

Supplemental Digital Content is available in the text.
Keywords: ATP-binding cassette transporter, cholesterol, high-density lipoprotein, metformin, sterol cardiovascular disease
Abstract
Objective—
The mechanisms underlying the cardiovascular benefit of the anti-diabetic drug metformin are poorly understood. Recent studies have suggested metformin may upregulate macrophage reverse cholesterol transport. The final steps of reverse cholesterol transport are mediated by the sterol transporters, ABCG5 (ATP-binding cassette transporter G5) and ABCG8 (ATP-binding cassette transporter G8), which facilitate hepato-biliary transport of cholesterol. This study was undertaken to assess the possibility that metformin induces Abcg5 and Abcg8 expression in liver and to elucidate the underlying mechanisms.
Approach and Results—
Metformin-treated mouse or human primary hepatocytes showed increased expression of Abcg5/8 and the bile salt export pump, Bsep. Administration of metformin to Western-type diet–fed mice showed significant upregulation of Abcg5/8 and Bsep. This resulted in increased initial clearance of 3H-cholesteryl ester HDL (high-density lipoprotein) from plasma. However, fecal 3H-cholesterol output was only marginally increased, possibly reflecting increased hepatic Ldlr (low-density lipoprotein receptor) expression, which would increase nonradiolabeled cholesterol uptake. Abcg5/8 undergo strong circadian variation. Available chromatin immunoprecipitation-Seq data suggested multiple binding sites for Period 2, a transcriptional repressor, within the Abcg5/8 locus. Addition of AMPK (5′ adenosine monophosphate-activated protein kinase) agonists decreased Period 2 occupancy, suggesting derepression of Abcg5/8. Inhibition of ATP citrate lyase, which generates acetyl-CoA from citrate, also decreased Period 2 occupancy, with concomitant upregulation of Abcg5/8. This suggests a mechanistic link between feeding-induced acetyl-CoA production and decreased cholesterol excretion via Period 2, resulting in inhibition of Abcg5/8 expression.
Conclusions—
Our findings provide partial support for the concept that metformin may provide cardiovascular benefit via increased reverse cholesterol transport but also indicate increased Ldlr expression as a potential additional mechanism. AMPK activation or ATP citrate lyase inhibition may mediate antiatherogenic effects through increased ABCG5/8 expression.
Metformin, the most widely used drug to treat diabetes mellitus, seems to offer moderate protection against atherosclerotic cardiovascular disease.1,2 The mechanism of this likely benefit is poorly understood. In metabolic studies, diabetics treated with metformin had a significantly lower VLDL (very low–density lipoprotein) cholesterol level in the postprandial state.3 However, metformin treatment has not been consistently associated with changes in VLDL/LDL (low-density lipoprotein) cholesterol levels in population studies.1,2,4 Thus, the mechanisms underlying its apparent cardiovascular benefit are unclear.5
Recent studies have shown that metformin increases HDL (high-density lipoprotein)-mediated reverse cholesterol transport (RCT), acting on macrophages to upregulate Abca1/g1 which mediate the efflux of cholesterol onto and apoA-1 and HDL particles.6 The final step in the RCT pathway is mediated by the cholesterol half-transporters ABCG5 (ATP-binding cassette transporter G5) and ABCG8 (ATP-binding cassette transporter G8),7–9 which reside on the canicular membrane of hepatocytes facilitating excretion of cholesterol and plant sterols into bile. Genome-wide association studies have identified SNPs (single nucleotide polymorphisms) in the ABCG5/8 locus associated with LDL cholesterol levels, total cholesterol levels, and coronary artery disease.10–13 Abcg5/8 are known to be transcriptionally upregulated by the liver X receptor alpha (LXRα, Nr1h3) and by FoxO (Forkhead box O) transcription factors.14–17 However, other modes of regulation, including by metformin, have not been well documented. In this study, we sought to determine whether the final step of RCT mediated by ABCG5/8 might also be upregulated by metformin. This led to the discovery that Period 2 (PER2), a circadian regulator, binds and represses Abcg5/8 mRNA expression. PER2 in turn seems to be oppositely regulated by the activities of AMPK (5′ adenosine monophosphate-activated protein kinase) and ATP citrate lyase (ACLY). These findings provide a mechanism to explain how biliary cholesterol excretion is linked to hepatic lipogenic activity and the circadian cycle.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Mice and In Vivo Experiments
We chose to use male mice for our studies as it has been previously reported in rodents that glucose tolerance and plasma insulin concentrations vary during the estrous cycle.18 C57BL/6J mice were purchased from Jackson Laboratory (Stock No. 000664).19 Albumin-Cre were purchased from Jackson Laboratory (Stock No. 003574)20 and bred with Prkaa1 (protein kinase AMP-activated catalytic subunit alpha 1) flox/ flox mice also purchased from Jackson Laboratory (Stock No. 014141)21 to produce mice with Prkaa1 knocked-out in liver (Alb-Cre(−) Prkaa1 fl/fl and Alb-Cre(+) Prkaa1). For chow fasting/refeeding model, C57Bl6/J mice were used. Mice were fasted for 14 hours, and then subsequently refed after injection with metformin (250 mg/kg) or saline control. After 4 hours of refeeding, mice were euthanized, and liver was collected and snap frozen in liquid nitrogen. For chronic metformin treatment, C57Bl6/J mice were fed a western-type diet (WTD; 42% calories from fat, 0.2% cholesterol, Envigo: TD.88132) for 3 months. Subsequently mice were split into 2 groups, one group receiving 250 mg/kg metformin or saline control (IP), daily, for 2 weeks. Mice were then euthanized in the fed state and livers were collected and snap frozen in liquid nitrogen and stored at −80°C. All protocols were approved by the Institutional Animal Care and Use Committee of Columbia University.
Primary Hepatocytes
Mouse hepatocytes were isolated using the Worthington Biochemical Corporation Hepatocyte Isolation Kit. In brief, mice were euthanized, and the livers were perfused with Hanks’ Balanced Salt solution, without calcium or magnesium. After perfusion, livers were switched to digestion media (L-15/MOPS solution) containing collagenase-elastase enzyme, supplemented with DNase1. After digestion, livers were removed and placed on a 10 cm plate and minced with 1 mL further of digestion media and placed at 37°C for 5 minutes. After incubation, cells were washed in excess Leibowitz Media containing 10% FBS and spun at 300g for 3 min. Cell pellet was subsequently washed 2× with Leibowitz buffer containing 0.2% FBS and 0.5% BSA. Cells were counted and seeded at a density of 0.3×106 cells per well of a six-well plate in DMEM (02.% FBS, 0.5% BSA). Forty-five minutes after plating, cells were washed with PBS, and new media (DMEM+0.2%FBS+0.5%BSA) was added containing AMPK activators or ACLY inhibitor as described in text. Phospho-ACC1 (acetyl-CoA carboxylase 1; Ser79) antibody was obtained from Cell Signaling (Clone D7D11).
RNA Isolation, cDNA, and Real-Time Quantitative Polymerase Chain Reaction
RNA was isolated using TriZol Reagent (Ambion) which was then combined with Zymogen RNA isolation kit (Quick-RNA MiniPrep). RNA was quantified using a NanoDrop 8000 Spectrophotometer (ThermoFisher). Five hundred nanogram of RNA was then used to synthesize cDNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific). cDNA was then diluted 10-fold for real-time quantitative polymerase chain reaction (PCR) analysis. Gene expression analysis was performed using a Step One Plus Real Time PCR system (Applied Biosciences). Fold change in gene expression was calculated using the ΔΔCt method. Real-time PCR primer sequences for mouse genes are as follows: Rplp0, 5′-gaaactgctgcctcacatccg, 3′-gctggcacagtgacctcacacg; Abcg5, 5′-atccaacacctctatgctaaatcac, 3′-tacattattggaccagttcagtcac; Abcg8, 5′-cctcatcattggcttccttac, 3′-attgacctctccgagtgacatt; bile salt export pump (Bsep), 5′-tctgactcagtgattcttcgca, 3′-cccataaacatcagccagttgt. Human real-time PCR primer sequences are as follows: CYCLOA, 5′-gccatccaaccactcagtct, 3′-atgtgtcagggtggtgacttc; ABCG5, 5′-tcctgaggagagtgacaagaaac, 3′-acgggaaacagattcacagc; ABCG8, 5′-ggaacccaggaatccttattctc, 3′-ggtcaggtccacatagaagtcag; BSEP, 5′-ttggctgatgtttgtgggaag, 3′-ccaaaaatgagtagcacgcct; ACLY, 5′-atcggttcaagtatgctcggg, 3′-aaggcatgctggactttgacta.
Chromatin Immunoprecipitation
Primary hepatocytes were isolated as described above, except that cells were plated on 15 cm collagen-coated plates at a density of 4.4×106 cells per plate. Cells were then treated with 0.5 mmol/L metformin, 10 μmol/L A-7, or 8 μmol/L BMS (iACLY) for 10 hours. Three to 4 animals were used for each chromatin immunoprecipitation (ChIP) and pooled for pull down. For RNA Pol II (Santa Cruz Biotechnology; H-224) and H3K4me3 (Abcam; ab8580) pull down, hepatocytes were washed 2 times with PBS and then cross-linked in 1% formaldehyde (Thermo Scientific, methanol-free; in PBS) for 15 min at room temperature. Cross-linking reaction was quenched with addition of 0.125 mol/L glycine and incubated for 10 min at room temperature. Quenching reaction was performed a subsequent time with fresh 0.125 mol/L glycine (in PBS) for another 10 min. Cells were then washed 3× with cold PBS and scrapped off plate in PBS and spun at 4000 rpm for 8 min. Supernatant was aspirated, and cells were resuspended in 3 mL of Nuclei Isolation Buffer (10 mmol/L Hepes [pH 7.4], 10 mmol/L KCl, 0.3% IGEPAL, Halt Protease and Phosphatase inhibitors [Thermo Scientific], 1 mmol/L PMSF, 1 mmol/L DTT, and 0.15 mmol/L spermine) and incubated on ice for 10 min. Samples were then transferred to a 10 mL dounce homogenizer and dounced 20 to 25 times to isolated nuclei. Crude nuclei were spun at 1500 rpm for 8 min and resuspended in 200 μL of micrococcal nuclease reaction buffer (20 mmol/L Tris-HCl [pH 8.0], 1% Triton-X100, 0.1% IGEPAL, 137 mmol/L NaCl). For PER2 (Alpha Diagnostic International, PER21-A) and cryptochrome 1 (CRY1; Alpha Diagnostic International, CRY11-A) pull down, cells were initially cross-linked with Di-(N-succinimidyl) glutarate (DSG, CovaChem; PBS, 12% DMSO) for 30 min at room temperature. Hepatocytes were washed with PBS and cross-linked with 1% formaldehyde for 15 min at room temperature and quenched and processed as described earlier.
Crude nuclei were then digested with micrococcal nuclease (New England Biolabs, M0247S) at 37°C to produce DNA fragments between 1 and 4 histone lengths (≈100–500 bp). Reactions were stopped by addition of excess EDTA (10 mmol/L final concentration). Samples were sonicated twice using a Fisher Scientific Sonic Dismembrator (Model FB120) at 40% amplitude for 15 s (30 s rest). Lysates were spun at 13 200 rpm for 2 min to remove insoluble fraction. Protein quantification of lysate was performed using a BCA Protein Assay Kit (Pierce). Forty microgram of protein was used for RNA Pol II and H3K4me3 pull down, while PER2 and CRY1 pull down was performed with 400 μg of protein.
IP reactions were diluted to 1 mL in MNase buffer (1 mmol/L PMSF, 1 mmol/L EDTA, Halt Protease and Phosphatase inhibitors). Lysates were cleared with Protein G Dynabeads (Invitrogen) for 1 hour at 4°C, after which time Dynabeads were removed (2.5% was removed for input), and 1 μg of anti-H3K4me3, 1 μg anti-RNA Pol II, 5 μg anti-PER2, or 5 μg anti-CRY1 were added to corresponding tubes along with IgG to control tubes. Immunoprecipitations were allowed to rotate overnight at 4°C. After overnight incubation, 30 μL of Protein G Dynabeads were added to each tube and allowed to incubate for another 2 hours. Dynabeads were separated using a DyanMag-2 (Invitrogen) magnet. Dynabeads were washed 2× with MNase Buffer, Wash Buffer 2 (20 mmol/L Tris-HCl [pH 8.0], 1% Triton-X100, 0.1% SDS, 500 mmol/L NaCl, 2 mmol/L EDTA), Wash Buffer 3 (20 mmol/L Tris-HCl [pH 8.0], 1% Triton-X100, 250 mmol/L LiCl, 2 mmol/L EDTA), and 1× with TE buffer (10 mmol/L Tris-HCl, 1 mmol/L EDTA).
To elute bound DNA, Dyanbeads were resuspended in 100 μL of SDS elution buffer (50 mmol/L NaHCO3, 1% SDS) and agitated for 15 min at room temperature. Supernatant was separated from beads using a magnet and placed in a new tube. A second elution was performed with 100 μL of elution buffer and incubated at 55°C for 15 min. Supernatant was again separated from beads using a magnet and was added to previous elution. NaCl was added to each sample to a final concentration of 250 mmol/L and incubated at 65°C overnight to reverse cross-link. Samples were then treated with RNaseA (10 μg/sample, 1 hour, 37°C) and then digested with Proteinase K (80 μg/sample, 55°C, 2 hours). DNA was isolated using QiaQuick columns (Qiagen) and eluted in ddH2O. Isolated DNA was stored at −80°C. Recovered DNA was quantified using Step One Plus Real Time PCR system (Applied Biosciences). Percent occupancy was calculated relative to input for each sample. ChIP primer sequences are as follows: Abcg5/8 promoter, 5′-gagcgttgacccatgtgaact, 3′-tatggcaagcgtagcgatct; Bsep promoter, 5′-gcactgctgtaagaccgttacc, 3′-gcttgggtagacgtgtgaagac; Dbp (D site of albumin promoter binding protein) enhancer, 5′-acacccgcatccgatagc, 3′-ccacttcgggccaatgag.
HDL 3H-Cholesteryl Ester Kinetics Studies
3H-Cholesteryl ester (3H-CE) HDL kinetics studies were performed as previously described.22–24 Briefly, 1mCi 3H-cholesterol was added to a 1:1 mixture of HDL and LCAT (lecithin cholesterol acyltransferase) and incubated for 3 hours at 37°C. 3H-HDL was recovered using ultracentrifugation, where it was added back to LDL (repeated twice) and incubated for 3 hours at 37°C. HDL was isolated once again using ultracentrifugation where it was then dialyzed against KBr overnight and subsequently sterile filtered through a 0.22 μm filter and stored at 4°C until injection. Mice were injected intravenously, and blood was taken at 0, 2, 4, 8, and 24 hours postinjection. Feces were collected over the course of 2 days. Feces were dried overnight in a 60°C oven, and the 3H-cholsterol was isolated using the Folch lipid extraction method.25
Plasmid and siRNA Transfections
HEK293 cells were transfected overnight with 500 ng of Flag-mPer2 plasmid and 10 ng of either siAcly (Dharmacon SMART Pool) or siRNA control using Lipofectamine 2000 in OptiMem media, which was replaced next day with DMEM+10% FBS. Forty-eight hours later, media was aspirated and replaced with DMEM+10% FBS containing 50 μg/mL cycloheximide. At indicated time points, cells were washed with PBS harvested in 250 μL of RIPA buffer (Halt Protease and Phosphatase inhibitors, 1 mmol/L PMSF, and 1 mmol/L EDTA). Cell lysates were sonicated 3 times at 50% amplitude for 15 s. Lysates were spun at 13 200 rpm for 3 min to remove precipitate. Protein was quantified using Pierce BCA Protein Assay Kit and run on an SDS-PAGE gel. For Flag-mPer2,26 Flag-mCry1,26 and Acly (S455D)27,28 cotransfections, HEK293 cells were transfected as described earlier, but 250 ng of each plasmid was used (250 ng Per2+250 ng Acly (S455D); 250 ng Cry1+Acly S455D). Where Acly (S455D) was not transfected, pCAG-RFP plasmid was used to make the total plasmid DNA transfected 500 ng per well. Anti-mouse anti-Flag (Sigma) was used to detect PER2 and CRY1. Flag-mPer2 (pP2BA) was a gift from Aziz Sancar (Addgene plasmid No. 31369). Flag-mCry (pMC1SG5) was a gift from Aziz Sancar (Addgene plasmid No 31282). Acly (S455D) was a gift from Kathryn Wellen (Addgene plasmid No 70768).
Statistical Analysis
All values are reported as mean±SD or SEM where indicated. Data were analyzed using Student t tests, unpaired t test with Welch’s correction, one-way analysis of variance, or 2-way analysis of variance with Tukey’s post hoc test where indicated. Tests were validated for normalcy using either D’Agostino & Pearson or Shapiro-Wilk normalcy tests, and variance was tested using the F test or Brown-Forsythe (analysis of variance). Statistical analysis was performed using Prism 7 (GraphPad) software.
Results
Metformin Induces Abcg5 and Abcg8 in Primary Hepatocytes and Liver
We treated mouse primary hepatocytes with 0.5 mmol/L metformin for 20 hours and found a marked upregulation of both Abcg5 and Abcg8 mRNAs along with the bile salt transporter, Bsep (Abcb11) (Figure 1A). Because metformin indirectly activates AMPK through interactions with mitochondrial proteins,29 we sought to establish whether the upregulation of Abcg5/8 was because of direct activation of AMPK. To this end, we used the AMPK-specific agonist A-76966230 in primary hepatocytes and found that in a dose-dependent manner A-769662 was able to induce Abcg5 and Abcg8 mRNA (Figure 1B). We observed similar effects on expression of ABCG5, ABCG8, and BSEP in primary human hepatocytes treated with AICAR (5-aminoimidazole-4-carboxamide ribonucleotide; Figure 1C), suggesting a conserved pathway between mice and humans. The relative levels of Abcg5 and Abcg8 mRNA decrease rapidly after initial plating (data not shown); however, Ct values were still in the range of 29 to 32 for Abcg5 and Abcg8 in our primary hepatocyte culture experiments (Table I in the online-only Data Supplement).
Figure 1.
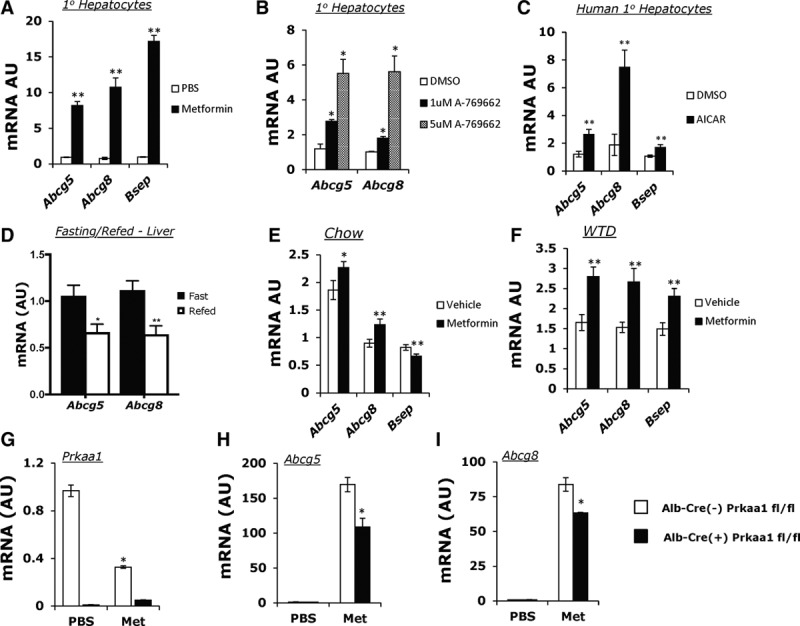
AMPK (5′ adenosine monophosphate-activated protein kinase) activation results in the upregulation of the liver cholesterol efflux genes, Abcg5/Abcg8 (ATP-binding cassette transporter G5/G8) in hepatocytes and liver. A, Real-time polymerase chain reaction (PCR) gene expression analysis of mouse primary hepatocytes treated with 0.5 mmol/L metformin for 20 hours in DMEM (0.2% FBS). B, Real-time PCR gene expression analysis of mouse primary hepatocytes treated with 2 doses, 1 and 5 μmol/L of A-7, an AMPK-specific agonist for 20 hours. C, Human primary hepatocytes treated with AICAR (5-aminoimidazole-4-carboxamide ribonucleotide; black bar; 0.5 μmol/L, 6 hours) or DMSO control (white bar). D, Abcg5 and Abcg8 expression in fasting (black bar) or refed (white bar) livers (n=4–5). E, Gene expression analysis of a fasting/refeeding protocol with a single 250 mg/kg metformin bolus during refeeding (n=15). F, Real-time PCR gene expression of the livers of mice fed a Western-type diet (WTD) for 3 months with a 2-week regiment of metformin (n=7; 250 mg/kg). F–H, Real-time quantitative PCR analysis of metformin treatment (0.5 μmol/L, 20 hours) in Alb-Cre(−)Prkaa1 fl/fl (white bar) and Alb-Cre(+) Prkaa1 fl/fl (black bar) primary hepatocytes looking at Prkaa1 (G), Abcg5 (H), and Abcg8 (I) expression. A, C, E, F, Data represents mean±SEM; *P<0.05, **P<0.01 using multiple t tests (FDR<0.05). B and D, Data represents n=3±SD; *P<0.05, **P<0.01 using unpaired Student t test. G–I, Data represents mean±SD; *P<0.05 using 2-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test.
Previous work has established the hormonal regulation of AMPK activity during fasting and feeding.31–34 Consistent with the cell culture results, we observed an increase in Abcg5/8 mRNA in fasting versus refed livers (Figure 1D). We also used a fasted/refed rodent model to test metformin induction of Abcg5/8 in vivo. After a bolus of metformin, there was a modest but significant upregulation of Abcg5 and Abcg8 in the refed group (n=15) compared with a saline control refed group (Figure 1E). AMPK activity has been shown to be downregulated in high-fat diet–fed mice,35,36 and therefore we sought to determine whether increasing AMPK signaling would upregulate Abcg5 and Abcg8 more prominently after feeding such a diet. We fed C57BL6/J mice a WTD (42% fat/cal, 0.2% Chol) for 3 months, then placed mice on metformin (250 mg/kg/d, IP) or saline intraperitoneally for 2 weeks and then euthanized them in the fed state. Consistent with our primary hepatocyte data, metformin prominently induced the expression of Abcg5, Abcg8, and Bsep compared with the saline control (Figure 1F; P<0.05).
Mice carry 2 isoforms of the catalytic subunit of AMPK, α1, α2 (Prkaa1, Prkaa2), which are thought to be similarly expressed in liver; however, Prkaa1 has been shown to rhythmically localize to the nucleus which may aid in regulating transcriptional activity.37 To examine the AMPK dependence of metformin-induced expression of Abcg5/8, we bred Albumin-Cre (Alb-Cre) with Prkaa1 floxed mice (fl/fl). In the Prkaa1 knockout (Alb-Cre(+) Prkaa1 fl/fl) hepatocytes, there was a significant reduction in Abcg5/8 mRNAs after metformin treatment compared with floxed controls (Alb-Cre(−) Prkaa1 fl/fl) (Figure 1G through 1-I; *P<0.05). This indicates that regulation of Abcg5/8 by metformin depends at least in part on intact AMPK signaling. The residual increase in Abcg5/8 expression in Alb-Cre(+) Prkaa1 fl/fl mice could reflect activity of Prkaa2 or an independent pathway, which has recently been demonstrated, where an AMPK β-specific subunit agonist induction of Abcg5/8 was in abolished α1/α2 knockout livers.38
Induction of Abcg5/8 Expression Does Not Require LXR or FoxO Transcription Factors
LXRα, the predominant isoform of LXR expressed in the liver,39–41 has been shown to mediate induction of Abcg5 and Abcg8 in response to feeding a high cholesterol diet.14 However, in primary hepatocytes lacking LXRα, the main isoform of LXR expressed in the liver, there was no difference in the induction of Abcg5/8 compared with wild-type controls (Figure I in the online-only Data Supplement). In addition, the FoxO family of transcription factors has been shown to induce Abcg5/8, providing a link between hepatic insulin resistance and gallstone formation.16 However, the induction of Abcg5/8 was actually more pronounced in response to AMPK activators in FoxO triple knockout (FoxO1/3/4, FoxO TKO) hepatocytes (Figure IIA and IIB in the online-only Data Supplement), indicating involvement of an alternative regulatory pathway. FoxO TKO hepatocytes did show reduced induction of Bsep when treated with AICAR compared with wild-type controls (Figure IIC in the online-only Data Supplement), consistent with previous reports.17
AMPK Activation Increases Abcg5/8 Transcriptional Activity
To determine if AMPK activators require new transcription to induce Abcg5/8 expression in primary hepatocytes, we treated cells with the transcription inhibitor actinomycin D42 in the presence or absence of AICAR. Interestingly, we found that actinomycin D alone was able to induce Abcg5/8 mRNA and that addition of AICAR had no further additive effect (Figure 2A). Moreover, inhibiting protein synthesis using CHX (cycloheximide) resulted in increased expression of Abcg5/8, as well as Bsep, and again there was no additional effect of AICAR in the presence of CHX (Figure 2B). These observations suggested that Abcg5/8 gene expression might be repressed by a short-lived transcriptional inhibitor. Furthermore, we saw an increase in RNA Polymerase II binding at the Abcg5/8 promoter, indicating enhanced transcription (Figure 2C), and a significant elevation in H3K4me3 at the promoters of Abcg5/8 and Bsep, an epigenetic mark suggesting more transcriptionally active chromatin at the Abcg5/8 locus (Figure 2D). Together these results indicate that AMPK activation increases transcription at the Abcg5/8 and Bsep loci, possibly via inhibition of the activity of a short-lived transcriptional repressor.
Figure 2.
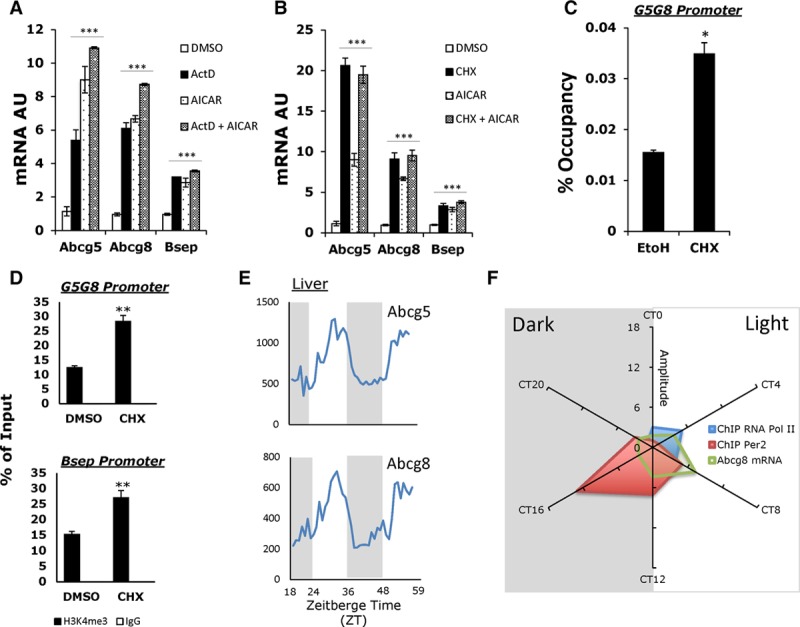
AMPK (5′ adenosine monophosphate-activated protein kinase) activation increases Abcg5/8 (ATP-binding cassette transporter G5/G8) transcriptional activity. A, Real-time quantitative polymerase chain reaction (PCR) analysis of Abcg5, Abcg8, and Bsep expression in primary hepatocytes treated with actinomycin (ActD), with or without AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) for 6 hours. B, Real-time qPCR analysis of Abcg5, Abcg8, and Bsep expression in primary hepatocytes treated with CHX (cycloheximide) with or without AICAR for 6 hours. C, RNA Pol II chromatin immunoprecipitation assay (ChIP) using primers against the Abcg5/8 bi-cistronic promoter. D, ChIP assay using either H3K4me3 antibody or IgG control in primary hepatocytes treated with CHX. Primers designed against the Abcg5/8 promoter and Bsep promoter. E, Liver microarray data of Abcg5 and Abcg8 expression over 2 days from Hughes et al.43 F, Radial plot of relative amplitude of RNA Pol II binding (blue), PER2 binding to Abcg5/8 promoter, and Abcg8 mRNA (green) as described in Koike et al.44 A and B, Data represents mean±SD; ***P<0.001 using 1-way analysis of variance (ANOVA) with Tukey’s post hoc test. C and D, Data represents mean±SD; *P<0.05, **P<0.01 using Student t test.
Binding of PER2 to the Abcg5/8 Locus Is Decreased by AMPK Activation
Many hepatic genes involved in metabolic processes are regulated in a circadian pattern.45 Mining microarray datasets,43 we observed a strong rhythmicity of Abcg5/8 with suppression of expression during the dark phase (Figure 2E), suggesting that the core circadian clock machinery could play a transcriptional regulatory role. Another core clock gene ChIP-seq database44 showed increased binding of Period 2, a repressor of transcription,46,47 over the Abcg8 promoter, paralleling the suppression of Abcg8 mRNA expression and Pol II binding during the dark phase with opposite findings during the light phase (Figure 2F).
AMPK signaling is known to decrease the stability of Period 2 indirectly through the action of a second kinase, CK1ε (casein kinase 1-epsilon) and also directly via phosphorylation of PER2 heterodimer partner, CRY1.37,48 PER2 protein levels are also rapidly decreased by CHX treatment, consistent with our data suggesting a short-lived transcriptional repressor (Figure 2B). We used a well characterized PER2 antibody49,50 to perform ChIP on primary hepatocytes treated with vehicle, metformin or A-769662. Using primers designed within the bicistronic promoter shared by Abcg5 and Abcg8, we found a significant decrease (P<0.01) in PER2 binding in the presence of metformin or A-769662 compared with vehicle control (Figure 3A). To validate the ChIP antibody for PER2, we used published ChIP primers44 against a known PER2 target, Dbp, showing the expected decrease (P<0.001) in PER2 binding within the Dbp upstream enhancer (Figure 3B). Additionally, CRY1 occupancy within the Abcg5/8 promoter was decreased (P<0.01) on activation of AMPK (Figure 3C).
Figure 3.
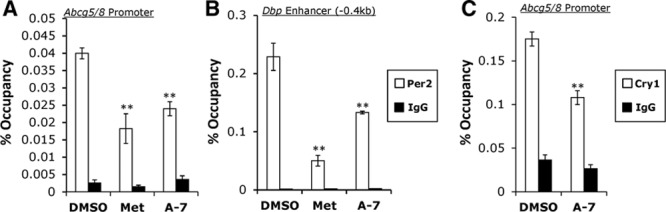
Period 2 (PER2) and cryptochrome 1 (CRY1) have decreased binding at the Abcg5/8 (ATP-binding cassette transporter G5/G8) promoter when treated with AMPK (5′ adenosine monophosphate-activated protein kinase) activating compounds, metformin, or A-769662. Chromatin immunoprecipitation (ChIP) assay pulling down endogenous Per2 (A and B) or Cry1 (C) at the Abcg5/8 promoter (A and C) or at the proximal Dbp promoter (B), a known PER2 binding locus. Data represents mean±SD; *P<0.05, **P<0.01 using Student t test.
Metformin Increases Clearance of 3H-CE in Mice on a Western-Type Diet
Given the role of Abcg5/8 in reverse cholesterol transport, we next sought to test whether metformin treatment increased cholesterol secretion to feces. We placed mice on a Western-type diet and injected them with metformin (250 mg/kg/d, IP) for 2 weeks at the end of which we injected with HDL containing3H-CE (intravenously). Three days before HDL injection, mice were given Ezetimibe (oral gavage, daily) to block cholesterol uptake via NPC1L1 in enterocytes. After injection, we took blood at 2, 4, 8, and 24 hours. Feces were also collected over a 2-day period of time, at which point mice were euthanized. Metformin-treated mice showed an increased rate of cholesterol clearance at 2 hours postinjection (Figure 4A; *P<0.05, two-way analysis of variance) compared with vehicle-treated mice. Using a 2-pool nonlinear model, we saw a significant increase in the initial clearance rate of 3H-CE (Figure 4B) in metformin-treated mice. We saw no difference in hepatic 3H-CE uptake (Figure IIIA in the online-only Data Supplement), while the fractional catabolic rate was similar between both groups (Figure IIIB in the online-only Data Supplement). The fractional flux to the side pool was also significantly increased in metformin-treated mice, while the turnover constant of the side pool remained similar between both groups (Figure IIIC and IIID in the online-only Data Supplement). However, while elevated, the excretion of 3H-cholesterol in feces was not significantly increased in metformin-treated mice (Figure 4C; P=0.20). One possible explanation for the latter finding could be an increase in uptake of LDL cholesterol in the liver of metformin-treated mice, decreasing the specific activity of hepatic 3H-cholesterol. There was a significant increase in Ldlr (low-density lipoprotein receptor) mRNA levels in WTD-fed mice treated with metformin (Figure IIIE in the online-only Data Supplement; *P<0.05). Another possibility is that metformin is increasing Abcg5/8 mRNA. However, there was no change in enterocyte Abcg5 or Abcg8 mRNA in metformin-treated mice (Figure IIIG in the online-only Data Supplement).
Figure 4.
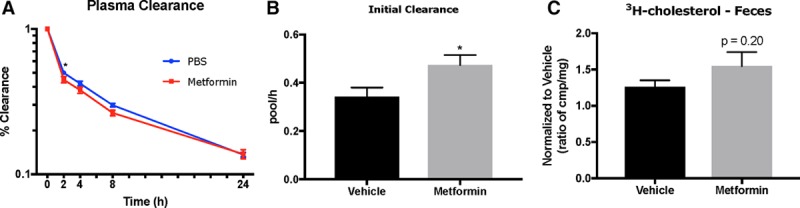
Metformin increases 3H-cholesterol clearance in plasma with trend of increased 3H-cholesterol in feces. A, Log-scale plot of percent clearance (% clearance) of 3H-cholesterol in vehicle (blue line) or metformin (red line) treated mice. B, Initial clearance of 3H-cholesterol using a 2-pool nonlinear modeling of individual plasma clearance curves. C, Measurement of 3H-cholesterol in feces of vehicle treated (black bar) or metformin treated (gray bar). Data are represented as mean±SEM of vehicle (n=10) or metformin (n=11). *P<0.05 using 2-way analysis of variance (ANOVA; A) or unpaired Student t test (B) or unpaired t test with Welch’s correction (C).
Inhibition of ACLY Induces Abcg5/8 Expression in Primary Hepatocytes
Given that PER2 is a transcriptional inhibitor of Abcg5/8 expression, and is opposed by AMPK activation, we sought additional evidence for a role of this pathway in the regulation of Abcg5/8 expression. PER2 is stabilized by the acetyl-CoA-driven acetylation of lysine residues, which inhibits ubiquitin-driven proteasomal degradation.51 Acetyl-CoA can be generated from mitochondrial-derived citrate by ACLY.52,53 It has been established in mice that PER2 protein is increased during the night phase of the circadian cycle51,54 corresponding to increased nutrient intake and increased acetyl-CoA generation. We therefore postulated that acetyl-CoA produced by ACLY could affect PER2 acetylation, as well as stability, and therefore modulate the expression of Abcg5/8. We used the ACLY inhibitor BMS 303141 (referred to in figures as iACLY)55,56 to treat primary hepatocytes over a time course similar to that of metformin treatment. This resulted in a significant upregulation of Abcg5, Abcg8, and Bsep in our primary hepatocyte model (Figure 5A), paralleled by a decreased occupancy of PER2 at the Abcg5/8 promoter (Figure 5B). There was also a significant decrease in PER2 binding at the Dbp locus (Figure 5C), consistent with a modification of PER2 rather than a locus-specific effect. The regulation of Abcg5/8 by inhibition of ACLY does not require intact AMPK signaling, as Prkaa1 KO hepatocytes responded similarly to wild-type hepatocytes (Figure 5D). Additionally, inhibition of ACLY did not result in increased ACC1 (Ser79) phosphorylation, a known target of AMPK, compared with metformin treatment (Figure 5E). This suggests that ACLY is not acting upstream of AMPK to regulate Abcg5/8 expression, but rather is acting through a parallel pathway or downstream of AMPK activity.
Figure 5.
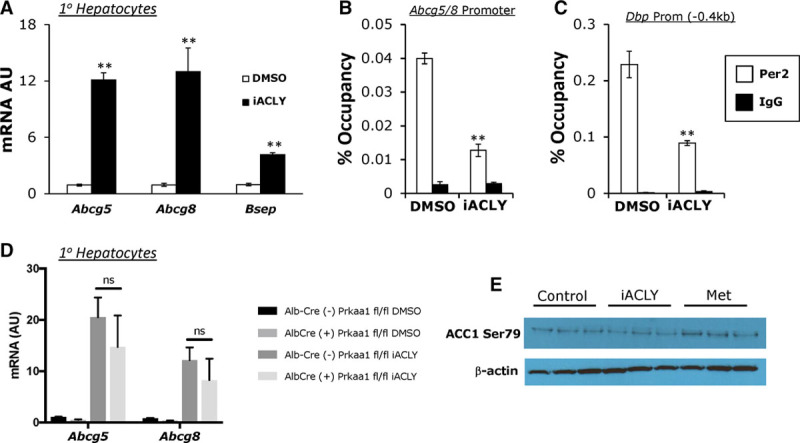
Inhibition of ATP citrate lyase (ACLY) results in the increase in Abcg5/8 (ATP-binding cassette transporter G5/G8) expression accompanying a reduction of period 2 (PER2) at Abcg5/8 promoter in primary hepatocytes. A, Real-time quantitative polymerase chain reaction (qPCR) analysis of Abcg5, Abcg8, and Bsep in primary hepatocytes treated with ACLY inhibitor, BMS 303141 (8 μmol/L), for 20 hours (n=3). B, Chromatin immunoprecipitation (ChIP) assay using endogenous Per2 antibody at the Abcg5/8 promoter or (C) Dbp promoter in primary hepatocytes treated with DMSO or iACLY (n=3–4 pooled). D, Real-time qPCR analysis of Abcg5 and Abcg8 in primary hepatocytes from Prkaa1 wild-type (WT) and Prkaa1 knockout (KO) animals treated with ACLY inhibitor, BMS 303141 (8 μmol/L) for 20 hours (n=3). E, Western blot of phosphorylation of ACC1 (acetyl-CoA carboxylase 1) at Ser 79 in cells treated with iACLY (8 μmol/L) or metformin (0.5 mmol/L) for 20 hours. Data represents mean±SD. *P<0.05, **P<0.01 using unpaired Student t test. B and C, Data represents mean±SD using ordinary 2-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test (D).
ACLY Activity Regulates Stability of PER2
Because we observed decreased PER2 binding at the Abcg5/8 locus with ACLY inhibition, we sought additional evidence that ACLY regulates PER2 stability. Thus, we knocked down Acly expression by siRNA (Figure 6A) in HEK293A cells expressing Flag-tagged PER226 and then treated cells with CHX (50 μg/mL). There was lower expression of PER2 protein at the 2-hour time point, suggesting that by limiting Acly we increased the turnover of PER2 (Figure 6B). Conversely, when we overexpressed a constitutively active form of ACLY, ACLY(S455D),27,28 we observed an increase in the overall levels of PER2, suggesting that increased production of acetyl-CoA increased PER2 stability (Figure 6C). Similarly, when we overexpressed ACLY(S455D) and Flag-tagged CRY1,26 we observed increased protein expression of CRY1 (Figure 6D). As CRY1 has not been shown to be acetylated, the increased protein levels of CRY1 could be because of increased endogenous PER2, which has been shown to block CRY1 interactions, the E3 ubiquitin ligases and subsequent proteasomal degradation.57–60
Figure 6.
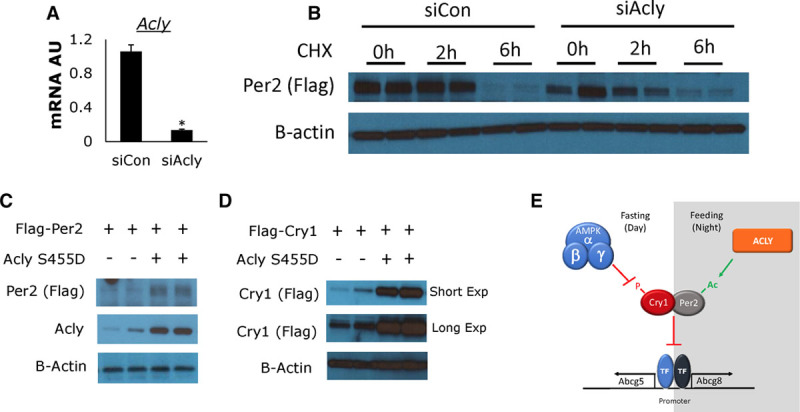
ATP citrate lyase (ACLY) activity regulates stability of Period 2 (PER2). A, Real-time quantitative polymerase chain reaction (qPCR) analysis of Acly expression in HEK293A cells transfected with siRNA against Acly or control, nonspecific siRNA. B, HEK293A cells cotransfected with Flag-tagged mPER2 with control siRNA or Acly siRNA, which were then treated with 50 μg/mL CHX (cycloheximide) for 0, 2, or 6 hours. C and D, HEK293A cells cotransfected with Flag-tagged mPER2 (C) or Flag-tagged mCRY1 (D), with or without a constitutively active form of ACLY, ACLY (S455D). E, Schematic representation of opposite effects of AMPK (5′ adenosine monophosphate-activated protein kinase) and ACLY on PER2/CRY1 repressive effects on Abcg5/8 (ATP-binding cassette transporter G5/G8) mRNA. Data represents mean±SD; *P<0.05, using unpaired Student t test.
Discussion
Our studies in mice and in primary mouse and human hepatocytes indicate that metformin and AMPK activation induce expression of Abcg5 and Abcg8, complementing recent studies from the Steinberg laboratory showing that metformin increases macrophage RCT in mice by upregulating Abca1/g1 expression in macrophages.6 Given the strong link between ABCG5/8 expression and coronary artery disease in humans,11–13 this suggests a novel mechanism to explain the apparent benefit of metformin treatment in cardiovascular disease. AMPK activation was shown to decrease the binding of a circadian transcriptional repressor, PER2, at the Abcg5/8 promoter, providing a mechanism to explain increased Abcg5/8 expression (and biliary cholesterol excretion) during fasting/daytime in mice. These observations are consistent with rodent models of circadian cholesterol efflux to bile. In rats, maximum cholesterol secretion into bile occurs at or near the day to night transition, while maximum bile salt output occurs in the dark phase, slightly lagging behind cholesterol secretion.61 In humans with the opposite circadian rhythm, bile becomes supersaturated with cholesterol (lithogenic) during fasting at night, and cholesterol crystals become prominent in bile with diets involving prolonged fasting.62–65 Interestingly, similar increases in Abcg5/8 expression in association with decreased PER2 binding to the Abcg5/8 promoter were observed in response to ACLY inhibition. Our studies suggest that increased ACLY activity (eg, during feeding) results in increased acetylation of PER2, thereby decreasing the expression of Abcg5/8, while AMPK activation (during fasting and the light phase) promotes the degradation of CRY1 and PER2 and increases Abcg5/8 expression (Figure 6E).
These findings may provide an AMPK-coordinated mechanism underpinning the balance of cholesterol disposition in hepatocytes between esterification/secretion in VLDL versus excretion into bile. AMPK acts to decrease fatty acid biosynthesis and to increase fatty acid oxidation,32,66 decreasing cholesterol esterification. The upregulation of Abcg5/8 in fasting liver resulting from AMPK activation may then facilitate removal of excess cholesterol (ie, not being incorporated into VLDL particles or esterified with fatty acids into cholesteryl esters) by excretion into bile. Similarly, inhibiting ACLY and therefore reducing de novo lipogenesis could result in an increase in Abcg5/8, which provides a mechanism to dispose of cholesterol by excretion. Additional transcriptional control of Abcg5/8 expression is known to be exerted via LXR and FOXO transcription factors. While the effects of AMPK activation were independent of LXR and FOXO1/3/4 in cultured hepatocytes, the more pronounced effect of metformin on Abcg5/8 expression in mice fed the WTD could be related to an effect of LXRs to open chromatin at the locus, increasing access to PER2. During refeeding, decreased nuclear FOXO1 and suppression of AMPK activity would act in parallel to decrease Abcg5/8 expression. Metformin treatment in the insulin resistant states might be expected to increase insulin action, decreasing nuclear FOXO1 and opposing the effects of increased AMPK activity. However, because metformin increased hepatic Abcg5/8 expression in WTD-fed mice, the predominant effect of metformin is presumably exerted by the mechanisms described herein.
We show that activation of AMPK and inhibition of ACLY decrease Period 2 transcriptional inhibitory activity at the Abcg5/8 locus. AMPK has been shown to decrease the stability of PER2 via an indirect pathway,48 as well as PER2 binding partner CRY1 via phosphorylation-mediated degradation.37 These findings link the negative limb of the circadian clock to Abcg5/8 expression via PER2. The positive arm of the core circadian clock pathway involving BMAL1 has also been implicated in the regulation of Abcg5/8 and cholesterol efflux. Pan et al67 found that knockout of Bmal1, globally or in liver specifically, decreased Abcg5/8 expression and consequently decreased cholesterol secretion into bile and feces and increased atherosclerosis. This regulation was indirect as BMAL1 regulated another transcription factor, Gata4/6, that directly bound to the Abcg5/8 bicistronic promoter.
Metformin is a preferred initial treatment for type 2 diabetes mellitus in part because of an apparent benefit on atherosclerotic cardiovascular disease. The UKPDS Study (United Kingdom Prospective Diabetes Study) demonstrated that patients treated with metformin had a reduced risk of myocardial infarction, as well as decreased risk of diabetes mellitus–related deaths and all-cause mortality compared with a conventional therapy.1 Moreover, patients with a history of coronary artery disease showed a significant decrease in cardiovascular events compared with patients given another class of anti-diabetic drug, glipizide.2 Early studies with metformin showed a decrease in total cholesterol, LDL cholesterol, and triglyceride levels compared with control groups, in addition to increased HDL levels.3,68–71 However, more recent studies with a longer follow-up duration have not shown the same effects on lipoproteins.2,4 While human relevance is uncertain, our studies raise the possibility that the cardiovascular benefit of metformin could be explained in part by upregulation of ABCG5/8. Genetic ablation of Abcg5/8 in mice resulted in decreased biliary cholesterol and increased VLDL cholesterol levels.9 Conversely, transgenic overexpression of human ABCG5/8 in mice resulted in increased cholesterol excretion to bile, decreased VLDL/LDL cholesterol, and decreased atherosclerotic lesion size in LDLR-deficient mice, indicating an LDLR-independent mechanism of LDL reduction.8 Our findings provide partial support for the hypothesis that the beneficial effects of metformin may be explained by increased RCT. We tested the RCT hypothesis by injecting 3H-CE HDL into WTD-fed mice given daily injections of metformin. There was a significant increase in the initial rapid clearance of 3H-CE from plasma. However, there was only a trend for increased excretion of 3H-cholesterol in feces (P=0.20). This could be because the 2 day fecal collection introduced too much variability to detect a transient initial effect. Alternatively, the increased Ldlr expression in liver could increase the uptake of nonradiolabeled LDL cholesterol, decreasing the specific activity of the hepatic and fecal 3H-cholesterol pool. The increased Ldlr mRNA may have arisen from increased expression of ABCG5/8, as well as inhibition of HMGCoA (3-hydroxy-3-methylglutaryl-CoA) reductase by AMPK activation.72,73Together the findings suggest that both LDL cholesterol lowering and increased RCT could represent benefits that accrue from metformin, AMPK activation, and ACLY inhibition via upregulation of ABCG5/8.
Targeting ACLY has been suggested as a treatment for cancer,27,74,75 and recently inhibition of ACLY has proved to be beneficial in models of high fat, high cholesterol feeding.76,77 Targeted disruption of ACLY using adenoviral shRNA-mediated knockdown resulted in decreases in VLDL TG and decreased expression of genes involved in lipogenesis and TG synthesis.76 In a hamster model on a high-fat high-cholesterol diet administration, an ACLY inhibitor, ETC-1002 (Bempedoic Acid78), resulted in decreases in LDL and VLDL cholesterol along with decreases in hepatic CE and free cholesterol.77 More recent studies with Bempedoic acid showed reduced atherosclerosis progression in an Ldlr−/− mouse model, and significant increases in hepatic Abcg5/8 mRNA expression were discovered by mRNA profiling.79 Our studies provide a mechanism to explain the link between ACLY inhibition and increased hepatic Abcg5/8 levels, involving the short-lived transcriptional repressor PER2. This could represent an important therapeutic benefit of this approach. Although increased biliary cholesterol secretion resulting from induction of Abcg5/8 by metformin, or ACLY inhibition, might be expected to increase gallstone formation, we found that Bsep was upregulated by a similar mechanism to Abcg5/8, suggesting a coordinated mechanism to dispose of cholesterol without favoring gallstone formation. Our studies thus suggest several potential ways to link diabetes mellitus treatments with a beneficial effect on cardiovascular disease without increasing gallstone risk.
Sources of Funding
This study was supported by National Institutes of Health (NIH) grant HL107653 (A.R. Tall), and HL 87123 (A.R. Tall, D. Accili). M.M. Molusky was also supported by the T32 training grant from the NIH (HL007343) and J. Hsieh by the Gilead Sciences Liver Scholar Program. R.A. Haeusler and S.X. Lee were supported by HL125649.
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- ABCG5/8
- ATP-binding cassette transporter G5/G8
- ACLY
- ATP citrate lyase
- Alb-Cre
- Cre recombinase driven by albumin promoter
- Alb-Cre(+)Prkaa1fl/fl
- hepatocyte-specific Prkaa1 knockout
- AMPK
- 5′ adenosine monophosphate-activated protein kinase
- BSEP
- bile salt export pump
- PRKAA1
- protein kinase AMP-activated catalytic subunit alpha 1
- ChIP
- chromatin immunoprecipitation
- CHX
- cycloheximide
- CRY1
- cryptochrome 1
- DBP
- D site of albumin promoter binding protein
- 3H-CE
- 3H cholesteryl ester
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- PER2
- period 2
- VLDL
- very low–density lipoprotein
- WTD
- Western-type diet
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311212/-/DC1.
Highlights
Metformin induces expression of the sterol transporters Abcg5/8 (ATP-binding cassette transporter G5/G8) by decreasing Period 2 occupancy on the Abcg5/8 promoter.
Our mechanistic data suggests a link between cholesterol excretion and hepatic lipogenic activity and circadian rhythm.
The findings suggest that both LDL (low-density lipoprotein) cholesterol lowering and increased reverse cholesterol transport could represent benefits that accrue from metformin, AMPK (5′ adenosine monophosphate-activated protein kinase) activation and ACLY inhibition via upregulation of ABCG5/8.
References
- 1.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–865. doi: 10.1016/S0140-6736(98)07037-8. [PubMed] [Google Scholar]
- 2.Hong J, Zhang Y, Lai S, et al. SPREAD-DIMCAD Investigators. Effects of metformin versus glipizide on cardiovascular outcomes in patients with type 2 diabetes and coronary artery disease. Diabetes Care. 2013;36:1304–1311. doi: 10.2337/dc12-0719. doi: 10.2337/dc12-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeppesen J, Zhou MY, Chen YD, Reaven GM. Effect of metformin on postprandial lipemia in patients with fairly to poorly controlled NIDDM. Diabetes Care. 1994;17:1093–1099. doi: 10.2337/diacare.17.10.1093. [DOI] [PubMed] [Google Scholar]
- 4.Preiss D, Lloyd SM, Ford I, McMurray JJ, Holman RR, Welsh P, Fisher M, Packard CJ, Sattar N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol. 2014;2:116–124. doi: 10.1016/S2213-8587(13)70152-9. doi: 10.1016/S2213-8587(13)70152-9. [DOI] [PubMed] [Google Scholar]
- 5.Ferrannini E, DeFronzo RA. Impact of glucose-lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J. 2015;36:2288–2296. doi: 10.1093/eurheartj/ehv239. doi: 10.1093/eurheartj/ehv239. [DOI] [PubMed] [Google Scholar]
- 6.Fullerton MD, Ford RJ, McGregor CP, LeBlond ND, Snider SA, Stypa SA, Day EA, Lhoták Š, Schertzer JD, Austin RC, Kemp BE, Steinberg GR. Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J Lipid Res. 2015;56:1025–1033. doi: 10.1194/jlr.M058875. doi: 10.1194/jlr.M058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 8.Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671–680. doi: 10.1172/JCI16001. doi: 10.1172/JCI16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci USA. 2002;99:16237–16242. doi: 10.1073/pnas.252582399. doi: 10.1073/pnas.252582399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teupser D, Baber R, Ceglarek U, et al. Genetic regulation of serum phytosterol levels and risk of coronary artery disease. Circ Cardiovasc Genet. 2010;3:331–339. doi: 10.1161/CIRCGENETICS.109.907873. doi: 10.1161/CIRCGENETICS.109.907873. [DOI] [PubMed] [Google Scholar]
- 13.IBC 50K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J Biol Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 15.van der Veen JN, Havinga R, Bloks VW, Groen AK, Kuipers F. Cholesterol feeding strongly reduces hepatic VLDL-triglyceride production in mice lacking the liver X receptor alpha. J Lipid Res. 2007;48:337–347. doi: 10.1194/jlr.M600170-JLR200. doi: 10.1194/jlr.M600170-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Biddinger SB, Haas JT, Yu BB, Bezy O, Jing E, Zhang W, Unterman TG, Carey MC, Kahn CR. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat Med. 2008;14:778–782. doi: 10.1038/nm1785. doi: 10.1038/nm1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haeusler RA, Pratt-Hyatt M, Welch CL, Klaassen CD, Accili D. Impaired generation of 12-hydroxylated bile acids links hepatic insulin signaling with dyslipidemia. Cell Metab. 2012;15:65–74. doi: 10.1016/j.cmet.2011.11.010. doi: 10.1016/j.cmet.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey CJ, Matty AJ. Glucose tolerance and plasma insulin of the rat in relation to the oestrous cycle and sex hormones. Horm Metab Res. 1972;4:266–270. doi: 10.1055/s-0028-1094063. doi: 10.1055/s-0028-1094063. [DOI] [PubMed] [Google Scholar]
- 19.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- 20.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 21.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagor WR, Brown RJ, Toh SA, Millar JS, Fuki IV, de la Llera-Moya M, Yuen T, Rothblat G, Billheimer JT, Rader DJ. Overexpression of apolipoprotein F reduces HDL cholesterol levels in vivo. Arterioscler Thromb Vasc Biol. 2009;29:40–46. doi: 10.1161/ATVBAHA.108.177105. doi: 10.1161/ATVBAHA.108.177105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Föger B, Santamarina-Fojo S, Shamburek RD, Parrot CL, Talley GD, Brewer HB., Jr Plasma phospholipid transfer protein. Adenovirus-mediated overexpression in mice leads to decreased plasma high density lipoprotein (HDL) and enhanced hepatic uptake of phospholipids and cholesteryl esters from HDL. J Biol Chem. 1997;272:27393–27400. doi: 10.1074/jbc.272.43.27393. [DOI] [PubMed] [Google Scholar]
- 24.Tollefson JH, Albers JJ. Isolation, characterization, and assay of plasma lipid transfer proteins. Methods Enzymol. 1986;129:797–816. doi: 10.1016/0076-6879(86)29106-5. [DOI] [PubMed] [Google Scholar]
- 25.Folch J, Lees M, Sloane stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 26.Ye R, Selby CP, Ozturk N, Annayev Y, Sancar A. Biochemical analysis of the canonical model for the mammalian circadian clock. J Biol Chem. 2011;286:25891–25902. doi: 10.1074/jbc.M111.254680. doi: 10.1074/jbc.M111.254680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JV, Carrer A, Shah S, et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20:306–319. doi: 10.1016/j.cmet.2014.06.004. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–416. doi: 10.1016/j.cmet.2006.05.005. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Hardie DG. The AMP-activated protein kinase pathway–new players upstream and downstream. J Cell Sci. 2004;117(pt 23):5479–5487. doi: 10.1242/jcs.01540. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 32.Assifi MM, Suchankova G, Constant S, Prentki M, Saha AK, Ruderman NB. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am J Physiol Endocrinol Metab. 2005;289:E794–E800. doi: 10.1152/ajpendo.00144.2005. doi: 10.1152/ajpendo.00144.2005. [DOI] [PubMed] [Google Scholar]
- 33.Dentin R, Benhamed F, Pégorier JP, Foufelle F, Viollet B, Vaulont S, Girard J, Postic C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J Clin Invest. 2005;115:2843–2854. doi: 10.1172/JCI25256. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 35.Woo SL, Xu H, Li H, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS One. 2014;9:e91111. doi: 10.1371/journal.pone.0091111. doi: 10.1371/journal.pone.0091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem. 2013;69:165–175. doi: 10.1007/s13105-012-0199-2. doi: 10.1007/s13105-012-0199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, Thompson CB, Evans RM. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326:437–440. doi: 10.1126/science.1172156. doi: 10.1126/science.1172156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esquejo RM, Salatto CT, Delmore J, et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine. 2018;31:122–132. doi: 10.1016/j.ebiom.2018.04.009. doi: 10.1016/j.ebiom.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 40.Repa JJ, Mangelsdorf DJ. Nuclear receptor regulation of cholesterol and bile acid metabolism. Curr Opin Biotechnol. 1999;10:557–563. doi: 10.1016/s0958-1669(99)00031-2. [DOI] [PubMed] [Google Scholar]
- 41.Apfel R, Benbrook D, Lernhardt E, Ortiz MA, Salbert G, Pfahl M. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol Cell Biol. 1994;14:7025–7035. doi: 10.1128/mcb.14.10.7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci USA. 1985;82:5328–5331. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 46.Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999;96:57–68. doi: 10.1016/s0092-8674(00)80959-9. [DOI] [PubMed] [Google Scholar]
- 47.Duong HA, Robles MS, Knutti D, Weitz CJ. A molecular mechanism for circadian clock negative feedback. Science. 2011;332:1436–1439. doi: 10.1126/science.1196766. doi: 10.1126/science.1196766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Um JH, Yang S, Yamazaki S, Kang H, Viollet B, Foretz M, Chung JH. Activation of 5’-AMP-activated kinase with diabetes drug metformin induces casein kinase Iepsilon (CKIepsilon)-dependent degradation of clock protein mPer2. J Biol Chem. 2007;282:20794–20798. doi: 10.1074/jbc.C700070200. doi: 10.1074/jbc.C700070200. [DOI] [PubMed] [Google Scholar]
- 49.Nishii K, Yamanaka I, Yasuda M, Kiyohara YB, Kitayama Y, Kondo T, Yagita K. Rhythmic post-transcriptional regulation of the circadian clock protein mPER2 in mammalian cells: a real-time analysis. Neurosci Lett. 2006;401:44–48. doi: 10.1016/j.neulet.2006.03.022. doi: 10.1016/j.neulet.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 50.Field MD, Maywood ES, O’Brien JA, Weaver DR, Reppert SM, Hastings MH. Analysis of clock proteins in mouse SCN demonstrates phylogenetic divergence of the circadian clockwork and resetting mechanisms. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- 51.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 52.Watson JA, Fang M, Lowenstein JM. Tricarballylate and hydroxycitrate: substrate and inhibitor of ATP: citrate oxaloacetate lyase. Arch Biochem Biophys. 1969;135:209–217. doi: 10.1016/0003-9861(69)90532-3. [DOI] [PubMed] [Google Scholar]
- 53.Linn TC, Srere PA. Binding of ATP citrate lyase to the microsomal fraction of rat liver. J Biol Chem. 1984;259:13379–13384. [PubMed] [Google Scholar]
- 54.Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14:2950–2961. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Z, Chu CH, Cheng D. A novel direct homogeneous assay for ATP citrate lyase. J Lipid Res. 2009;50:2131–2135. doi: 10.1194/jlr.D900008-JLR200. doi: 10.1194/jlr.D900008-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li JJ, Wang H, Tino JA, et al. 2-hydroxy-N-arylbenzenesulfonamides as ATP-citrate lyase inhibitors. Bioorg Med Chem Lett. 2007;17:3208–3211. doi: 10.1016/j.bmcl.2007.03.017. doi: 10.1016/j.bmcl.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 57.Gatfield D, Schibler U. Physiology. Proteasomes keep the circadian clock ticking. Science. 2007;316:1135–1136. doi: 10.1126/science.1144165. doi: 10.1126/science.1144165. [DOI] [PubMed] [Google Scholar]
- 58.Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152:1106–1118. doi: 10.1016/j.cell.2013.01.054. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- 59.Yoo SH, Mohawk JA, Siepka SM, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–1105. doi: 10.1016/j.cell.2013.01.055. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmalen I, Reischl S, Wallach T, Klemz R, Grudziecki A, Prabu JR, Benda C, Kramer A, Wolf E. Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell. 2014;157:1203–1215. doi: 10.1016/j.cell.2014.03.057. doi: 10.1016/j.cell.2014.03.057. [DOI] [PubMed] [Google Scholar]
- 61.Ho KJ, Drummond JL. Circadian rhythm of biliary excretion and its control mechanisms in rats with chronic biliary drainage. Am J Physiol. 1975;229:1427–1437. doi: 10.1152/ajplegacy.1975.229.5.1427. doi: 10.1152/ajplegacy.1975.229.5.1427. [DOI] [PubMed] [Google Scholar]
- 62.Metzger AL, Adler R, Heymsfield S, Grundy SM. Diurnal variation in biliary lipid composition. Possible role in cholesterol gallstone formation. N Engl J Med. 1973;288:333–336. doi: 10.1056/NEJM197302152880702. doi: 10.1056/NEJM197302152880702. [DOI] [PubMed] [Google Scholar]
- 63.Northfield TC, Hofmann AF. Biliary lipid secretion in gallstone patients. Lancet. 1973;1:747–748. doi: 10.1016/s0140-6736(73)92130-2. [DOI] [PubMed] [Google Scholar]
- 64.Bloch HM, Thornton JR, Heaton KW. Effects of fasting on the composition of gallbladder bile. Gut. 1980;21:1087–1089. doi: 10.1136/gut.21.12.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaffer EA, Braasch JW, Small DM. Bile composition at and after surgery in normal persons and patients with gallstones. Influence of cholecystectomy. N Engl J Med. 1972;287:1317–1322. doi: 10.1056/NEJM197212282872603. doi: 10.1056/NEJM197212282872603. [DOI] [PubMed] [Google Scholar]
- 66.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 67.Pan X, Bradfield CA, Hussain MM. Global and hepatocyte-specific ablation of Bmal1 induces hyperlipidaemia and enhances atherosclerosis. Nat Commun. 2016;7:13011. doi: 10.1038/ncomms13011. doi: 10.1038/ncomms13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giugliano D, De Rosa N, Di Maro G, Marfella R, Acampora R, Buoninconti R, D’Onofrio F. Metformin improves glucose, lipid metabolism, and reduces blood pressure in hypertensive, obese women. Diabetes Care. 1993;16:1387–1390. doi: 10.2337/diacare.16.10.1387. [DOI] [PubMed] [Google Scholar]
- 69.Hollenbeck CB, Johnston P, Varasteh BB, Chen YD, Reaven GM. Effects of metformin on glucose, insulin and lipid metabolism in patients with mild hypertriglyceridaemia and non-insulin dependent diabetes by glucose tolerance test criteria. Diabete Metab. 1991;17:483–489. [PubMed] [Google Scholar]
- 70.Reaven GM, Johnston P, Hollenbeck CB, Skowronski R, Zhang JC, Goldfine ID, Chen YD. Combined metformin-sulfonylurea treatment of patients with noninsulin-dependent diabetes in fair to poor glycemic control. J Clin Endocrinol Metab. 1992;74:1020–1026. doi: 10.1210/jcem.74.5.1569149. doi: 10.1210/jcem.74.5.1569149. [DOI] [PubMed] [Google Scholar]
- 71.Robinson AC, Burke J, Robinson S, Johnston DG, Elkeles RS. The effects of metformin on glycemic control and serum lipids in insulin-treated NIDDM patients with suboptimal metabolic control. Diabetes Care. 1998;21:701–705. doi: 10.2337/diacare.21.5.701. [DOI] [PubMed] [Google Scholar]
- 72.Beg ZH, Allmann DW, Gibson DM. Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem Biophys Res Commun. 1973;54:1362–1369. doi: 10.1016/0006-291x(73)91137-6. [DOI] [PubMed] [Google Scholar]
- 73.Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- 74.Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005;24:6314–6322. doi: 10.1038/sj.onc.1208773. doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- 75.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 76.Wang Q, Li S, Jiang L, Zhou Y, Li Z, Shao M, Li W, Liu Y. Deficiency in hepatic ATP-citrate lyase affects VLDL-triglyceride mobilization and liver fatty acid composition in mice. J Lipid Res. 2010;51:2516–2526. doi: 10.1194/jlr.M003335. doi: 10.1194/jlr.M003335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pinkosky SL, Filippov S, Srivastava RA, Hanselman JC, Bradshaw CD, Hurley TR, Cramer CT, Spahr MA, Brant AF, Houghton JL, Baker C, Naples M, Adeli K, Newton RS. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J Lipid Res. 2013;54:134–151. doi: 10.1194/jlr.M030528. doi: 10.1194/jlr.M030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pinkosky SL, Newton RS, Day EA, Ford RJ, Lhotak S, Austin RC, Birch CM, Smith BK, Filippov S, Groot PH, Steinberg GR, Lalwani ND. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat Commun. 2016;7:13457. doi: 10.1038/ncomms13457. doi: 10.1038/ncomms13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Samsoondar JP, Burke AC, Sutherland BG, Telford DE, Sawyez CG, Edwards JY, Pinkosky SL, Newton RS, Huff MW. Prevention of diet-induced metabolic dysregulation, inflammation, and atherosclerosis in Ldlr-/- mice by treatment with the ATP-citrate lyase inhibitor bempedoic acid. Arterioscler Thromb Vasc Biol. 2017;37:647–656. doi: 10.1161/ATVBAHA.116.308963. doi: 10.1161/ATVBAHA.116.308963. [DOI] [PubMed] [Google Scholar]