

Abstract
Bromodomain-containing proteins (BDPs) are involved in the regulation of eukaryotic gene expression. Compounds that bind and/or inhibit BDPs are of interest as tools to better understand epigenetic regulation, and as possible drug leads for different diseases, including malaria. In this study, we assessed the activity of 42 compounds demonstrated or predicted (using virtual screening of a pharmacophore model) to bind/inhibit eukaryotic BDPs for activity against Plasmodium falciparum malaria parasites. In silico docking studies indicated that all compounds are predicted to participate in a typical hydrogen bond interaction with the conserved asparagine (Asn1436) of the P. falciparum histone acetyltransferase (PfGCN5) bromodomain and a conserved water molecule. Only one compound (the dimethylisoxazole SGC-CBP30; a selective inhibitor of CREBBP (CBP) and EP300 bromodomains) is also predicted to have a salt-bridge between the morpholine nitrogen and Glu1389. When tested for in vitro activity against asynchronous asexual stage P. falciparum Dd2 parasites, all compounds displayed 50% growth inhibitory concentrations (IC50) >10 μM. Further testing of the three most potent compounds using synchronous parasites for 72 h showed that SGC-CBP30 was the most active (IC50 3.2 μM). In vitro cytotoxicity assays showed that SGC-CBP30 has ∼7-fold better selectivity for the parasites versus a human cell line (HEK 293). Together these data provide a possible starting point for future investigation of these, or related compounds, as tools to understand epigenetic regulation or as potential new drug leads.
Keywords: Plasmodium falciparum, Anti-plasmodial activity, Bromodomain protein inhibitor
Graphical abstract

Highlights
-
•
42 demonstrated or predicted BDP binders/inhibitors investigated.
-
•
In silico docking predicts all have hydrogen bond interaction with conserved Asn1436 of PfGCN5 bromodomain.
-
•
Pan-bromodomain inhibitor SGC-CBP30 also has predicted salt-bridge between morpholine nitrogen and Glu1389.
-
•
SGC-CBP30 has most potent in vitro activity against asexual-stage P. falciparum (IC50 3.2 μM).
-
•
SGC-CBP30 has ∼7-fold better selectivity for P. falciparum versus HEK 293 cell.
1. Introduction
Bromodomains are conserved acetyl-lysine-specific protein-interaction modules (Filippakopoulos and Knapp, 2014). Bromodomain-containing proteins (BDPs) have been shown to play important roles in regulating eukaryotic gene expression and mutations or changes in the expression of BDPs has been linked to human diseases, including cancer, inflammation, neurological disorders, cardiovascular disease and diabetes (Ferri et al., 2016; Jung et al., 2015). As a result of these links, BDP inhibitors are under investigation as drug leads, with the hope that interfering with lysine acetylation mediated signalling may be therapeutic (Papavassiliou and Papavassiliou, 2014).
Similar to higher eukaryotes, the epigenetic regulation of gene expression is important in human protozoan pathogens. For example, parasites such as Toxoplasma, Trypanosoma and Plasmodium that cause toxoplasmosis, typanosomiasis and malaria, respectively, rely on epigenetic modifications to regulate gene expression (Jeffers et al., 2017). BDPs have also been identified in all of these parasites, and have been hypothesized to be potential drug targets (reviewed in (Jeffers et al., 2017)). Seven BDP encoding genes have been annotated in Plasmodium falciparum (Jeffers et al., 2017), with two partially characterised to date. P. falciparum histone acetyltransferase GCN5 (PfGCN5; PF3D7_0823300) has been shown to have lysine acetyltransferase (KAT) activity (Fan et al., 2004; Josling et al., 2012) and P. falciparum bromodomain protein 1 (PfBDP1; PF3D7_1033700) has been shown to be involved in the regulation of invasion-related genes in asexual stage parasites (Josling Gabrielle et al., 2015). Both of these BDPs appear to be essential for parasite growth (Josling Gabrielle et al., 2015; Cui et al., 2007). While the crystal structure of PfGCN5 bromodomain in complex with a triazolophthalazine-based small molecule inhibitor (L-45/L-Moses) has been reported (Moustakim et al., 2017), there is a gap in our knowledge with respect to studies investigating the growth inhibitory effect of BDP binders/inhibitors against malaria parasites (reviewed in (Jeffers et al., 2017)). In this study, a panel of 42 potential BDP binders/inhibitors, including 38 identified by an in silico pharmacophore screen, were examined for predicted binding to P. falciparum BDP/bromodomains and for in vitro growth inhibitory activity against asexual-stage P. falciparum infected erythrocytes. The three most potent anti-plasmodial compounds were assessed in an additional P. falciparum growth inhibition assay, and for cytotoxicity against a mammalian cell line.
2. Methods
2.1. Compounds
The anti-plasmodial control drug chloroquine diphosphate salt (Sigma-Aldrich, USA) was prepared as a 10–20 mM stock in phosphate buffered saline (PBS). The BDP binders/inhibitors bromosporine, CPI-203, PFI-4 and SGC-CBP30 (all from Selleck Chemicals, USA) were prepared as 10–20 mM stocks in DMSO. A further 38 compounds (Table 1) were obtained from the Princeton Biomolecular Research, Inc. (Princeton, NJ, USA) compound library, and prepared as 10–20 mM stocks in DMSO. These 38 compounds were selected based on virtual screening of a pharmacophore model of the bromodomain of PF3D7_0110500 (PDB ID 4PY6), selected as it was the only P. falciparum bromodomain/BDP in the Protein Databank (Berman et al., 2000) crystallized in complex with an inhibitor (the PLK1 kinase/BRD4 dual inhibitor BI-2536) (Chen et al., 2015). Based on the crystal structure of PF3D7_0110500 (PDB ID 4PY6) in complex with BI-2536, a pharmacophore model was generated using the program LigandScout 3.1 (Wolber and Langer, 2005). Residues of the protein binding pocket were assigned as excluded volume features. The model was manually curated: the hydrophobic feature generated for the ethyl moiety of the inhibitor was removed and a hydrophobic feature was added for the methyl-group of the dihydropteridine core. The pharmacophore model was screened against the Princeton Biomolecular Research, Inc. compound collection (multiconformational format) using the iscreen module implemented in LigandScout 3.1, using default settings.
Table 1.
In vitro activity of BDP inhibitors against asexual stage P. falciparum Dd2 parasites.
| Compound | Structure | cLog P | IC50PfDd2 (μM) | Compound | Structure | cLog P | IC50PfDd2 (μM) |
|---|---|---|---|---|---|---|---|
| Chloroquine | 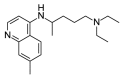 |
4.63 | 0.11 (±0.04) | OSSL_258894b |  |
2.29 | 33.87 (±4.52) |
| Bromosporinea | 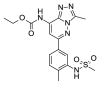 |
1.67 | 26.33 (±7.13) | OSSL_258896b | 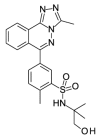 |
3.16 | 33.98 (±21.38) |
| CPI-203a |  |
3.28 | 81.43 (±13.68) | OSSL_258897b | 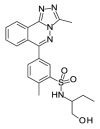 |
3.37 | 35.00 (±0.00) |
| PFI-4a | 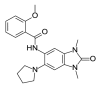 |
2.35 | 26.43 (±3.82) | OSSL_258891b | 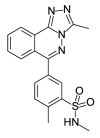 |
2.99 | 40.78 (±28.87) |
| SGC-CBP30a | 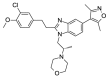 |
5.09 | 10.03 (±0.32) | OSSL_258895b |  |
2.75 | 47.43 (±8.06) |
| OSSK_842646b | 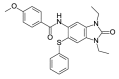 |
4.83 | 11.28 (±2.00) | OSSL_258907b |  |
2.43 | 71.77 (±20.41) |
| OSSK_764253b |  |
3.07 | 32.45 (±15.98) | OSSL_258903b |  |
4.77 | >100 |
| OSSK_764205b |  |
3.65 | 20.23 (±1.60) | OSSL_258893b |  |
3.32 | >100 |
| OSSK_995759b |  |
2.77 | 43.24 (±12.43) | OSSL_158302b |  |
4.78 | 14.70 (±0.00) |
| OSSL_308235b |  |
1.73 | 53.83 (±17.10) | OSSK_711135b |  |
3.20 | >100 |
| OSSK_764265b |  |
1.64 | 57.30 (±19.34) | OSSK_711274b |  |
4.63 | 36.93 (±20.19) |
| OSSK_764219b | 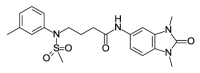 |
1.35 | 72.40 (±3.35) | OSSK_711212b | 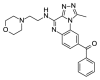 |
2.35 | 87.35 (±1.67) |
| OSSK_764195b | 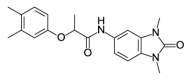 |
2.79 | 72.88 (±8.79) | OSSK_711203b | 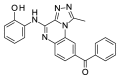 |
4.33 | >100 |
| OSSK_764277b |  |
1.86 | 81.25 (±12.64) | OSSL_094251b |  |
4.59 | 23.25 (±6.15) |
| OSSK_842567b |  |
4.21 | >100 | OSSK_442833b | 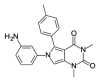 |
3.46 | 48.06 (±6.79) |
| OSSK_711132b |  |
2.29 | >100 | OSSL_094246b |  |
2.90 | >100 |
| OSSK_447894b | 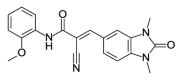 |
1.91 | >100 | OSSK_287503b | 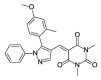 |
3.12 | >100 |
| OSSL_326023b |  |
2.79 | >100 | OSSK_695521b | 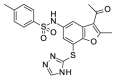 |
3.61 | 59.60 (±26.44) |
| OSSL_258906b |  |
3.54 | 11.80 (±3.06) | OSSL_094264b | 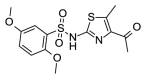 |
1.57 | >100 |
| OSSL_258905b |  |
5.24 | 28.82 (±4.58) | OSSK_446201b | 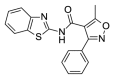 |
3.91 | 97.20 (±0.00) |
| OSSL_258904b | 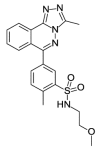 |
2.90 | 26.03 (±2.24) | OSSK_310407b | 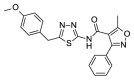 |
4.40 | >100 |
| OSSL_258898b | 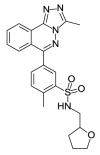 |
3.56 | 28.17 (±4.79) |
Purchased from Selleck Chemicals.
Purchased from Princeton Biomolecular Research Inc.
2.2. Docking studies
Pharmacophore hits identified by virtual screening were prepared for docking using the LigPrep tool as implemented in Schrödinger's software (Anonymous, 2012), where all possible tautomeric forms as well as stereoisomers were generated and energy minimized using the OPLS force field. The PfGCN5-bromodomain (PDB ID 4QNS) and PfBDP1 (PDB ID 3FKM) crystal structures were retrieved from the PDB. The protein structures were superposed and subsequently prepared with Schrödinger's Protein Preparation Wizard: Hydrogen atoms were added and the hydrogen bond network was subsequently optimized. The protonation states at pH 7.0 were predicted using the PROPKA tool within the Schrödinger program. The structures were finally subjected to a restrained energy minimization step using the OPLS2005 force field (RMSD of the atom displacement for terminating the minimization 0.3 Å). The four conserved water molecules lining the bottom of the binding cavity were retained. Receptor grid preparation for the docking procedure was carried out by assigning the conserved asparagine residue as the centroid of the grid box. The ligands were docked into the prepared protein structures using GLIDE (Schrödinger Inc, New York, USA) in the Standard Precision mode.
2.3. P. falciparum in vitro culture and growth inhibition assays
P. falciparum multi-drug resistant Dd2 parasites were cultured in O positive human erythrocytes in RPMI 1640 media (Gibco, USA) supplemented with 10% heat-inactivated pooled human sera and 5 μg/mL gentamicin. Cells were cultured at 37 °C in 5% O2 and 5% CO2 in N2, essentially as previously described (Trager and Jensen, 1976). Growth inhibitory activity of compounds was tested in vitro against asexual intraerythrocytic stage parasites over 48 h starting with asynchronous parasites or over 72 h starting with ring-stage parasites, using [3H]-hypoxanthine-uptake growth inhibition assays, as previously described (Chua et al., 2017). At least three independent assays, each in triplicate wells, were carried out and 50% inhibitory concentrations (IC50's), determined by log-linear interpolation (Huber and Koella, 1993). Data are presented as mean IC50 (±SD). The antimalarial drug chloroquine served as a positive control.
2.4. Cytotoxicity assays
Cytotoxicity assays were carried out using human embryonic kidney cells (HEK 293), as previously described (Engel et al., 2015). All assays were carried out in triplicate wells on three separate occasions. Data are presented as mean IC50 (±SD), with IC50's calculated determined by log-linear interpolation (Huber and Koella, 1993).
3. Results and discussion
To investigate the anti-plasmodial activity of potential BDP binders/inhibitors (hereafter termed BDPi), a panel of 42 compounds (Table 1) was tested. Compounds included four known BDPi (bromosporine, CPI-203, PFI-4 and SGC-CBP30; Table 1) with different mammalian BDP specificities. Bromosporine (Picaud et al., 2016) is a pan-BDP inhibitor, while CPI-203 (Filippakopoulos et al., 2010), PFI-4 (Demont et al., 2014) and SGC-CBP30 (Gallenkamp et al., 2014) each have specificity for different mammalian BDPs. A further 38 compounds were selected by virtual screening of the Princeton Biomolecular Research Inc. compound library. These compounds were selected based on in silico screening of a pharmacophore-model (Supplementary Figure S1) obtained using the crystal structure of PF3D7_0110500 (PDB ID 4PY6) which, at commencement of this study, was the only available structure of a P. falciparum bromodomain crystallized in complex with an inhibitor (the PLK1 kinase/BRD4 dual inhibitor BI-2536) (Chen et al., 2015). The 38 compounds identified as potential inhibitors/binders by this virtual screen (Table 1) span different chemotypes, including some known BDPi scaffolds such as benzimidazolone (Demont et al., 2014; Bamborough et al., 2016) and triazolophthalazine (Fedorov et al., 2014).
Docking studies were carried out on all 42 compounds with the available crystal structures of bromodomains of PfGCN5 (PDB ID 4QNS) and PfBDP1 (PDB ID 3FKM). Each compound is predicted to participate in a typical hydrogen bond interaction with the conserved asparagine of PfGCN5 (Asn1436) and a conserved water molecule (Supplementary Figure S2; SGC-CBP30, OSSL_258906, OSSL_158302 and OSSK_842646 shown). Additionally, SGC-CBP30 is predicted to have a salt-bridge between the morpholine nitrogen and Glu1389 (Supplemental Figure S2b). This salt-bridge is also observed in the previously published crystal structure of the bromodomain of PfGCN5 in complex with triazolophthalazine (Moustakim et al., 2017) (Supplemental Figure S2a). The salt-bridge interaction was not observed for any of the other compounds assessed for anti-plasmodial activity in this study. Docking in the available crystal structure of PfBDP1 (PDB ID 3FKM) was, however, more problematic, since the crystal structure is in apo form, and part of the ZA loop (residues 362–366) is missing. This ZA loop is a highly flexible loop which is known to constitute an important part of the inhibitor-binding pocket of bromodomains (Filippakopoulos et al., 2010; Muller et al., 2011). Docking studies revealed that the compounds can form hydrogen bond interactions with the conserved Asn413 of PfBDP1, and SGC-CBP30 can also profit from an additional salt bridge with Asp361 (Supplemental Figure S2f). While these docking studies provide interesting preliminary predictions regarding interactions with P. falciparum BDP(s), future studies are needed to experimentally determine if these specific compounds bind/inhibit P. falciparum BDP(s) and, if so, with what specificity. Since the selective CPB/EP300 inhibitor SGC-CBP30 showed the highest antiplasmodial activity among the tested compounds, a BLAST search (Altschul et al., 1990) was conducted in order to find the closest homologues of CBP and EP300 bromodomains in P. falciparum (Supplemental Figure S3). This revealed that PfBDP1 shares the highest homology with CPB and EP300.
In vitro 48 h activity assays with asynchronous asexual intraerythrocytic stage P. falciparum line Dd2 parasites demonstrated that all 42 BDPi lack potent anti-Plasmodium activity (IC50 ≥ 10 μM; Table 1). The three most potent compounds were the CPB/EP300 bromodomain inhibitor SGC-CBP30 (IC50 10.03 (±0.32)), the diethylbenzimidazolone OSSK_842646 (IC50 11.28 (±2.00)) and the triazolophthalazine OSSL_258906 (IC50 11.80 (±3.06)). The activity of these compounds was confirmed in a 72 h assay starting with synchronous ring-stage parasites. The activity of OSSK_842646, OSSL_258906 and the control drug chloroquine was not significantly different between 48 h and 72 h assays (P > 0.05; Table 2). While the dimethylisoxazole SGC-CBP30 showed greater activity in the 72 versus 48 h assay (Table 2; p = 0.006), potency was still low (μM range). Although cytotoxicity studies with SGC-CBP30 suggest low selectivity for the parasite versus HEK 293 cells (Table 2; ∼7-fold), an IC50 value for OSSK_842646, OSSL_258906 against HEK 293 cells was not achieved (IC50 > 50 μM) so selectivity indices could not be accurately determined (Table 2; >4-fold).
Table 2.
Comparative anti-plasmodial activity of selected compounds in 48 h versus 72 h growth inhibition assays and cytotoxicity activity against HEK 293 cells.
| Compounds |
PfDd2 IC50 (μM) |
p value | HEK 293 IC50 (μM) | SIa | |
|---|---|---|---|---|---|
| 48 h | 72 h | ||||
| SGC-CBP30 | 10.03 (±0.32) | 3.16 (±1.94) | 0.006 | 22.09 (±2.00) | 2–7 |
| OSSK_842646 | 11.28 (±2.00) | 6.35 (±2.45) | 0.073 | >50 | >4 |
| OSSL_258906 | 11.80 (±3.06) | 5.96 (±1.19) | 0.051 | >50 | >4 |
| CQ | 0.11 (±0.04) | 0.05 (±0.02) | 0.052 | 18.58 (±1.95) | 169–372 |
SI – Selectivity index (mammalian cell IC50/PfDd2 IC50).
The current study describes the anti-plasmodial activity of a panel of compounds known or predicted to bind/inhibit mammalian BDP(s). While data suggest that these compounds have low potency against the P. falciparum parasites, they provide a possible starting point for the investigation of rationally designed analogues with improved activity against malaria parasites. In addition, as LC-MS data show that BDPs’ are also expressed in sexual stages of Plasmodium parasite development (Silvestrini et al., 2010), investigating the activity of these compounds against different Plasmodium life cycle stages may be warranted. Further studies on the potential specificity of these compounds for P. falciparum BDPs may also validate these inhibitors as chemical tools to study Plasmodium epigenetic regulatory processes.
Acknowledgements
We thank the Australian Red Cross Blood Service for the provision of human blood and sera. We thank the Australian National Health and Medical Research Council (APP1074016 to KTA) for research support and Griffith University for scholarship support (GUIPRS and GUPRS to MJC). This project was carried out in part under the A-PARADDISE program funded under the European Union's Seventh Framework Programme (to KTA and WS).
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ijpddr.2018.03.001.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Anonymous . 2012. Schrödinger Suite 2012: Glide, Version 5.8. Schrödinger, Llc, New York, NY. [Google Scholar]
- Bamborough P., Barnett H.A., Becher I., Bird M.J., Chung C.W., Craggs P.D., Demont E.H., Diallo H., Fallon D.J., Gordon L.J., Grandi P., Hobbs C.I., Hooper-Greenhill E., Jones E.J., Law R.P., Le Gall A., Lugo D., Michon A.M., Mitchell D.J., Prinjha R.K., Sheppard R.J., Watson A.J., Watson R.J. GSK6853, a chemical probe for inhibition of the BRPF1 bromodomain. ACS Med. Chem. Lett. 2016;7:552–557. doi: 10.1021/acsmedchemlett.6b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Yap J.L., Yoshioka M., Lanning M.E., Fountain R.N., Raje M., Scheenstra J.A., Strovel J.W., Fletcher S. BRD4 structure-activity relationships of dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536. ACS Med. Chem. Lett. 2015;6:764–769. doi: 10.1021/acsmedchemlett.5b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua M.J., Arnold M.S.J., Xu W., Lancelot J., Lamotte S., Späth G.F., Prina E., Pierce R.J., Fairlie D.P., Skinner-Adams T.S., Andrews K.T. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol.: Drugs and Drug Resistance. 2017;7:42–50. doi: 10.1016/j.ijpddr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L., Miao J., Cui L. Cytotoxic effect of curcumin on malaria parasite Plasmodium falciparum: inhibition of histone acetylation and generation of reactive oxygen species. Antimicrob. Agents Chemother. 2007;51:488–494. doi: 10.1128/AAC.01238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demont E.H., Bamborough P., Chung C.W., Craggs P.D., Fallon D., Gordon L.J., Grandi P., Hobbs C.I., Hussain J., Jones E.J., Le Gall A., Michon A.M., Mitchell D.J., Prinjha R.K., Roberts A.D., Sheppard R.J., Watson R.J. 1,3-Dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 bromodomain. ACS Med. Chem. Lett. 2014;5:1190–1195. doi: 10.1021/ml5002932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J.A., Jones A.J., Avery V.M., Sumanadasa S.D., Ng S.S., Fairlie D.P., Skinner-Adams T., Andrews K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int J Parasitol Drugs Drug Resist. 2015;5:117–126. doi: 10.1016/j.ijpddr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q., An L., Cui L. Plasmodium falciparum histone acetyltransferase, a yeast GCN5 homologue involved in chromatin remodeling. Eukaryot. Cell. 2004;3:264–276. doi: 10.1128/EC.3.2.264-276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O., Lingard H., Wells C., Monteiro O.P., Picaud S., Keates T., Yapp C., Philpott M., Martin S.J., Felletar I., Marsden B.D., Filippakopoulos P., Muller S., Knapp S., Brennan P.E. [1,2,4]triazolo[4,3-a]phthalazines: inhibitors of diverse bromodomains. J. Med. Chem. 2014;57:462–476. doi: 10.1021/jm401568s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri E., Petosa C., McKenna C.E. Bromodomains: structure, function and pharmacology of inhibition. Biochem. Pharmacol. 2016;106:1–18. doi: 10.1016/j.bcp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P., Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., Philpott M., Munro S., McKeown M.R., Wang Y., Christie A.L., West N., Cameron M.J., Schwartz B., Heightman T.D., La Thangue N., French C.A., Wiest O., Kung A.L., Knapp S., Bradner J.E. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallenkamp D., Gelato K.A., Haendler B., Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- Huber W., Koella J.C. A comparison of three methods of estimating EC50 in studies of drug resistance of malaria parasites. Acta Trop. 1993;55:257–261. doi: 10.1016/0001-706x(93)90083-n. [DOI] [PubMed] [Google Scholar]
- Jeffers V., Yang C., Huang S., Sullivan W.J., Jr. Bromodomains in Protozoan parasites: evolution, function, and opportunities for drug development. Microbiol. Mol. Biol. Rev. 2017;81 doi: 10.1128/MMBR.00047-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josling G.A., Selvarajah S.A., Petter M., Duffy M.F. The role of bromodomain proteins in regulating gene expression. Genes. 2012;3:320–343. doi: 10.3390/genes3020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josling Gabrielle A., Petter M., Oehring Sophie C., Gupta Archna P., Dietz O., Wilson Danny W., Schubert T., Längst G., Gilson Paul R., Crabb Brendan S., Moes S., Jenoe P., Lim Shu W., Brown Graham V., Bozdech Z., Voss Till S., Duffy Michael F. A Plasmodium falciparum bromodomain protein regulates invasion gene expression. Cell Host Microbe. 2015;17:741–751. doi: 10.1016/j.chom.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Jung M., Gelato K.A., Fernandez-Montalvan A., Siegel S., Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7:487–501. doi: 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- Moustakim M., Clark P.G., Trulli L., Fuentes de Arriba A.L., Ehebauer M.T., Chaikuad A., Murphy E.J., Mendez-Johnson J., Daniels D., Hou C.D., Lin Y.H., Walker J.R., Hui R., Yang H., Dorrell L., Rogers C.M., Monteiro O.P., Fedorov O., Huber K.V., Knapp S., Heer J., Dixon D.J., Brennan P.E. Discovery of a PCAF bromodomain chemical probe. Angew Chem. Int. Ed. Engl. 2017;56:827–831. doi: 10.1002/anie.201610816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Filippakopoulos P., Knapp S. Bromodomains as therapeutic targets. Expet Rev. Mol. Med. 2011;13 doi: 10.1017/S1462399411001992. e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavassiliou K.A., Papavassiliou A.G. Bromodomains: pockets with therapeutic potential. Trends Mol. Med. 2014;20:477–478. doi: 10.1016/j.molmed.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Picaud S., Leonards K., Lambert J.P., Dovey O., Wells C., Fedorov O., Monteiro O., Fujisawa T., Wang C.Y., Lingard H., Tallant C., Nikbin N., Guetzoyan L., Ingham R., Ley S.V., Brennan P., Muller S., Samsonova A., Gingras A.C., Schwaller J., Vassiliou G., Knapp S., Filippakopoulos P. Promiscuous targeting of bromodomains by bromosporine identifies BET proteins as master regulators of primary transcription response in leukemia. Sci Adv. 2016;2 doi: 10.1126/sciadv.1600760. e1600760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestrini F., Lasonder E., Olivieri A., Camarda G., van Schaijk B., Sanchez M., Younis Younis S., Sauerwein R., Alano P. Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics. 2010;9:1437–1448. doi: 10.1074/mcp.M900479-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W., Jensen J.B. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Wolber G., Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.