

Supplemental Digital Content is available in the text.
Keywords: alcohol dependence, disease comorbidity, genome-wide association studies, major depressive disorder, polygenic risk scores, psychiatric genomics consortium
Abstract
The clinical comorbidity of alcohol dependence (AD) and major depressive disorder (MDD) is well established, whereas genetic factors influencing co-occurrence remain unclear. A recent study using polygenic risk scores (PRS) calculated based on the first-wave Psychiatric Genomics Consortium MDD meta-analysis (PGC-MDD1) suggests a modest shared genetic contribution to MDD and AD. Using a (∼10 fold) larger discovery sample, we calculated PRS based on the second wave (PGC-MDD2) of results, in a severe AD case–control target sample. We found significant associations between AD disease status and MDD-PRS derived from both PGC-MDD2 (most informative P-threshold=1.0, P=0.00063, R2=0.533%) and PGC-MDD1 (P-threshold=0.2, P=0.00014, R2=0.663%) meta-analyses; the larger discovery sample did not yield additional predictive power. In contrast, calculating PRS in a MDD target sample yielded increased power when using PGC-MDD2 (P-threshold=1.0, P=0.000038, R2=1.34%) versus PGC-MDD1 (P-threshold=1.0, P=0.0013, R2=0.81%). Furthermore, when calculating PGC-MDD2 PRS in a subsample of patients with AD recruited explicitly excluding comorbid MDD, significant associations were still found (n=331; P-threshold=1.0, P=0.042, R2=0.398%). Meanwhile, in the subset of patients in which MDD was not the explicit exclusion criteria, PRS predicted more variance (n=999; P-threshold=1.0, P=0.0003, R2=0.693%). Our findings replicate the reported genetic overlap between AD and MDD and also suggest the need for improved, rigorous phenotyping to identify true shared cross-disorder genetic factors. Larger target samples are needed to reduce noise and take advantage of increasing discovery sample size.
Introduction
The co-occurrence of alcohol dependence (AD) and major depressive disorder (MDD) has been well established, and epidemiological assessments of AD and MDD have linked AD to higher risk of depression and vice versa (Woodruff et al., 1973; Kessler et al., 1996; Swendsen and Merikangas, 2000; Crum et al., 2008; Foulds et al., 2015). Formal genetic studies indicate that AD and MDD share common genetic factors (Winokur and Coryell, 1991; Maier et al., 1994; Kendler et al., 1995; Prescott et al., 2000; Nurnberger et al., 2002; Lyons et al., 2006). Genome-wide association studies (GWAS) have the ability to identify genetic loci associated with complex disorders [e.g. AD (Treutlein et al., 2009; Kapoor et al., 2014; Zuo et al., 2014; Gelernter et al., 2014) and MDD (Ripke et al., 2013; Wray and Sullivan, 2017)] and allow the study of genetic risk shared between complex genetic disorders (Wray et al., 2014). So far, however, only limited support for specific genes contributing to the two illnesses on the level of individual genetic variation has been found (Edwards et al., 2012).
The polygenic risk score (PRS) approach is a statistical method that enables the assessment of additive effects of multiple common genome-wide genetic variations on risk for a disorder, and is well suited to characterize shared genetic etiology of complex disorders (Purcell et al., 2009). A recent study (Andersen et al., 2017) used this approach to examine the genetic overlap between AD and MDD. They calculated MDD-PRS based on the results of the first-wave meta-analysis of the MDD Working Group of the Psychiatric Genomics Consortium (PGC) (PGC-MDD1: cases, n=9240; controls, n=9519) GWAS (Ripke et al., 2013), finding associations with increased risk for AD in four independent AD-GWAS data sets (cases ranged from 317 to 2135), explaining from 0.18 to 2.6% of the variance in AD (Nagelkerke’s R2). Mentioned as a limitation was the small size of the MDD discovery sample used, proposing that increased sample sizes would improve MDD-PRS predictive ability.
Here, we sought to substantiate the findings of Andersen et al. (2017) in an independent sample of patients having severe AD while calculating MDD-PRS based on the much larger PGC-MDD2 discovery sample (n=59 265 cases, n=112 092 controls; Wray and Sullivan, 2017). For context, we also calculated PRS using an MDD target sample. Furthermore, we examined whether any association would be observed using a subset of this AD sample whose patients had been recruited explicitly excluding comorbid MDD.
Materials and methods
Target samples
The target sample comprised 1333 male patients with German ancestry having severe AD requiring hospitalization and 1307 population-based controls, previously described in Treutlein et al. (2009) and Frank et al. (2012) [i.e. the German Study on the Genetics of Addiction (Alcoholism), GESGA]. Controls overlapping between the GESGA sample and PGC-MDD discovery samples were removed before analysis. In summary, all patients fulfilled the AD DSM-IV criteria and were recruited from consecutive admissions to psychiatry and addiction medicine departments of psychiatric hospitals participating in the German addiction research network.
A subset of the cases (PREDICT subsample; Mann et al., 2009) comprising 332 patients with AD was recruited explicitly excluding comorbid MDD.
The MDD target sample comprised cases (n=597) from the Bonn/Mannheim (BoMa) MDD study and German population-based controls (n=1292), described previously (Rietschel et al., 2010; Ripke et al., 2013).
All participants provided written informed consent, and procedures used were approved by the respective local ethics committees and in accordance with the Declaration of Helsinki.
Discovery samples
The PGC-MDD2 discovery sample comprised 59 265 cases and 112 092 controls (leave-one-out meta-analysis omitting the BoMa-MDD sample included in the original PGC-MDD2 (Wray and Sullivan, 2017) meta-analysis).
The PGC-MDD1 discovery sample comprised 8148 cases and 7955 controls (leave-one-out meta-analysis omitting the BoMa-MDD and RADIANT-German samples included in the original PGC-MDD1 (Ripke et al., 2013) meta-analysis which had overlapping controls with the GESGA sample). The PGC-MDD1 data set is a subset of the PGC-MDD2 data set.
Genotyping and quality control
Detailed information on genotyping and QC is available in Frank et al. (2012). In summary, filtering for uncommon SNPs (minor allele frequency<0.1), individual missingness (>0.01), low-quality genotyping (missingness >0.02), and Hardy–Weinberg equilibrium (1.0×10-6) was performed. After QC and excluding overlapping samples, the final GESGA sample comprised 1330 cases and 1051 controls (n=382001 SNPs). Of these cases, 331 were from the PREDICT study and 999 were not. Additional minor allele frequency filtering was performed for each subsample (PREDICT: n=381453 SNPs; non-PREDICT: n=381914 SNPs). After QC and removing overlap, the BoMa-MDD sample comprised 586 cases and 1062 controls (n=3523389 SNPs).
Polygenic risk score calculation
PRSs were calculated using PRSice v1.25 (Euesden et al., 2015). We calculated MDD-PRS in the GESGA sample using the PGC-MDD2 results following previously published methods (Ripke et al., 2014).
In summary, linkage disequilibrium (LD) clumping was carried out, retaining the variant with the smallest P value from each LD block and discarding all variants with r2 greater than or equal to 0.1 located within 500 kb around that variant. The major histocompatibility complex of chromosome 6 was excluded, as frequently done when calculating PRS owing to long-range LD, making linkage equilibrium difficult to contain (Euesden et al., 2015). PRS were calculated at a range of P value thresholds (P=5×10−8, 1×10−6, 1×10−4, 0.001, 0.01, 0.05, 0.1, 0.2, 0.5, 1.0).
Regression analyses were performed on AD case–control status with the first 10 principal components as covariates. The proportion of variance in case–control status explained by MDD-PRS was assessed by Nagelkerke’s pseudo R2 derived from the difference between the full regression model (PRS+covariates) and the null model (only covariates) (Purcell et al., 2009; Power et al., 2015). For comparison, we calculated MDD-PRS in the GESGA sample using the PGC-MDD1 discovery sample.
For context, we further analyzed the association of the MDD-PRS with MDD phenotype using the same parameters in the BoMa-MDD target sample, using both PGC-MDD1 and PGC-MDD2 results, serving as a positive control.
To examine whether existing AD/MDD comorbidity might be driving association, we conducted several additional analyses. Using the PGC-MDD2 results as the discovery sample, we analyzed PRS separately in PREDICT subsample cases (n=331) and non-PREDICT cases (n=999).
Results
Tables S1-3 show P value thresholds, significance (P values), R2, and number of informative SNPs, which were included at each P value threshold for each PRS analysis.
We found significant associations between AD disease status and MDD-PRS derived from both PGC-MDD2 (most informative P-threshold=1.0, P=0.00063, R2=0.533%; Fig. 1a) and PGC-MDD1 (P-threshold=0.2, P=0.00014, R2=0.663%; Fig. 1b) meta-analyses; the larger discovery sample did not yield additional predictive power.
Fig. 1.
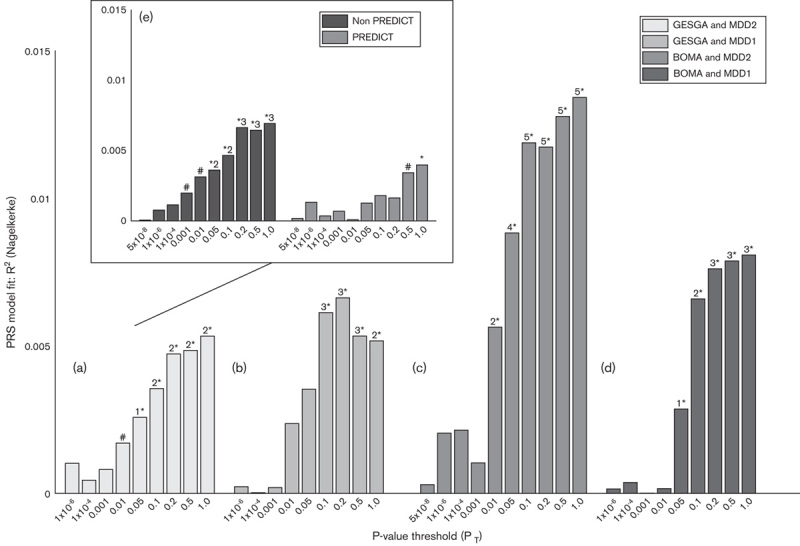
Polygenic risk score model fit. (a) GESGA target sample based on PGC-MDD2 (n=59 265 cases; n=112 092 controls); (b) PGC-MDD1 (n=8148 cases; n=7955 controls) discovery samples; (c) BoMa-MDD target sample based on PGC-MDD2 and (d) PGC-MDD1; and inset (e) non-PREDICT (left) and PREDICT (right) GESGA target subsamples based on PGC-MDD2. MDD, major depressive disorder; PGC, Psychiatric Genomics Consortium. #P<0.10; 1*P<0.05; 2*P<0.01; 3*P<0.001; 4*P<0.0001; 5*P<0.00001.
In contrast, calculating PRS in a MDD target sample yielded increased power when using PGC-MDD2 (P-threshold=1.0, P=0.000038, R2=1.34%; Fig. 1c) versus PGC-MDD1 (P-threshold=1.0, P=0.0013, R2=0.81%; Fig. 1d).
When calculating PGC-MDD2 PRS in the PREDICT subsample (excluding comorbid MDD), significant associations were still found (P-threshold=1.0, P=0.042, R2=0.398%). Meanwhile, PRS in non-PREDICT cases (i.e. not explicitly excluding MDD comorbidity) predicted more variance (P-threshold=1.0, P=0.0003, R2=0.693%; Fig. 1e, inset).
Discussion
Our analysis confirms the contribution of shared genetic risk for AD and MDD long suggested by formal genetics studies that was only recently detected using a molecular approach in Andersen et al. (2017).
Determining shared genetic etiology in comorbid disorders is necessarily faced with the problem of ‘enrichment’ of the comorbid disorders in both discovery and target samples. Our analysis of the PREDICT sample alone revealed that even in AD cases expressly excluding comorbid MDD, genetic overlap is observed; a higher proportion of explained variance was observed using only non-PREDICT cases. These findings are consistent with those of Andersen et al. (2017), who showed that significant genetic overlap remained when calculating PRS in the AD-GWAS data sets after adjusting for MDD status, and also when using a MDD GWAS data set without comorbid MDD-AD cases (Andersen et al., 2017). These results suggest that although PGC-MDD-GWAS samples are likely to contain individuals with AD, these are not fully responsible for the associations observed. For our current analysis, comorbidity information with AD in the PGC-MDD2 discovery sample was not available, nor was MDD status in the full GESGA sample; it should be noted that the possibility of enrichment nevertheless remains. Moreover, these findings underscore the need for rigorous phenotyping and improved characterization of samples, and in particular detailed assessment of disease comorbidity, symptomatology, and severity, all of which will be vital in the effort to understand shared genetic risk of complex diseases.
Interestingly, our use of a substantially larger discovery sample did not demonstrate increased predictive power in the AD sample. One reason for this is that the effect itself is modest in size and less robust against noise. In contrast, the larger discovery sample did yield increased predictive power in the MDD sample; the effects were much stronger. One consideration with respect to our findings is that patients in our AD sample were all males, whereas the discovery sample in addition contained females, potentially affecting predictability. Meanwhile, the MDD target sample contained both males and female patients. However, additional analysis using male-only MDD-PRS, and statistically controlling for sex in the MDD-BoMa samples, did not indicate a substantial influence of sex on our results (see Supplementary Material, Supplemental digital content 1, http://links.lww.com/PG/A204).
Another recent study using a polygenic approach has shown that the level and risk of AD and MDD comorbidity may be linked to neuropsychiatric traits and brain volumes (Zhou et al., 2017). Further research is needed to decipher this pleiotropy and to assess causality. Although not possible here owing to the relatively small size of the target sample, the application of techniques to dissect pleiotropy, such as BUHMBOX (Han et al., 2016) or Mendelian randomization (Davey Smith and Hemani, 2014), will lead to better understanding of this disease comorbidity. Another approach which can be utilized with larger target samples is LD score regression to estimate genetic correlation across diseases and subgroups (Bulik-Sullivan et al., 2015).
Ongoing increases in discovery sample size will lead to continued increases in the ability to explain variance in mental disorders, augmenting the ability to further dissect the shared pathophysiology reflected in the genetic overlap between comorbid diseases. Larger target samples are needed to reduce noise and take advantage of increasing discovery sample size.
Our findings replicate the genetic overlap between AD and MDD and suggest the need for improved, rigorous phenotyping to identify true shared cross-disorder genetic factors. Once assessed, future efforts in the field will be able to take advantage of symptomatology and precise comorbidity information to inform analyses. Importantly, this will also lead to both improved patient stratification and corresponding personalization of care in clinical settings.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.psychgenetics.com.
Acknowledgements
MR and MMN were supported by the German Federal Ministry of Education and Research (BMBF) through grants BMBF 01ZX1311A (to MR and MMN), through grants 01ZX1314A (to MMN) and 01ZX1314G (to MR) within the e:Med research program, and by the German Research Foundation via the Excellence Cluster ImmunoSensation, NO246/10-1 (to MMN) and RI 908/11-1 (to MR).
Norbert Wodarz has received funding from the German Research Foundation (DFG) and Federal Ministry of Education and Research Germany (BMBF); he has received speaker’s honoraria and travel funds from Janssen-Cilag and Indivior. He took part in industry sponsored multicenter randomized trials by D&A Pharma and Lundbeck. Norbert Scherbaum received honoraria for the participation in advisory boards and for holding lectures by the companies AbbVie, Sanofi-Aventis, Mundipharma, Indivior, and Lundbeck. Monika Ridinger received compensation from Lundbeck Switzerland and Lundbeck institute for advisory boards and expert meeting, and from Lundbeck and Lilly Suisse for workshops and presentations. Karl Mann received honoraria from Lundbeck, Pfizer, Novartis, and AbbVie. Markus M. Nöthen received funding from the BMBF and DFG; he has received honoraria from the Lundbeck Foundation, the Robert Bosch Foundation and from the Deutsches Ärzteblatt, and a salary from the Life & Brain GmbH.
Conflicts of interest
There are no conflicts of interest.
Footnotes
*Jerome C. Foo and Fabian Streit contributed equally to the writing of this article.
Members of Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium are listed in Supplementary Material (Supplemental digital content 1, http://links.lww.com/PG/A204)
Contributor Information
Collaborators: Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium
References
- Andersen AM, Pietrzak RH, Kranzler HR, Ma L, Zhou H, Liu X, et al. (2017). Polygenic scores for major depressive disorder and risk of alcohol dependence. JAMA Psychiatry 74:1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. (2015). An atlas of genetic correlations across human diseases and traits. Nat Genet 47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crum RM, Green KM, Storr CL, Chan YF, Ialongo N, Stuart EA, Anthony JC. (2008). Depressed mood in childhood and subsequent alcohol use through adolescence and young adulthood. Arch Gen Psychiatry 65:702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey Smith G, Hemani G. (2014). Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AC, Aliev F, Bierut LJ, Bucholz KK, Edenberg H, Hesselbrock V, et al. (2012). Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr Genet 22:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euesden J, Lewis CM, O’Reilly PF. (2015). PRSice: Polygenic Risk Score software. Bioinformatics 31:1466–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds JA, Adamson SJ, Boden JM, Williman JA, Mulder RT. (2015). Depression in patients with alcohol use disorders: systematic review and meta-analysis of outcomes for independent and substance-induced disorders. J Affect Disord 185:47–59. [DOI] [PubMed] [Google Scholar]
- Frank J, Cichon S, Treutlein J, Ridinger M, Mattheisen M, Hoffmann P, et al. (2012). Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol 17:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. (2014). Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry 19:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Pouget JG, Slowikowski K, Stahl E, Lee CH, Diogo D, et al. (2016). A method to decipher pleiotropy by detecting underlying heterogeneity driven by hidden subgroups applied to autoimmune and neuropsychiatric diseases. Nat Genet 48:803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Wang JC, Wetherill L, Le N, Bertelsen S, Hinrichs AL, et al. (2014). Genome-wide survival analysis of age at onset of alcohol dependence in extended high-risk COGA families. Drug Alcohol Depend 142:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Walters EE, Neale MC, Kessler RC, Heath AC, Eaves LJ. (1995). The structure of the genetic and environmental risk factors for six major psychiatric disorders in women. Phobia, generalized anxiety disorder, panic disorder, bulimia, major depression, and alcoholism. Arch Gen Psychiatry 52:374–383. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Nelson CB, Mcgonagle KA, Edlund MJ, Frank RG, Leaf PJ. (1996). The epidemiology of co-occurring addictive and mental disorders: implications for prevention and service utilization. Am J Orthopsychiatry 66:17–31. [DOI] [PubMed] [Google Scholar]
- Lyons MJ, Schultz M, Neale M, Brady K, Eisen S, Toomey R, et al. (2006). Specificity of familial vulnerability for alcoholism versus major depression in men. J Nerv Ment Dis 194:809–817. [DOI] [PubMed] [Google Scholar]
- Maier W, Lichtermann D, Minges J. (1994). The relationship between alcoholism and unipolar depression – a controlled family study. J Psychiatr Res 28:303–317. [DOI] [PubMed] [Google Scholar]
- Mann K, Kiefer F, Smolka M, Gann H, Wellek S, Heinz A. (2009). Searching for responders to acamprosate and naltrexone in alcoholism treatment: rationale and design of the PREDICT study. Alcohol Clin Exp Res 33:674–683. [DOI] [PubMed] [Google Scholar]
- Nurnberger JI, Jr, Foroud T, Flury L, Meyer ET, Wiegand R. (2002). Is there a genetic relationship between alcoholism and depression? Alcohol Res Health 26:233–240. [PMC free article] [PubMed] [Google Scholar]
- Power RA, Steinberg S, Bjornsdottir G, Rietveld CA, Abdellaoui A, Nivard MM, et al. (2015). Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat Neurosci 18:953–955. [DOI] [PubMed] [Google Scholar]
- Prescott CA, Aggen SH, Kendler KS. (2000). Sex-specific genetic influences on the comorbidity of alcoholism and major depression in a population-based sample of US twins. Arch Gen Psychiatry 57:803–811. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietschel M, Mattheisen M, Frank J, Treutlein J, Degenhardt F, Breuer R, et al. (2010). Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry 68:578–585. [DOI] [PubMed] [Google Scholar]
- Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, et al. (2013). A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 18:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JT, Farh K-H, Holmans PA, et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swendsen JD, Merikangas KR. (2000). The comorbidity of depression and substance use disorders. Clin Psychol Rev 20:173–189. [DOI] [PubMed] [Google Scholar]
- Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P, et al. (2009). Genome-wide association study of alcohol dependence. Arch Gen Psychiatry 66:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winokur G, Coryell W. (1991). Familial alcoholism in primary unipolar major depressive disorder. Am J Psychiatry 148:184–188. [DOI] [PubMed] [Google Scholar]
- Woodruff RA, JR, Guze SB, Clayton PJ, Carr D. (1973). Alcoholism and depression. Arch Gen Psychiatry 28:97–100. [DOI] [PubMed] [Google Scholar]
- Wray NR, Sullivan PF. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics 50:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. (2014). Research review: polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry 55:1068–1087. [DOI] [PubMed] [Google Scholar]
- Zhou H, Polimanti R, Yang BZ, Wang Q, Han S, Sherva R, et al. (2017). Genetic risk variants associated with comorbid alcohol dependence and major depression. JAMA Psychiatry 74:1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L, Lu L, Tan Y, Pan X, Cai Y, Wang X, et al. (2014). Genome-wide association discoveries of alcohol dependence. Am J Addict 23:526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.psychgenetics.com.