3 and doubly labeled product in the case of entry 2 (see Supplementary Information).
Scheme 4. Mechanistic Studies.

a Percentages in parenthesis indicate the amount of doubly labeled product. b Reactions run using an autoclave (see footnote 16). c The percentage refers to yields determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-triisopropylbenzene as internal standard.
At this stage, we are uncertain whether O2 reacts directly to oxidize the alkylpalladium(II) intermediate to a Pd(III) or Pd(IV) species18 or whether O2 is first converted in situ to a reactive oxygen species, such as a peroxide.4c,4d The combination of hydrogen peroxide and DMBQ under nitrogen atmosphere under standard conditions was found to provide 37% of the aminohydroxylated product (Scheme 4C, entry 1), indicating that H2O2 is a competent oxidant. Other peroxides, such as DTBP, were ineffective under similar conditions, unless used in the presence of oxygen (see Supplementary Information). Based on the results described above, we propose the mechanism shown in Scheme 5, where the oxygen atom in the product is introduced from oxygen atmosphere (Scheme 5).
Scheme 5.

Proposed Labeling Mechanism
In conclusion, we have developed a palladium(II)-catalyzed intermolecular aminohydroxylation reaction of non-conjugated alkenes bearing a removable 8-aminoquinoline directing group with DMBQ/O2/HFIP as the oxidant. This reaction has allowed us to achieve intermolecular β,γ-selective aminooxygenation of 3-butenoic acid derivatives, using oxygen as the OH source. The reaction proceeded smoothly with a range of phthalimide-type nucleophiles and various substituted alkene substrates. The reaction was amenable to scale up, and the 8-aminoquinoline directing group could be easily removed via hydrolysis to provide bioactive compounds. Future investigations will focus on characterizing the reactive O-atom electrophile and elucidating the mechanistic details of the oxidation and C–O reductive elimination steps. Additionally, subsequent studies will seek to and expand the nucleophile and electrophile scope of this approach to alkene difunctionalization. These results will be reported in due course.
Supplementary Material
Table 2.
Reaction Substrate Scope.a

|
Reaction conditions: 1a (0.1 mmol), 2a (1.5 equiv), Pd(OAc)2 (10 mol %), DMBQ (2 equiv), K2CO3 (1 equiv), HFIP (0.2 mL), 100 °C, O2 (1 atm), 12–16 h. Percentages refer to isolated yields.
Intramolecular cyclized compound was also recovered (see footnote 13).
Pd(OAc)2 (15 mol%), 120 °C, 2 d.
N-(but-3-en-1-yl)picolinamide was used as the alkene.
Acknowledgments
This work was financially supported by TSRI, Pfizer, Inc., the National Institutes of Health (1R35GM125052) and Bristol-Myers Squibb (Unrestricted Grant). We thank Dr. Yi Hsiao and Dr. Shulin Wu (BMS) for assistance in performing reactions in the autoclave, Dr. Jason S. Chen for assistance with product purification and Dr. Milan Gembicky and Dr. Arnold L. Rheingold (UCSD) for X-ray crystallographic analysis.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Experiment details, spectra data, copies of 1H and 13C NMR spectra, and X-ray crystallographic data. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li G, Chang HT, Sharpless KB. Angew Chem Int Ed Enl. 1996;35:451–454. [Google Scholar]
- 2.For selected reviews: Reiser O. Angew Chem Int Ed Engl. 1996;35:1308–1309.O’Brien P. Angew Chem Int Ed Engl. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T.Bodkin JA, McLeod MD. J Chem Soc, Perkin Trans 1. 2002:2733–2746.Bergmeier SC. Tetrahedron. 2000:2561.Knappke CEI, von Wangelin AJ. ChemCatChem. 2010;2:1381–1383.Heravi MM, Lashaki TB, Fattahi B, Zadsirjan V. RSC Adv. 2018;8:6634–6659. doi: 10.1039/c7ra12625e.
- 3.(a) Bruncko M, Schlingloff G, Sharpless KB. Angew Chem Int Ed Engl. 1997;36:1483–1486. [Google Scholar]; (b) Tao B, Schlingloff G, Sharpless KB. Tetrahedron Lett. 1998;39:2507–2510. [Google Scholar]; (c) Streuff J, Osterath B, Nieger M, Muñiz K. Tetrahedron: Asymmetry. 2005;16:3492–3496. [Google Scholar]
- 4.(a) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (b) Borsini E, Broggini G, Fasana A, Gallai S, Khansaa M, Piarulli U, Rigamonti M. Adv Synth Catal. 2011;353:985–994. [Google Scholar]; (c) Zhu H, Chen P, Liu G. Org Lett. 2015;17:1485–1488. doi: 10.1021/acs.orglett.5b00373. [DOI] [PubMed] [Google Scholar]; (d) Kou X, Li Y, Zhang X, Yang G, Zhang W. Org Lett. 2015;17:5566–5569. doi: 10.1021/acs.orglett.5b02703. [DOI] [PubMed] [Google Scholar]; (e) Li J, Grubbs RH, Stoltz BM. Org Lett. 2016;18:5449–5451. doi: 10.1021/acs.orglett.6b02722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (b) Desai LV, Sanford MS. Angew Chem Int Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454. [DOI] [PubMed] [Google Scholar]; (c) Martínez C, Wu Y, Weinstein AB, Stahl SS, Liu G, Muñiz K. J Org Chem. 2013;78:6309–6315. doi: 10.1021/jo400671q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martínez C, Pérez EG, Iglesias Á, Escudero-Adán EC, Muñiz K. Org Lett. 2016;18:2998–3001. doi: 10.1021/acs.orglett.6b01368. [DOI] [PubMed] [Google Scholar]
- 6.(a) Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]; (b) Michaelis DJ, Ischay MA, Yoon TP. J Am Chem Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. [DOI] [PubMed] [Google Scholar]; (c) DePorter SM, Jacobsen AC, Partridge KM, Williamson KS, Yoon TP. Tetrahedron Lett. 2010;51:5223–5225. doi: 10.1016/j.tetlet.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sanjaya S, Chiba S. Org Lett. 2012;14:5342–5345. doi: 10.1021/ol302525m. [DOI] [PubMed] [Google Scholar]; (e) Herrera C, Madrid-Rojas M, López JJ, Cañete Á, Hermosilla-Ibáñez P, Pérez EG. Chem Cat Chem. 2016;8:2015–2018. [Google Scholar]
- 7.(a) Williamson KS, Yoon TP. J Am Chem Soc. 2010;132:4570–4571. doi: 10.1021/ja1013536. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Williamson KS, Yoon TP. J Am Chem Soc. 2012;134:12370–12373. doi: 10.1021/ja3046684. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu GS, Zhang YQ, Yuan YA, Xu H. J Am Chem Soc. 2013;135:3343–3346. doi: 10.1021/ja311923z. [DOI] [PubMed] [Google Scholar]; (d) Zhang YQ, Yuan YA, Liu GS, Xu H. Org Lett. 2013;15:3910–3913. doi: 10.1021/ol401666e. [DOI] [PubMed] [Google Scholar]; (e) Lu DF, Zhu CL, Jia ZX, Xu H. J Am Chem Soc. 2014;136:13186–13189. doi: 10.1021/ja508057u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Legnani L, Morandi B. Andew Chem Int Ed. 2016;55:2248. doi: 10.1002/anie.201507630. [DOI] [PubMed] [Google Scholar]
- 8.Muñiz K, Iglesias A, Fang Y. Chem Commun. 2009:5591–5593. doi: 10.1039/b912139k. [DOI] [PubMed] [Google Scholar]
- 9.(a) Mahoney JM, Smith CR, Johnston JN. J Am Chem Soc. 2005;127:1354–1355. doi: 10.1021/ja045608c. [DOI] [PubMed] [Google Scholar]; (b) Moriyama K, Izumisawa Y, Togo H. J Org Chem. 2012;77:9846–9851. doi: 10.1021/jo301523a. [DOI] [PubMed] [Google Scholar]; (c) Xu F, Zhu L, Zhu S, Yan X, Xu HC. Chem Eur J. 2014;20:12740–12744. doi: 10.1002/chem.201404078. [DOI] [PubMed] [Google Scholar]; (d) Jeon H, Kim D, Lee JH, Song J, Lee WS, Dang DW, Kang S, Lee SB, Choi S, Hong KB. Adv Synth Catal. 2018;360:779–783. [Google Scholar]
- 10.(a) Gurak JA, Jr, Yang KS, Liu Z, Engle KM. J Am Chem Soc. 2016;138:5805–5808. doi: 10.1021/jacs.6b02718. [DOI] [PubMed] [Google Scholar]; (b) Yang K, Gurak JA, Jr, Liu Z, Engle KM. J Am Chem Soc. 2016;138:14705–14712. doi: 10.1021/jacs.6b08850. [DOI] [PubMed] [Google Scholar]; (c) Liu Z, Zeng T, Yang KS, Engle KM. J Am Chem Soc. 2016;138:15122–15125. doi: 10.1021/jacs.6b09170. [DOI] [PubMed] [Google Scholar]; (d) Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM. J Am Chem Soc. 2017;139:11261–11270. doi: 10.1021/jacs.7b06520. [DOI] [PubMed] [Google Scholar]; (e) Liu Z, Ni HQ, Zeng T, Engle KM. J Am Chem Soc. 2018;140:3223–3227. doi: 10.1021/jacs.8b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tran VT, Gurak JA, Jr, Yang KS, Engle KM. Nat Chem. 2018 doi: 10.1038/s41557-018-0110-z. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For representative review describing the discovery and development of bidentate directing groups in C–H activation, see: Daugulis O, Roane J, Tran LD. Acc Chem Res. 2015;48:1053–1064. doi: 10.1021/ar5004626.
- 12.See Supplementary Information for other unsuccessful oxidant screens.
-
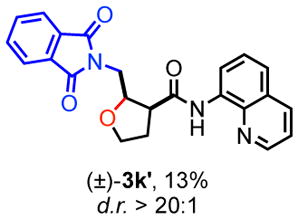
13.Benzyl-deprotected byproduct 3k′ was also formed in 13% yield.
- 14.Deguchi T, Xin H-L, Morimoto H, Ohshima T. ACS Catal. 2017;7:3157–3161. [Google Scholar]
- 15.Exner K, Herizmann M, Yang F, Kegel M, Keller M, Knothe L, Großmann B, Heinze J, Prinzbach H. Eur J Org Chem. 2005;7:1311–1331. [Google Scholar]
- 16.(a) Zhang D, Pan ZH, Awouluyi M, Lipton SA. Trends in Pharmacological Science. 2001;22(3):121–132. doi: 10.1016/s0165-6147(00)01625-4. [DOI] [PubMed] [Google Scholar]; (b) Borowicz KK, Zadrozniak M, Czuczwar SJ. Pharmacol Rep. 2005;57:121–123. [PubMed] [Google Scholar]
- 17.This reaction was performed in an autoclave rather than in the standard reaction vessel (Schleck tube, which was charged with O2 using a balloon). The autoclave was used in this case in order to minimize the volume of 18O2 that is wasted during the reaction setup. See the Supporting Information for an experimental procedure. We believe that the lower yield with the autoclave is due to difference in the optimized experimental setup, as the yield of an identical reaction in the autoclave under 16O2 atmosphere was 33%.
- 18.(a) Zhang J, Khaskin E, Anderson NP, Zavalij PY, Vedernikov AN. Chem Commun. 2008;0:3625–3627. doi: 10.1039/b803156h. [DOI] [PubMed] [Google Scholar]; (b) Zhang YH, Yu JQ. J Am Chem Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n. [DOI] [PubMed] [Google Scholar]; (c) Khusnutdinova JR, Qu R, Zhang Y, Rath NP, Mirica LM. Organometallics. 2012;31:4627–4630. [Google Scholar]; (d) Tang F, Zhang Y, Rath NR, Mirica LM. Organometallics. 2012;31:6690–6696. [Google Scholar]; (e) Qu F, Khusnutdinova JR, Rath NP, Mirica LM. Chem Commun. 2014;50:3036–3039. doi: 10.1039/c3cc49387c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.