

Abstract
Objectives:
To investigate efficacy and safety of a single-tablet regimen of darunavir/cobicistat/emtricitabine/tenofovir alafenamide (D/C/F/TAF) 800/150/200/10 mg vs. darunavir/cobicistat plus emtricitabine/tenofovir disoproxyl fumarate (TDF) (control) in antiretroviral-treatment-naive, HIV-1-infected adults.
Design:
Phase-3, randomized, active-controlled, double-blind, international, multicenter, noninferiority study (NCT02431247).
Methods:
Seven hundred and twenty-five participants were randomized (1 : 1) to D/C/F/TAF (362) or control (363). The primary objective was to demonstrate noninferiority of D/C/F/TAF vs. control for percentage viral load less than 50 copies/ml (FDA-snapshot analysis) at 48 weeks (10% margin).
Results:
At week 48, D/C/F/TAF was noninferior to control (91.4 vs. 88.4% achieved viral load <50 copies/ml, respectively; difference 2.7%; 95% CI −1.6 to 7.1; P < 0.0001), with 4.4 vs. 3.3% of patients, respectively, having viral load greater or equal to 50 copies/ml. No treatment-emergent mutations associated with darunavir or TAF/TDF resistance were observed in either group. One patient (D/C/F/TAF) was identified with M184I/V conferring resistance to emtricitabine. Incidences of grades 3 and 4 adverse events (5 vs. 6%), serious adverse events (5 vs. 6%) and adverse event-related discontinuations (2 vs. 4%) were low and similar between groups. Mean decrease in urine protein/creatinine ratio was greater with D/C/F/TAF than control (−22.42 vs. −10.34 mg/g, P = 0.033). Mean percentage change in bone mineral density with D/C/F/TAF vs. control was 0.21 vs. −2.73%, P < 0.0001 (hip), −0.68 vs. −2.38%, P = 0.004 (lumbar spine), and −0.26 vs. −2.97%, P < 0.0001 (femoral neck). Median change from baseline in total cholesterol/HDL-cholesterol ratio was 0.20 vs. 0.08, P = 0.036.
Conclusion:
D/C/F/TAF achieved a high virologic suppression rate (91.4%) and was noninferior to darunavir/cobicistat with F/TDF. D/C/F/TAF also demonstrated the bone and renal safety advantages of TAF in combination with darunavir/cobicistat.
Keywords: darunavir/cobicistat/emtricitabine/tenofovir alafenamide, efficacy, once daily, safety, single-tablet HIV-1 regimen
Introduction
Combination antiretroviral therapy (ART) regimens for HIV-1-infected patients are now more effective, safe and convenient. However, treatment adherence, emergence of resistant virus with virologic failure, and tolerability remain important challenges [1]. Convenient once-daily, single-tablet regimens (STR) can facilitate treatment adherence and improve treatment effectiveness [2,3].
Since its initial approval in 2006, substantial clinical trial data and clinical experience with darunavir have accumulated, demonstrating the potent and durable virologic response, high genetic barrier to resistance, and favorable safety profile in ART-naive, HIV-1-infected patients [4,5]. A substantial proportion of newly diagnosed patients in the United States and Europe are treated with a boosted protease inhibitor [6,7], and darunavir is the recommended protease inhibitor in treatment guidelines [8–10]. United States guidelines recommend two nucleoside or nucleotide analogue reverse transcriptase inhibitors (NRTIs) combined with an integrase strand transfer inhibitor (INSTI), or in certain clinical situations boosted darunavir 800 mg once daily or a nonnucleoside reverse transcriptase inhibitor (NNRTI) [8,9]. Boosted darunavir is recommended for patients with uncertain adherence, those who require a regimen with a high-resistance barrier, or those patients without available resistance results [8]. European guidelines recommend two NRTIs combined with either an INSTI, boosted darunavir or an NNRTI for all ART-naive patients [10], with both darunavir and atazanavir as recommended protease inhibitors in the BHIVA guidelines [11].
Phase-3 studies have established the noninferior antiviral efficacy and improved renal and bone safety of ART regimens containing tenofovir alafenamide (TAF), a newer tenofovir prodrug, vs. tenofovir disoproxil fumarate (TDF), combined with different third agents [12,13], making TAF an optimal backbone component.
Darunavir/cobicistat/emtricitabine/tenofovir alafenamide (D/C/F/TAF) 800/150/200/10 mg is the first and only once-daily protease inhibitor-containing STR in development, combining the antiviral efficacy, and resistance barrier of darunavir with the safety of TAF. D/C/F/TAF was approved for use in Europe in September 2017, and is investigational and currently undergoing regulatory review in the United States. D/C/F/TAF is being evaluated in two international, randomized, phase-3 studies: AMBER (NCT02431247) in ART-naive, HIV-1-infected adults, and EMERALD (NCT02269917) in treatment-experienced adults with virologically suppressed HIV infection [14]. We present the 48-week primary analysis of AMBER, which evaluated D/C/F/TAF vs. darunavir/cobicistat in combination with emtricitabine/TDF (F/TDF).
Methods
Study design
AMBER (TMC114FD2HTX3001; ClinicalTrials.gov NCT02431247; EudraCT 2015–000754–38) is a phase-3, randomized, active-controlled, double-blind, noninferiority study being conducted at 121 sites across 10 countries in North America (USA, Canada) and Europe (Belgium, France, Germany, Italy, Poland, Russia, Spain, UK). The trial included a ∼30-day screening period (up to ≤6 weeks) and a 48-week treatment period. In addition, all patients continue to receive D/C/F/TAF in an open-label, single-arm treatment phase up to week 96, and then in a roll-over extension phase.
Participants were randomized (1 : 1) using a computer-generated interactive web-response system to receive D/C/F/TAF 800/150/200/10 mg (q.d.) daily or darunavir/cobicistat 800/150 mg fixed-dose combination (FDC) co-administered with F/TDF 200/300 mg FDC daily (control). Participants received placebo tablets matching the alternative treatment – three tablets in total – and were instructed to take all study drugs and matching placebo tablets with food at approximately the same time each morning. Randomization was stratified by screening viral load (≤ or >100 000 copies/ml) and CD4+ cell count (< or ≥200 cells/μl).
The trial was conducted in accordance with the principles of Good Clinical Practice and Declaration of Helsinki. The protocol and amendments were reviewed and approved by an institutional review board or independent ethics committee. All study participants provided written informed consent.
Study population
Eligible patients were treatment-naive, HIV-1-infected adults (≥18 years) with a screening plasma viral load at least 1000 copies/ml, CD4+ cell count greater than 50 cells/μl, genotypic sensitivity to darunavir, emtricitabine, and tenofovir (GenoSure MG HIV-1 protease/reverse transcriptase genotype assay; Monogram Biosciences, South San Francisco, California, USA), and an estimated glomerular filtration rate based on serum creatinine (eGFRcr) at least 70 ml/min (Cockcroft–Gault formula) [15]. Main exclusion criteria included diagnosis of a new AIDS-defining condition within 30 days prior to screening, hepatitis B or C coinfection, clinically significant disease (e.g. malignancy, severe infections), and pregnancy or breastfeeding in women. Medications or herbal supplements known or suspected to have drug interactions with the investigational medications were disallowed.
Main study assessments and outcomes
Study visits were at baseline, weeks 2, 4, 8, and 12, and then every 12 weeks until week 96. Adverse events were graded according to the Division of AIDS grading table [16] and coded using the Medical Dictionary for Regulatory Activities (version 19.1). At each visit, urine and blood samples were collected for plasma viral load (COBAS AmpliPrep/COBAS TaqMan HIV-1 Test, V2.0; Roche Diagnostics, Basel, Switzerland) and CD4+ cell count determinations, biochemistry, hematology, urinalysis and urine chemistry, serum cystatin C for calculating eGFRcyst [Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula] [17], and serum creatinine for calculating eGFRcr (Cockcroft–Gault formula and CKD-EPI formula) [15,17]. Treatment adherence was monitored at each visit (except week 2) by drug accountability (pill count and patient log booklet). The renal proteinuria biomarkers, urinary retinol-binding protein (RBP), and beta-2-microglobulin were measured at baseline, weeks 2, 4, 12, 24, and 48 in the fasted state. Fasted metabolic profile assessments (total, high-density lipoprotein [HDL]-cholesterol and low-density lipoprotein [LDL]-cholesterol, triglycerides) were performed at baseline, weeks 24 and 48. Pharmacokinetic sampling was performed at weeks 2, 4, 8, 12, 24, 36, 48, and study endpoint.
Protocol-defined virologic failure (PDVF) was defined as virologic nonresponse (viral load <1 log10 reduction from baseline and ≥50 copies/ml at week 8, confirmed at next visit) or virologic rebound (confirmed viral load ≥50 copies/ml after confirmed, consecutive viral load <50 copies/ml or confirmed viral load >1 log10 increase from the nadir) and/or viremia at the final time point (viral load ≥400 copies/ml at study endpoint or study discontinuation after week 8). Post screening resistance testing (PhenoSense GT) was performed on samples from patients with PDVF and viral load greater or equal to 400 copies/ml at time of failure (preferably confirmed, or otherwise unconfirmed) or at later time points.
The primary objective was the noninferiority evaluation of D/C/F/TAF vs. darunavir/cobicistat co-administered with F/TDF in the proportion of patients with viral load less than 50 copies/ml (response rate) by the Food and Drug Administration (FDA)-snapshot analysis at week 48.
Secondary outcomes included proportion of patients with viral load <20 and <200 copies/ml (FDA-snapshot analysis) and viral load<50 copies/ml (time-to-loss-of-virologic-response algorithm) at week 48; changes from baseline in log10 viral load and CD4+ cell count; antiretroviral resistance development in PDVFs; safety and tolerability through 48 weeks; changes from baseline at week 48 in serum creatinine, eGFRcr, eGFRcyst, and ratios of total urine protein, urine albumin, urine RBP, and beta-2-microglobulin to creatinine (UPCR, UACR, RPB:Cr, and B2M:Cr, respectively).
Bone investigation substudy
The bone investigation substudy was performed at selected study sites in consenting participants from both randomization groups. Endpoints at weeks 24 and 48 were percentage changes from baseline in spine, hip, and femoral neck bone mineral density (BMD; measured by dual-energy X-ray absorptiometry scans); changes in associated T score (normal BMD defined as a T score ≥−1; osteopenia as a T score from ≥−2.5 to <−1; and osteoporosis as a T score <−2.5); and changes in bone biomarkers, alkaline phosphatase (ALP), procollagen type N-terminal propeptide (P1NP), C-type collagen sequence (CTX), parathyroid hormone (PTH), and 25-hydroxy vitamin (25[OH]D), measured in the fasted state.
Statistical analysis
The week-48 primary analysis was performed on the intent-to-treat population (constituting all patients who were randomized and received at least one dose of study drug). A per-protocol analysis was also performed, excluding patients with major protocol violations or other predefined criteria that potentially affected efficacy. Data analysis was performed using SAS software (SAS Institute, Inc, Cary, North Carolina, USA) version 9.2.
Assuming a response rate of 80% at week 48 (FDA-snapshot analysis) for both treatment groups, 335 patients needed to be enrolled in each group to establish noninferiority of D/C/F/TAF to control, with a noninferiority margin of 10% at 90% power and a one-sided significance level of 2.5%. For the bone investigation substudy, at least 85 patients per treatment group were required to detect an absolute difference between groups in BMD of at least 2% with 90% power, assuming a 4% inter-subject variability and a one-sided significance level of 2.5%.
Noninferiority of D/C/F/TAF to control would be demonstrated if the lower limit of the two-sided 95% confidence interval (CI) of the stratum-adjusted (viral load ≤100 000 or >100 000 copies/ml and CD4+ cell count <200 or ≥200 cells/μl) Mantel–Haenszel difference between treatment groups (D/C/F/TAF minus control) in the week-48 response rate was greater than −10%. Superiority would be established if the lower limit of the 95% CI was greater than 0.
The difference between groups in least square mean (LSM) change from baseline at week 48 in CD4+ cell count and associated 95% CIs were constructed using analysis of covariance (ANCOVA), including CD4+ cell count at baseline as a continuous covariate. In patients who discontinued, CD4+ cell count values after discontinuation were imputed with the baseline value (noncompleter = failure). For other missing values, the last observation was carried forward.
Baseline and postbaseline HIV-1 genotypes were analyzed for protease resistance-associated mutations (RAMs) [including International Antiviral Society (IAS)–USA primary PI RAMs] and reverse transcriptase RAMs (including IAS-USA NRTI RAMs and IAS–USA NNRTI RAMs), as well as specific RAMs to the study drugs [18]. Antiretroviral sensitivity, based on the genotype/phenotype report, was also assessed.
Within-treatment comparisons of mean changes from baseline in renal and bone biomarkers, and fasting lipids were performed using the Wilcoxon signed-rank test. Between-treatment comparisons were assessed using the Wilcoxon rank-sum test. Between-treatment differences in change from baseline in serum creatinine, eGFR, and BMD were tested using ANCOVA, including treatment as a factor and corresponding baseline values as covariates.
Results
Patient disposition and baseline characteristics
The study began on 6 July 2015, and the cut-off date for the week-48 primary analysis was 13 March 2017. Of 866 screened patients, 725 were randomized and included in the intent-to-treat population (Fig. 1); 362 received D/C/F/TAF and 363 received darunavir/cobicistat with F/TDF.
Fig. 1.
Patient disposition through 48 weeks.

AE, adverse event; Control regimen, darunavir/cobicistat with emtricitabine/tenofovir disoproxil fumarate once daily; D/C/F/TAF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide once daily. aReceived at least one dose of study medication. bBased upon the ‘Trial Termination’ electronic case report form page as reported by the investigators. Reasons for discontinuation may not match those reported in Supplementary Table 1, because patients may have viral load data within the week-48 window, which was used to determine the FDA-snapshot category. cOccurred in the follow-up phase (11 days after last study drug intake). FDA, Food and Drug Administration.
Through 48 weeks, 93.6% (339/362) of patients in the D/C/F/TAF group and 92.3% (335/363) in the control group completed therapy (Fig. 1). The most common reasons for discontinuing the study, as reported by the investigators, were adverse events, withdrawn consent, and loss to follow-up.
Baseline characteristics were balanced between the two groups (Table 1). Median age was 34 years, 88% were men, 83% were white, and 18% had viral load at least 100 000 copies/ml. Median baseline CD4+ cell count was 453 cells/μl.
Table 1.
Patient baseline demographics and disease characteristics.
| Demographics, n (%), unless stated | D/C/F/TAF 800/150/200/10 mg once daily, N = 362 | Control regimen, N = 363 | Total, N = 725 |
| Median age (IQR), years | 34 (27–42) | 34 (27–42) | 34 (27–42) |
| More than 50 | 36 (10) | 32 (9) | 68 (9) |
| Gender | |||
| Female | 44 (12) | 41 (11) | 85 (12) |
| Male | 318 (88) | 322 (89) | 640 (88) |
| Race | |||
| White | 300 (83) | 300 (83) | 600 (83) |
| Black/African-American | 40 (11) | 40 (11) | 80 (11) |
| Other | 22 (6) | 23 (6) | 45 (6) |
| Ethnicity | |||
| Hispanic or Latino | 50 (14) | 45 (12) | 95 (13) |
| Baseline disease characteristics | |||
| Median (IQR) time since diagnosis, months | 5.73 (2.53–25.59) | 4.30 (2.07–17.74) | 4.83 (2.33–21.62) |
| Median (IQR) log10 viral load, copies/ml | 4.44 (4.03–4.82) | 4.57 (4.15–4.88) | 4.52 (4.10–4.87) |
| Viral load at least 100 000 copies/ml, n (%) | 60 (17) | 70 (19) | 130 (18) |
| Median (IQR) CD4+ cell count, cells/μl | 461.5 (342–617) | 440.0 (325–594) | 453.0 (333–601) |
| CD4+ cell count less than 200 cells/μl, n (%) | 22 (6) | 29 (8) | 51 (7) |
| Median (IQR) eGFRcr, ml/min (Cockcroft–Gault) | 119.3 (104.8–135.2) | 118.4 (103.2–138.4) | 119.1 (104.4–136.5) |
| Genotypea at screening, n (%) [18] | N = 361b | N = 362b | N = 723 |
| At least one darunavir resistance-associated mutation | 3 (1) | 4 (1) | 7 (1)c |
| At least one primary protease inhibitor resistance-associated mutation | 7 (2) | 8 (2) | 15 (2) |
| At least one NRTI resistance-associated mutation | 18 (5) | 16 (4) | 34 (5)d |
| At least one NNRTI resistance-associated mutation | 55 (15) | 63 (17) | 118 (16)e |
Control regimen, darunavir/cobicistat plus emtricitabine/tenofovir disoproxil fumarate once daily; D/C/F/TAF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide once daily; eGFRcr, estimated glomerular rate based on serum creatinine; IQR, interquartile range; NNRTI, nonnucleoside analogue reverse transcriptase inhibitor; NRTI, nucleoside or nucleotide analogue reverse transcriptase inhibitor.
aGenoSureMG.
bOne patient in each group had failed screening genotypes and were enrolled based on local genotypes.
cSix V11I, one L33F.
dThe most prevalent NRTI mutation: A62V: 21/725 (2.9%).
eThe most prevalent NNRTI mutation: K103N 26/725 (3.6%).
As depicted by the protocol, at screening, all enrolled participants demonstrated genotypic sensitivity to darunavir, emtricitabine, and tenofovir based on the genotype report. Few had viruses with at least one darunavir RAMs (1%) or primary PI RAMs (2%) (Table 1). No RAMs related to emtricitabine or TDF/TAF were detected. NNRTI and NRTI RAMs were detected in 16 and 5% of patients, respectively (Table 1).
Efficacy
In the primary analysis of virologic response at week 48 (FDA-snapshot analysis), noninferiority of D/C/F/TAF [91.4% (331/362)] vs. control [88.4% (321/363)] was demonstrated (difference 2.7%; 95% CI −1.6 to 7.1; P < 0.0001; Fig. 2a and Supplemental Table S1). A low proportion of participants in the D/C/F/TAF group [4.4% (16/362)] and control group [3.3% (12/363)] had a viral load greater or equal to 50 copies/ml at week 48 (FDA-snapshot analysis).
Fig. 2.
Week-48 Food and Drug Administration-snapshot analysis (<50 copies/ml).
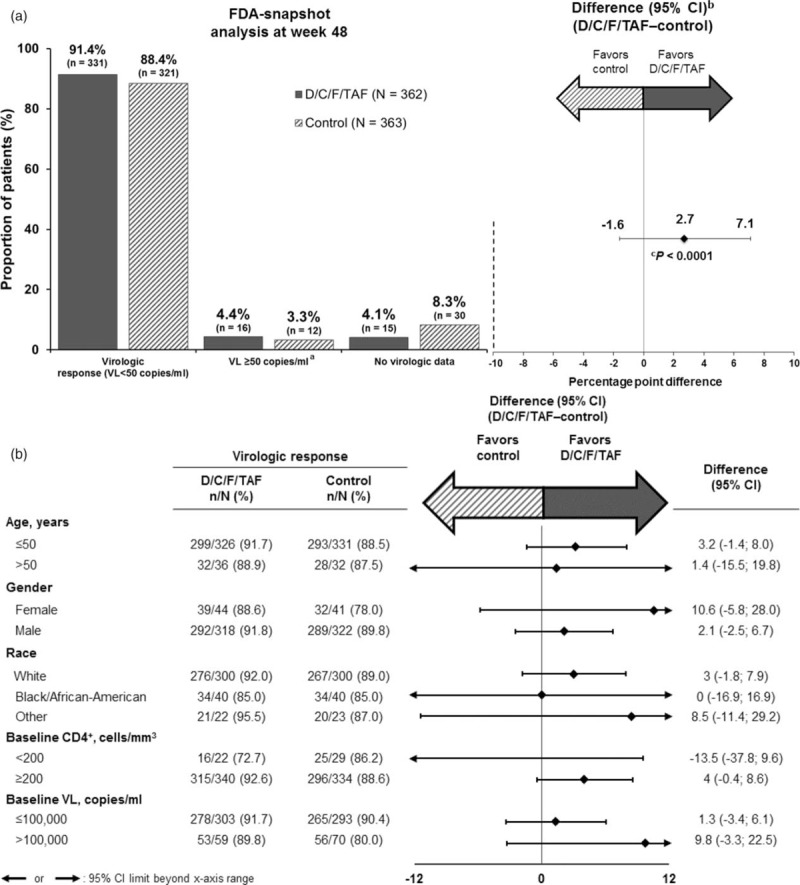
(a) Virologic outcomes overall and (b) subgroup analyses of week-48 response rates. CI, confidence interval; control regimen, darunavir/cobicistat with emtricitabine/tenofovir disoproxil fumarate once daily; D/C/F/TAF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide once daily; VL, viral load. aLast viral load in the week-48 window at least 50 copies/ml, or discontinuations for efficacy reasons, or premature discontinuations not because of efficacy, adverse events or death with a last viral load at least 50 copies/ml. bCalculated with Mantel–Haenszel test adjusting for screening viral load (≤ or >100 000 copies/ml) and CD4+ cell count (< or ≥200 cells/μl). cP value for noninferiority at 10% margin.
Results from the per-protocol analysis confirmed noninferiority of D/C/F/TAF [94% (327/348)] to control [92.2% (317/344)] (difference 1.5%; 95% CI −2.3 to 5.2; P < 0.0001), as did other sensitivity analyses (Supplementary Table 2). Week-48 response rates (FDA-snapshot analysis) were consistent across a range of patient subgroups (Fig. 2b).
A similar proportion of patients in each group also achieved a viral load <200 or <20 copies/ml (FDA-snapshot analysis) at week 48 (Supplementary Table 2). LSM increases (P < 0.0001) from baseline in CD4+ cell count (noncompleter = failure) at week 48 were 190.5 cells/μl for D/C/F/TAF vs. 172.0 cells/μl for control (P = 0.213 between groups; Supplementary Table 2).
Virology
Through week 48, eight (D/C/F/TAF) and six (control) participants had PDVF, with paired screening and postbaseline on-treatment genotypes available for seven vs. two patients, respectively. No darunavir, primary protease inhibitor, or TDF/TAF RAMs emerged in any patient. An M184V/I mutation associated with phenotypic resistance to emtricitabine and lamivudine was identified in one patient receiving D/C/F/TAF. This patient harbored a K103N mutation at screening, indicating transmitted NNRTI (efavirenz and nevirapine) resistance. Although the patient appeared to have good adherence (≥95% based on pill count), darunavir plasma concentrations were low [32–192 ng/ml, except at week 4 (1440 ng/ml)], indicating nonadherence that resulted in the patient being discontinued from the study after week 48. All other participants had virus that remained susceptible to all drugs in the treatment regimens.
Adherence
Through week 48, 88.3% (264/299) vs. 88.3% (271/307) of patients in the D/C/F/TAF and control groups, respectively, were at least 95% adherent as measured by pill count (all patients took three tablets daily based on the study design).
Safety
Safety profiles were similar between groups (Table 2). Most adverse events regardless of causality were grade 1 or 2. The most common (≥5% in either group) study drug-related adverse events through week 48 were diarrhea, rash, and nausea (Table 2). All episodes of study drug-related diarrhea were mild or moderate (grade 1 or 2) and mostly transient in duration. Only one patient in each group (0.3%) discontinued the study because of diarrhea. There were no nervous system study drug-related adverse events greater than 5% nor discontinuations in either group.
Table 2.
Treatment-emergent adverse events and laboratory abnormalities through 48 weeks.
| D/C/F/TAF 800/150/200/10 mg once daily, N = 362 | Control regimen, N = 363 | |
| Any adverse event regardless of causality | 312 (86) | 307 (85) |
| Any study drug-related adverse event | 126 (35) | 151 (42) |
| Any grade 3 or 4 adverse event regardless of causality | 19 (5) | 22 (6) |
| Any serious adverse event regardless of causalitya | 17 (5) | 21 (6) |
| Adverse events leading to permanent discontinuationb | 7c (2) | 16 (4) |
| Deathd | 0 | 0 |
| Most common adverse events regardless of causality (≥5% of patients in either group) | ||
| Diarrheae | 71 (20) | 66 (18) |
| Headache | 47 (13) | 32 (9) |
| Nasopharyngitis | 40 (11) | 31 (9) |
| Rash | 32 (9) | 25 (7) |
| Nausea | 28 (8) | 45 (12) |
| Upper respiratory tract infection | 20 (6) | 21 (6) |
| Fatigue | 19 (5) | 18 (5) |
| Syphilis | 17 (5) | 19 (5) |
| Osteopenia | 17 (5) | 27 (7) |
| Bronchitis | 14 (4) | 19 (5) |
| Adverse events at least possibly related to study drug (≥5% of patients in either group) | ||
| Diarrheae | 31 (9) | 40 (11) |
| Rash | 22 (6) | 14 (4) |
| Nausea | 20 (6) | 36 (10) |
| Median (IQR) change from baseline in fasting lipids at week 48 | ||
| Total cholesterol (mg/dl) | 28.6 (12.8 to 47.2)f | 10.4 (−8.0 to 29.8) |
| HDL-cholesterol (mg/dl) | 4.3 (−1.2 to 12.0)f | 1.5 (−3.9 to 8.1) |
| LDL-cholesterol (mg/dl) | 17.4 (2.9 to 32.9)f | 5.0 (−10.8 to 19.0) |
| Triglycerides (mg/dl) | 23.9 (−3.0 to 58.5)g | 14.2 (−12.0 to 40.7) |
| Total cholesterol/HDL-cholesterol ratio | 0.20 (−0.28 to 0.67)h | 0.08 (−0.41 to 0.53) |
Data are n (%) unless otherwise stated.
Control regimen, darunavir/cobicistat plus emtricitabine/tenofovir disoproxil fumarate once daily; D/C/F/TAF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide once daily; HDL, high-density lipoprotein; IQR, interquartile range; LDL, low-density lipoprotein.
aConsidered study drug-related in zero (D/C/F/TAF group) vs. six patients (1.7% control; rash and toxic skin eruption in two patients each, and bone marrow edema and Stevens–Johnson syndrome in one patient each).
bD/C/F/TAF (n = 7): rash (n = 4), generalized rash, maculopapular rash, diarrhea (n = 1 each); control (n = 16): rash/erythema (n = 7), toxic skin eruption (n = 2), neoplasms (n = 2), Stevens–Johnson syndrome, diarrhea, bone marrow edema, increased beta-2-microglobulin, and arthralgia (n = 1 each).
cOne fewer patient in the D/C/F/TAF group (compared with Fig. 1) had an adverse event assessed as leading to discontinuation as data are taken from the adverse event electronic case report form (whether or not the drug was withdrawn), and the patient had interrupted treatment.
dOne death occurred in the control arm, but in follow-up (not considered related to study drug).
eThe majority of episodes of diarrhea were mild: grade 1: 16 vs. 13% (related: 7 vs. 9%) and grade 2: 4 vs. 5% (related: 2 vs. 2%).
fP < 0.0001 (total cholesterol, HDL-cholesterol, LDL-cholesterol).
gP = 0.001 (triglycerides).
hP = 0.036 (total cholesterol/HDL-cholesterol ratio) for D/C/F/TAF group vs. control group.
Renal adverse events regardless of causality occurred in 2% (7/362) of D/C/F/TAF vs. 6% (21/363) of control patients. No renal adverse events were suggestive of treatment-emergent proximal renal tubulopathy and no renal adverse events led to discontinuation.
Grades 3 and 4 adverse events regardless of causality, serious adverse events, and adverse event-related discontinuations were rare (Table 2). The only grade 4 adverse event reported for at least two patients was suicide attempt, reported in two (0.6%) patients in the control group. There were no deaths during the treatment phase in either group (Table 2). However, one patient in the control group died following grade 4 sepsis in the follow-up phase (11 days after last study drug intake), which was not considered related to study drug (Fig. 1). Incidences and types of laboratory abnormalities were similar in both treatment groups, being mostly grade 1 or 2.
Median changes from baseline at week 48 for fasting lipid parameters were higher for D/C/F/TAF than control (Table 2 and Supplementary Figure 1). Changes in HDL-cholesterol favored D/C/F/TAF and remaining lipid increases favored control, with a small, statistically significant difference in the change from baseline in total cholesterol/HDL-cholesterol ratio between groups. Six (1.7%) vs. two (0.6%) patients, respectively, initiated a lipid-lowering drug during the treatment period (P = 0.1770 between groups).
Serum creatinine increased from baseline to week 48 in the D/C/F/TAF group (4.8 μmol/l), consistent with cobicistat inhibition of creatinine tubular secretion [19], but less so than in the control group (8.2 μmol/l; P < 0.0001, ANCOVA D/C/F/TAF vs. control). Consequently, the mean decrease in eGFRcr (CKD-EPI formula) at week 48 was less for D/C/F/TAF than control (−5.9 vs. −9.3 ml/min per 1.73 m2, respectively; P < 0.0001, ANCOVA; Fig. 3a), although mean eGFRcr was within normal limits. However, mean eGFRcyst (CKD-EPI formula) actually increased at week 48, and the increase was greater for D/C/F/TAF than control (5.3 vs. 2.9 ml/min per 1.73 m2, respectively; P = 0.001, ANCOVA) (Fig. 3b).
Fig. 3.
Mean change from baseline to week 48 in kidney and bone parameters.
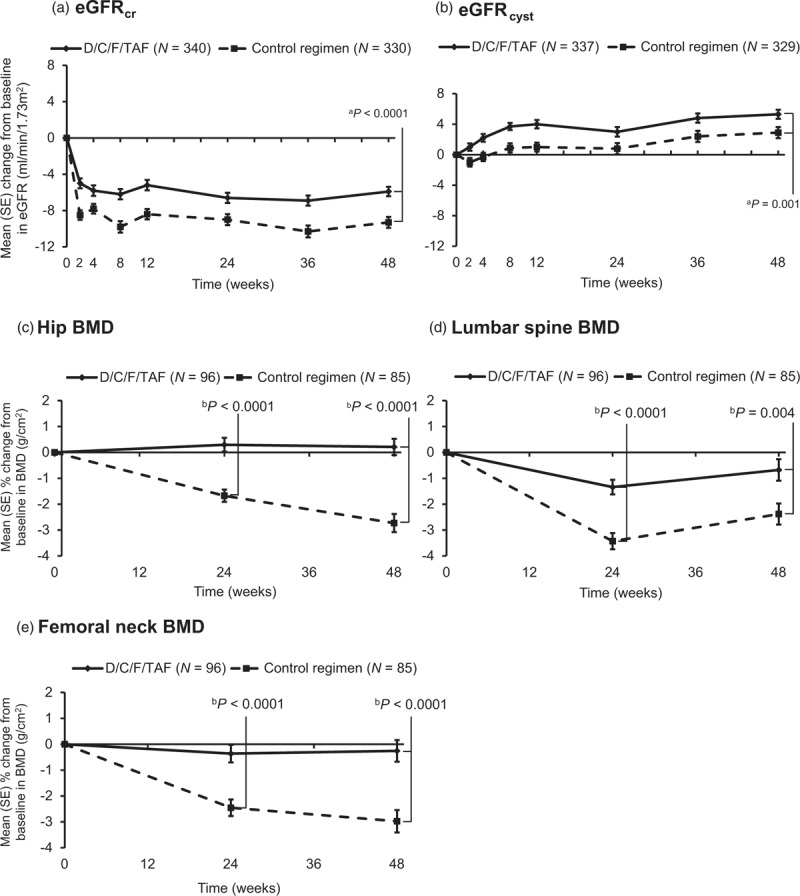
Mean change in (a) eGFRcr and (b) eGFRcyst was based on serum concentrations and the Kidney Disease Epidemiology Collaboration formula. BMD of the (c) hip, (d) lumbar spine, and (e) femoral neck was analyzed with dual energy X-ray absorptiometry. Bars show SE. ANCOVA, analysis of covariance; BMD, bone mineral density; Control regimen, darunavir/cobicistat with emtricitabine/tenofovir disoproxil fumarate once daily; D/C/F/TAF, darunavir/cobicistat/emtricitabine/tenofovir alafenamide once daily; eGFRcr, estimated glomerular filtration rate based on serum creatinine; eGFRcyst, estimated glomerular filtration rate based on serum cystatin C; SE, standard error. aP-value for between-treatment comparison estimated using ANCOVA, including treatment as a factor and corresponding baseline eGFR as a covariate; within-group changes from baseline at week 48, P < 0.0001 (both groups for eGFRcr and eGFRcyst). bP value for between-treatment comparison estimated using ANCOVA, including treatment as a factor and baseline BMD as a covariate; Within-group changes from baseline at week 48 at each site, P < 0.0001 (D/C/F/TAF); P = nonsignificant (control).
At week 48, all quantitative measures demonstrated less proteinuria for D/C/F/TAF vs. control, as determined by mean changes from baseline in UPCR [−22.42 mg/g (SD 71.98) vs. −10.34 mg/g (118.18), respectively; P = 0.033], UACR [−2.45 mg/g (23.81) vs. −0.58 mg/g (68.93); P = 0.003], RBP:Cr [16.84 μg/g (317.31) vs. 401.12 μg/g (2688.91); P < 0.0001], and B2M:Cr [−100.58 μg/g (788.60) vs. 837.63 μg/g (6122.87); P < 0.0001].
Baseline characteristics in the bone investigation substudy were well balanced between the D/C/F/TAF (N = 113) and control (N = 99) groups (Supplementary Table 3). Hip, lumbar spine, and femoral neck BMD from baseline to week 48 were stable with D/C/F/TAF (mean percentage change 0.21, −0.68, and −0.26% at each site, respectively; Fig. 3), whereas they decreased significantly at week 48 in the control group [−2.73, −2.38, and −2.97%, respectively; P < 0.0001 (hip and femoral neck) and P = 0.004 (spine) for between-treatment comparisons]. Fewer patients receiving D/C/F/TAF had at least 3% decreases from baseline in BMD at each site than in the control group. More patients had at least 3% increases in the D/C/F/TAF group (Supplementary Table 4). A similar trend was seen for at least 5 and at least 7% increases or decreases in BMD (Supplementary Table 4). At week 48, a greater proportion of participants receiving D/C/F/TAF had improvements in T score at each site than in the control group, and a smaller proportion of participants receiving D/C/F/TAF had worsening BMD status (Supplementary Table 4). Fractures occurred infrequently and were not different between groups [1.1% (4/362) D/C/F/TAF vs. 0.6% (2/363) control; P = 0.451]; all were traumatic and none were suspected to be osteoporotic. New antiosteoporotic treatment was started by 9/362 (2.5%) vs. 16/363 (4.4%) patients, respectively, during the treatment phase. Changes from baseline in bone biomarker levels (ALP, P1NP, CTX, and PTH) suggested less bone turnover for D/C/F/TAF than control (Supplementary Figure 2). 25[OH]D levels increased from baseline in both groups.
Discussion
In this investigational phase-3, double-blinded, randomized, controlled trial, the D/C/F/TAF once-daily STR was virologically noninferior to darunavir/cobicistat co-administered with F/TDF in ART-naive patients. Response rates were similar across age, sex, race, and baseline HIV characteristics including CD4+ cell count less than 200 cells/μl and viral load greater than 100 000 copies/ml. Although INSTI-based regimens have rapidly moved up in global treatment guidelines [8–10], there are still many patients who might benefit from the established characteristics of the protease inhibitor darunavir, such as high genetic barrier to resistance, efficacy in the face of resistance and uncertain adherence, provider comfort, and experience. Well powered, phase-3, double-blinded, randomized studies provide the most rigorous evidence to drive treatment guidelines. The week-48 virologic response rate (FDA-snapshot analysis) of 91.4% for D/C/F/TAF was among the highest achieved by a STR in phase-3 trials (range 80–93%) of ART-naive patients [12,20–26], and higher than in prior phase-3 trials with darunavir [4,23,27,28].
No treatment-emergent mutations associated with darunavir or tenofovir resistance were observed. Only one patient (D/C/F/TAF) was found to have M184I/V, conferring resistance to emtricitabine; this patient also had a transmitted K103N mutation at screening. M184V was detected pretreatment by deep sequencing (Illumina MiSeq) as a minority variant (9.4%). In addition, for this patient, darunavir plasma concentrations were low and much lower than the steady-state predose concentration (∼692 ng/ml), indicating potential nonadherence, which in fact resulted in discontinuation from the study. The observation of no darunavir phenotypic resistance and the genotypic results are consistent with previous darunavir studies [4,5,27,29], confirming the high resistance barrier of darunavir-based initial ART with no emergence of DRV resistance. D/C/F/TAF is the only STR in development that combines the high barrier to resistance of darunavir with the F/TAF backbone. In this context, D/C/F/TAF may have an important role for treating patients with uncertain adherence or who plan to start treatment prior to the availability of resistance-testing results [8]. Patients with transmitted NNRTI and NRTI resistance were included in the study. As D/C/F/TAF does not require HLA B∗5701 screening or hepatitis or resistance testing before treatment initiation, it is currently being evaluated in a rapid initiation protocol (NCT03227861). These characteristics suggest D/C/F/TAF is a highly feasible option in a test and treat setting or for very early treatment-naive patients where rapid combination ART initiation could be warranted.
Safety profiles were similar between the two treatment groups. However, adverse event-related discontinuations were lower for D/C/F/TAF (2%) than control (4%), and similar to those reported in phase-3 studies of other recently approved STRs [12,20–26]. The low incidences and similar types of adverse events, grade 3 or 4 adverse events, and serious adverse events between groups reflects the well characterized safety profiles for darunavir and cobicistat reported previously [4,27,29]. Given the low incidence of nervous system adverse events, D/C/F/TAF may be an important treatment option for ART-naive patients at risk of nervous system adverse events, such as insomnia and depression.
Less renal tubular proteinuria, and more favorable hip and spine BMD for D/C/F/TAF compared with control are consistent with TAF vs. TDF effects [12,13,29–31]. The improvement in eGFRcyst could reflect ART-related improvement in HIV-associated renal impairment, as was seen in the START study [32]. The favorable renal tubular and BMD outcomes at the 48-week time point are reassuring, given the fact that the cumulative adverse effects of TDF on renal and bone outcomes have been greater whenever TDF was combined with boosted protease inhibitors [33]. Median increases from baseline in fasting lipids were higher for D/C/F/TAF vs. control, with the increase in HDL-cholesterol favoring D/C/F/TAF and remaining lipid increases favoring control. There was a small, statistically significant difference in the total cholesterol/HDL-cholesterol ratio between groups. Differences in lipid profiles were likely because of the loss of the lipid-lowering effect of TDF rather than an adverse effect of TAF or any other of the components on lipids [12,13].
As in other recent phase-3 trials in ART-naive patients [20–26], study limitations were inclusion of more than 80% white patients and a comparatively small proportion of female or older (>50 years) participants or who had high viral loads. The latter most likely reflects earlier initiation of ART based on current guideline recommendations [8–11]. Phase-3 studies often lack power to detect rare clinical safety events; however, the large clinical safety database for darunavir and substantial clinical experience counterbalance this limitation. Renal and bone safety were assessed using surrogate markers rather than clinical events, and bone safety was assessed in a smaller number of patients.
In conclusion, D/C/F/TAF was noninferior to a regimen of darunavir/cobicistat co-administered with F/TDF at week 48, with a high virologic response (91.4%) in ART-naive, HIV-1-infected adults. D/C/F/TAF was associated with a better bone and renal safety profile than control, with few moderate, severe, or serious adverse events. Changes in HDL-cholesterol favored D/C/F/TAF and remaining lipid increases favored control. D/C/F/TAF is a novel STR that combines the known efficacy and high-genetic barrier to resistance of darunavir with the safety advantages of TAF to provide a new option for the treatment of ART-naive, HIV-1-infected patients.
Acknowledgements
This study was sponsored by Janssen.
We thank the patients and their families for their participation and support during the study, the central and local Janssen AMBER study teams, study center staff, and the principal investigators:
Belgium: S. De Wit, E. Florence, L. Vandekerckhove, B. Vandercam; Canada: J. Brunetta, M. Klein, D. Murphy, A. Rachlis, S. Walmsley; France: F. Ajana, L. Cotte, P.-M. Girard, C. Katlama, J.-M. Molina, I. Poizot-Martin, F. Raffi, D. Rey, J. Reynes, E. Teicher, Y. Yazdanpanah; Germany: K. Arastéh, M. Bickel, J. Bogner, S. Esser, G. Faetkenheuer, H. Jessen, W. Kern, J. Rockstroh, C. Spinner, H.-J. Stellbrink, A. Stoehr; Italy: A. Antinori, F. Castelli, A. Chirianni, A. De Luca, A. Di Biagio, M. Galli, A. Lazzarin, F. Maggiolo, R. Maserati, C. Mussini; Poland: A. Garlicki, J. Gasiorowski, W. Halota, A. Horban, M. Parczewski, A. Piekarska; Russia: E. Belonosova, O. Chernova, N. Dushkina, V. Kulagin, E. Ryamova, A. Shuldyakov, N. Sizova, O. Tsybakova, E. Voronin, A. Yakovlev; Spain: A. Antela, J.R. Arribas, J. Berenguer, J. Casado, V. Estrada, M.J. Galindo, M. Garcia Del Toro, J.M. Gatell, M. Gorgolas, F. Gutierrez, M.D.M. Gutierrez, E. Negredo, J.A. Pineda, D. Podzamczer, J. Portilla Sogorb, A. Rivero, R. Rubio, P. Viciana, I. De Los Santos; UK: A. Clarke, B.G. Gazzard, M.A. Johnson, C. Orkin, I. Reeves, L. Waters; United States: P. Benson, L. Bhatti, F. Bredeek, G. Crofoot, D. Cunningham, E. DeJesus, J. Eron, F. Felizarta, R. Franco, J. Gallant, D. Hagins, K. Henry, D. Jayaweera, C. Lucasti, C. Martorell, C. McDonald, J. McGowan, A. Mills, J. Morales-Ramirez, D. Prelutsky, M. Ramgopal, B. Rashbaum, P. Ruane, J. Slim, A. Wilkin, J. deVente.
We also thank other Janssen staff members for their input into this manuscript. We acknowledge Ian Woolveridge of Zoetic Science, an Ashfield company, Macclesfield, UK, for assistance in drafting the manuscript and coordinating and collating author contributions, which was funded by Janssen. Week-48 data were presented in part at the 16th European AIDS Conference, 25–27 October 2017, Milan, Italy.
Contributors: J.G., C.O., J.-M.M., E.N., A.A., A.M., J.E., and J.R. were investigators in the study and reported data for the patients. E.V.L., E.L., V.H., J.J., S.V., and M.O. were involved in the data analyses. All authors had full access to the data, were involved in the development of the primary manuscript and interpretation of data, have read and approved the final version, and have met the criteria for authorship as established by the ICMJE. The corresponding author had final responsibility to submit the manuscript for publication.
Conflicts of interest
Source of funding: This study was sponsored by Janssen. The funder was involved in the study design, study conduct, data collection, and data analysis.
J.E. has received research grants from Janssen, Gilead Sciences, and ViiV Healthcare, and has served as a consultant to Bristol Myers Squibb (BMS), Merck, Janssen, Gilead Sciences, and ViiV Healthcare. C.O. has received speaker honoraria or consulting fees for attending speakers’ bureaus or advisory boards and research grants from Janssen, Merck, ViiV Healthcare, and Gilead Sciences. J.G. has received consulting fees or advisory board honoraria from BMS, Gilead Sciences, Merck, ViiV Healthcare, and Theratechnolgies, and research grants from AbbVie, BMS, Gilead Sciences, Janssen Therapeutics, Merck, Sangamo Biosciences, and ViiV Healthcare/GlaxoSmithKline. He has since become a full-time employee of Gilead Sciences. J.-M.M. has participated in advisory boards for Merck, Gilead Sciences, Janssen, ViiV Healthcare, BMS, and Teva, and a speakers’ bureau for Gilead. He has received research grants from Merck and Gilead Sciences. E.N. has received speaker honoraria or consulting fees from ViiV Healthcare, Merck, Janssen Cilag, BMS, Gilead Sciences, and AbbVie. A.A. has served as a consultant to BMS, Gilead Sciences, Janssen Cilag, Merck, ViiV Healthcare, and AbbVie. He has received institutional research grants from BMS, Gilead Sciences, Janssen Cilag, and ViiV Healthcare. A.M. has received grants and personal fees from Gilead Sciences, ViiV Healthcare, Janssen and Merck, and grants from BMS and Sangamo. J.R. has received grants and personal fees from Janssen, Gilead Sciences, Merck, and ViiV Healthcare. E.V.L., E.L., V.H., J.J., S.V., and M.O. are all full-time employees of Janssen and potential stockholders of Johnson and Johnson.
Supplementary Material
Contributor Information
Collaborators: on behalf of the AMBER study group
References
- 1.Nachega JB, Marconi VC, van Zyl GU, Gardner EM, Preiser W, Hong SY, et al. HIV treatment adherence, drug resistance, virologic failure: evolving concepts. Infect Disord Drug Targets 2011; 11:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clay PG, Nag S, Graham CM, Narayanan S. Meta-analysis of studies comparing single and multitablet fixed dose combination HIV treatment regimens. Medicine (Baltimore) 2015; 94:e1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cotte L, Ferry T, Pugliese P, Valantin MA, Allavena C, Cabié A, et al. Dat’AIDS Study Group Effectiveness and tolerance of single tablet versus once daily multiple tablet regimens as first-line antiretroviral therapy - results from a large french multicenter cohort study. PLoS One 2017; 12:e0170661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin C, DeJesus E, Khanlou H, Stoehr A, Supparatpinyo K, Lathouwers E, et al. Final 192-week efficacy and safety of once-daily darunavir/ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naïve patients in the ARTEMIS trial. HIV Med 2013; 14:49–59. [DOI] [PubMed] [Google Scholar]
- 5.Lathouwers E, Wong EY, Luo D, Seyedkazemi S, De Meyer S, Brown K. HIV-1 resistance rarely observed in patients using darunavir once-daily regimens across clinical studies. HIV Clin Trials 2017; 18:196–204. [DOI] [PubMed] [Google Scholar]
- 6.Borges ÁH, Lundh A, Tendal B, Bartlett JA, Clumeck N, Costagliola D, et al. Nonnucleoside reverse-transcriptase inhibitor- vs ritonavir-boosted protease inhibitor-based regimens for initial treatment of HIV infection: a systematic review and metaanalysis of randomized trials. Clin Infect Dis 2016; 63:268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahlich J, Groß M, Kuhlmann A, Bogner J, Heiken H, Stoll M. The choice between a ritonavir-boosted protease inhibitor- and a nonnucleoside reverse transcriptase inhibitor-based regimen for initiation of antiretroviral treatment - results from an observational study in Germany. J Pharm Policy Pract 2016; 9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DHHS guidelines. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. 1–239 (updated May 30, 2018). Available at: https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf [Accessed 6 June 2018] [Google Scholar]
- 9.Günthard HF, Saag MS, Benson CA, del Rio C, Eron JJ, Gallant JE, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2016 recommendations of the International Antiviral Society-USA Panel. JAMA 2016; 316:191–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.EACS. European AIDS Clinical Society Guidelines. Version 9.0. October 2017. Available at: http://www.eacsociety.org/files/guidelines_9.0-english.pdf [Accessed 15 January 2018] [Google Scholar]
- 11.BHIVA guidelines for the treatment of HIV-1-positive adults with ART 2015 (2016 interim update). Available at: http://www.bhiva.org/documents/Guidelines/Treatment/2016/treatment-guidelines-2016-interim-update.pdf [Accessed 15 January 2018] [Google Scholar]
- 12.Sax PE, Wohl D, Yin MT, Post F, DeJesus E, Saag M, et al. GS-US-292-0104/0111 Study Team Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, noninferiority trials. Lancet 2015; 385:2606–2615. [DOI] [PubMed] [Google Scholar]
- 13.Gallant JE, Daar ES, Raffi F, Brinson C, Ruane P, DeJesus E, et al. Efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate given as fixed-dose combinations containing emtricitabine as backbones for treatment of HIV-1 infection in virologically suppressed adults: a randomised, double-blind, active-controlled phase 3 trial. Lancet HIV 2016; 3:e158–e165. [DOI] [PubMed] [Google Scholar]
- 14.Orkin C, Molina J-M, Negredo E, Arribas JR, Gathe J, Eron JJ, et al. Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine/tenofovir disoproxil fumarate regimens to the once-daily complete HIV-1 regimen of darunavir/cobicistat/emtricitabine/tenofovir alafenamide (D/C/F/TAF) in virologically suppressed, HIV-1-infected adults through 48 weeks (EMERALD): a phase 3, randomized, noninferiority trial. Lancet HIV 2018; 5:e23–e34. [DOI] [PubMed] [Google Scholar]
- 15.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976; 16:31–41. [DOI] [PubMed] [Google Scholar]
- 16.NIAID. Division of AIDS table for grading the severity of adult and pediatric adverse events – version 2. November 2014. Available at: http://rsc.tech-res.com/docs/default-source/safety/daids_ae_grading_table_v2_nov2014.pdf [Accessed 15 January 2018] [Google Scholar]
- 17.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, et al. CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, et al. 2014 update of the drug resistance mutations in HIV-1. Top Antivir Med 2014; 22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 19.German P, Liu HC, Szwarcberg J, Hepner M, Andrews J, Kearney BP, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr 2012; 61:32–40. [DOI] [PubMed] [Google Scholar]
- 20.Cohen C, Wohl D, Arribas JR, Henry K, Van Lunzen J, Bloch M, et al. Week 48 results from a randomized clinical trial of rilpivirine/emtricitabine/tenofovir disoproxil fumarate vs. efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV-1-infected adults. AIDS 2014; 28:989–997. [DOI] [PubMed] [Google Scholar]
- 21.Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. GS-US-236-0102 study team Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet 2012; 379:2439–2448. [DOI] [PubMed] [Google Scholar]
- 22.Walmsley SL, Antela A, Clumeck N, Duiculescu D, Eberhard A, Gutiérrez F, et al. SINGLE Investigators Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med 2013; 369:1807–1818. [DOI] [PubMed] [Google Scholar]
- 23.Clotet B, Feinberg J, van Lunzen J, Khuong-Josses MA, Antinori A, Dumitru I, et al. ING114915 study team Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naive adults with HIV-1 infection (FLAMINGO): 48 week results from the randomised open-label phase 3b study. Lancet 2014; 383:2222–2231. [DOI] [PubMed] [Google Scholar]
- 24.Sax PE, Pozniak A, Montes ML, Koenig E, DeJesus E, Stellbrink HJ, et al. Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380-1490): a randomised, double-blind, multicentre, phase 3, noninferiority trial. Lancet 2017; 390:2073–2082. [DOI] [PubMed] [Google Scholar]
- 25.Gallant J, Lazzarin A, Mills A, Orkin C, Podzamczer D, Tebas P, et al. Bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir, abacavir, and lamivudine for initial treatment of HIV-1 infection (GS-US-380-1489): a double-blind, multicentre, phase 3, randomised controlled noninferiority trial. Lancet 2017; 390:2063–2072. [DOI] [PubMed] [Google Scholar]
- 26.Squires KE, Molina J-M, Sax PE, Wong WW, Orkin C, Sussmann O, et al. Fixed dose combination of doravirine/lamivudine/TDF is noninferior to efavirenz/emtricitabine/TDF in treatment-naïve adults with HIV-1 infection: week 48 results of the Phase 3 DRIVE-AHEAD study [abstract TUAB0104LB]. 9th IAS Conference on HIV Science, 23–26 July 2017. [Google Scholar]
- 27.Tashima K, Crofoot G, Tomaka FL, Kakuda TN, Brochot A, Van de Casteele T, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a Phase IIIb, open-label single-arm trial. AIDS Res Ther 2014; 11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lennox JL, Landovitz RJ, Ribaudo HJ, Ofotokun I, Na LH, Godfrey C, et al. ACTG A5257 Team Efficacy and tolerability of 3 nonnucleoside reverse transcriptase inhibitor-sparing antiretroviral regimens for treatment-naive volunteers infected with HIV-1: a randomized, controlled equivalence trial. Ann Intern Med 2014; 161:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills A, Crofoot G, Jr, McDonald C, Shalit P, Flamm JA, Gathe J, Jr, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate in the first protease inhibitor-based single-tablet regimen for initial HIV-1 therapy: a randomized Phase 2 study. J Acquir Immune Defic Syndr 2015; 69:439–445. [DOI] [PubMed] [Google Scholar]
- 30.Orkin C, DeJesus E, Ramgopal M, Crofoot G, Ruane P, LaMarca A, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, noninferiority study. Lancet HIV 2017; 4:e195–e204. [DOI] [PubMed] [Google Scholar]
- 31.DeJesus E, Ramgopal M, Crofoot G, Ruane P, LaMarca A, Mills A, et al. Switching from efavirenz, emtricitabine, and tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, noninferiority study. Lancet HIV 2017; 4:e205–e213. [DOI] [PubMed] [Google Scholar]
- 32.Mocroft A, Achra AC, Ross M, Ryom L, Avihingsanon A, Bakowska E, et al. Deferred antiretroviral therapy is associated with lower estimated glomerular filtration rate in HIV-positive individuals with high CD4 counts [abstract WEPDB0101]. 21st International AIDS Conference, 18–22 July 2016. [Google Scholar]
- 33.Morlat P, Vivot A, Vandenhende M-A, Dauchy FA, Asselineau J, Déti E, et al. the Groupe D’epidémiologie Clinique du Sida en Aquitaine (Gecsa) Role of traditional risk factors and antiretroviral drugs in the incidence of chronic kidney disease, ANRS CO3 Aquitaine Cohort, France, 2004–2012. PLoS One 2013; 8:e66223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.