

Supplemental Digital Content is available in the text.
Keywords: abdominal aortic aneurysm, chemokine, endothelial cell, G protein coupled receptor, neutrophil recruitment
Abstract
Objective—
Chemokine-mediated neutrophil recruitment contributes to the pathogenesis of abdominal aortic aneurysm (AAA) and may serve as a promising therapeutic target. FAM3D (family with sequence similarity 3, member D) is a recently identified novel chemokine. Here, we aimed to explore the role of FAM3D in neutrophil recruitment and AAA development.
Approach and Results—
FAM3D was markedly upregulated in human AAA tissues, as well as both elastase- and CaPO4-induced mouse aneurysmal aortas. FAM3D deficiency significantly attenuated the development of AAA in both mouse models. Flow cytometry analysis indicated that FAM3D−/− mice exhibited decreased neutrophil infiltration in the aorta during the early stage of AAA formation compared with their wild-type littermates. Moreover, application of FAM3D-neutralizing antibody 6D7 through intraperitoneal injection markedly ameliorated elastase-induced AAA formation and neutrophil infiltration. Further, in vitro coculture experiments with FAM3D-neutralizing antibody 6D7 and in vivo intravital microscopic analysis indicated that endothelial cell–derived FAM3D induced neutrophil recruitment. Mechanistically, FAM3D upregulated and activated Mac-1 (macrophage-1 antigen) in neutrophils, whereas inhibition of FPR1 (formyl peptide receptor 1) or FPR2 significantly blocked FAM3D-induced Mac-1 activation, indicating that the effect of FAM3D was dependent on both FPRs. Moreover, specific inhibitors of FPR signaling related to Gi protein or β-arrestin inhibited FAM3D-activated Mac-1 in vitro, whereas FAM3D deficiency decreased the activation of both FPR-Gi protein and β-arrestin signaling in neutrophils in vivo.
Conclusions—
FAM3D, as a dual agonist of FPR1 and FPR2, induced Mac-1-mediated neutrophil recruitment and aggravated AAA development through FPR-related Gi protein and β-arrestin signaling.
Abdominal aortic aneurysm (AAA) is a life-threatening vascular disease characterized by permanent dilatation of the aorta with extremely high mortality on rupture.1,2 Although not fully understood, the pathogenesis of AAA includes extracellular matrix degradation, vascular smooth muscle cell (VSMC) dedifferentiation/apoptosis, reactive oxygen species accumulation, and inflammatory cell infiltration.3 Open surgery or endovascular repair is currently the first therapeutic choice, but those 2 treatments have high postoperative mortality and ineffective extension of long-term survival, respectively.4 Thus, further exploration of the underlying mechanism of AAA could yield substantial benefits by promoting the development of effective medical treatment and improving interventions and therapies for AAA.
Inflammatory cell infiltration is one of the key pathological features of AAA. Among inflammatory cells, neutrophils are the first cell population recruited to the site of inflammation in inflammatory vascular diseases.5 Depletion of circulating neutrophils via antineutrophil antibody treatment inhibits neutrophil and macrophage infiltration and reduces elastase-induced AAA formation in mice, indicating the pathogenic role of neutrophils in AAA.6 Chemokines released from endothelial cells (ECs), such as CXCL8 (C-X-C motif chemokine ligand 8) and CXCL1, recruit neutrophils, activate neutrophil adhesion molecules (eg, VLA-4 [very late antigen-4], Mac-1 [macrophage-1 antigen]), and cause the interaction between ECs and neutrophils, thereby initiating vascular inflammation.7,8 Targeting of these chemokines may favor the inhibition of neutrophil recruitment and relieve vascular inflammatory diseases. Although application of neutralizing antibody against chemokine receptor CXCR2 rescued acute aortic dissection rupture9 and attenuated elastase-induced AAA development,10 it remains unclear whether direct intervention against chemokines mediating neutrophil recruitment could ameliorate AAA development.
FAM3D (family with sequence similarity 3, member D) belongs to a cytokine-like family composed of 4 members, referred to as FAM3A, FAM3B, FAM3C, and FAM3D, which share a 4-helix-bundle structure.11 The FAM3D protein consists of 223 amino acids, and its molecular weight is 33 kDa. FAM3D is constitutively expressed in the gastrointestinal tract, and the regulation of FAM3D expression and production is linked to nutritional regulation, that is, FAM3D is upregulated after intake of a high-fat diet both in mice and in human.12 Our previous study identified FAM3D as a new chemokine-like factor lacking the typical cysteine residue structural domain of classic chemokines.13 However, the role of FAM3D in the pathogenesis of AAA remains elusive. Here, we found that FAM3D induced neutrophil recruitment through FPR (formyl peptide receptor) signaling–mediated activation of Mac-1, thereby aggravating AAA development.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Reagents
Antibodies against FAM3D (AF3027 and MAB2869) were purchased from R&D Systems (Minneapolis, MN). Type I porcine pancreatic elastase (E1250, lot No. SLBN4280V) and Ang II (angiotensin II; A9525) were purchased from Sigma-Aldrich (St Louis, MO). PE-conjugated antibody against human CD11b (CBRM1/5, 12-0113-41) for flow cytometry was purchased from eBioscience (San Diego, CA); PE anti-human CD29 (556049) and PerCP/Cy5.5 anti-mouse Ly-6C (128011) for flow cytometry and anti-CD31 (553370) used in en face staining were purchased from BD Biosciences (San Diego, CA); fluorescein isothiocyanate (FITC) anti-mouse CD45 (103107), APC anti-mouse CD11b (101211), PE anti-mouse Ly-6G/C (108407), PE anti-mouse Ly6G (127607), APC anti-mouse CD86 (105011), and PE anti-mouse F4/80 (123109) used in flow cytometry as well as Ly6G (1A8) antibodies (127601) applied for immunohistochemical and immunofluorescent staining were purchased from BioLegend (San Diego, CA). Alexa Fluor 546 goat anti-rat IgG (A11081) and Alexa Fluor 488 donkey anti-rabbit IgG (A21206), as well as the dye DiI (D3911) for staining neutrophils, were purchased from Life Technologies (San Diego, CA). Antibodies against phosphorylated (9101s) and total (9102s) ERK1/2 (extracellular regulated MAP kinase), phosphorylated (9371s) and total (2058s) PKC (protein kinase C), phosphorylated (9271s) and total (4691p) AKT (PKB, protein kinase B), phosphorylated Src (2101s), and phosphorylated (4511s) and total p38 (8690s) p38MAPK (mitogen-activated kinase-like protein), caspase 3 (9662s), cleaved caspase 3 (9661s), Bax (2772s), as well as the Hoechst (4082s) used to stain cell nuclei, were purchased from Cell Signaling Technology (Boston, MA). Antibodies against Src (AB21267a), smooth muscle α-actin (α-actin, ab5694), neutrophil elastase (ab21595) and proteinase-3 (ab103632) was purchased from Abcam (Cambridge, UK).
Mouse Models of AAA
All animal studies and experimental procedures were approved by the Institutional Animal Care and Use Committee of Peking University Health Science Center (LA2016–182), an ethical organization authorized by the Beijing Municipal Science & Technology Commission. C57BL/6 mice were purchased from Vital River (Beijing, People’s Republic of China).
As male mice are more susceptible than female mice to AAA,14–16 male mice were used for AAA induction. Elastase was used to induce AAA in 11- to 12-week-old male mice. The mice were anesthetized, and the infrarenal aortas were exposed, isolated, and wrapped with sterile cotton, which was soaked with 30 μL of elastase (68.68 IU/mL, lot No. SLBN4280V) for 40 minutes. Then, the elastase-soaked cotton was removed, and 0.9% NaCl was used to perfuse the abdominal cavity before suturing. Aortas were collected 14 days after surgery.17 Additionally, CaPO4 was used to induce AAA in 11- to 12-week-old male mice as well. In brief, the mice were anaesthetized, and the infrarenal aortas were exposed, isolated, and wrapped with gauze presoaked in 0.5 mol/L CaCl2 for 10 minutes. Then, the CaCl2-soaked gauze was removed, and the aortas were wrapped with gauze presoaked in PBS for another 5 minutes to form CaPO4. The abdominal cavities were washed with 0.9% NaCl before suturing. The aortas were collected 14 days after surgery.18 Four-month-old male ApoE−/− mice were used for Ang II infusion. Mice were infused with Ang II (1000 ng/kg/min) or saline via subcutaneously implanted osmotic mini-pumps (1004, Alzet, Cupertino, CA).19 The suprarenal abdominal aortas were collected 7 days after implantation surgery.
Western Blotting
Tissues or cells were lysed in RIPA buffer. Protein concentrations were evaluated using a BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Equal amounts of total protein were separated using 10%, 12%, or 15% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membranes were incubated with primary antibodies and appropriate IRDye-conjugated secondary antibodies (Rockland Inc., Limerick, PA). The immunofluorescent signal was detected by an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). The information of dilution and working concentrations of the primary antibodies were referred to in Table III in the online-only Data Supplement.
Real-Time Polymerase Chain Reaction
Total RNA was extracted from infrarenal abdominal aortas using TRIzol Reagent (APPLYGEN, Beijing, People’s Republic of China). Equal amounts (2 μg) of total RNA were reverse transcribed to cDNA using 5× All-In-One RT master mix (G490; Applied Biological Materials Inc., Richmond, Canada). Real-time polymerase chain reaction (PCR) amplification was performed using SYBR Green I 2× PCR mix (TransGen Biotech, Beijing, People’s Republic of China) and recorded under an Mx3000 Multiplex Quantitative PCR System (Stratagene, La Jolla, CA). The PCR program consisted of 94°C for 5 minutes; 40 cycles of 94°C for 30 s, 56°C to 58°C for 30 s, and 72°C for 30 s, followed by a final extension step at 72°C for 5 minutes. The data were analyzed using the ΔΔCT method. The primer sequence information is included in Table IV in the online-only Data Supplement.
Generation of FAM3D−/− Mice
FAM3D−/− mice were generated on a C57BL/6 background by TALEN-mediated gene targeting. We designed target vectors specific to exon 3 of FAM3D. These vectors were transcribed into mRNA in vitro, after which they were microinjected into the fertilized eggs of C57BL/6 mice. The heterozygotes were identified and intercrossed to generate FAM3D−/− mice with heterozygous (FAM3D+/−) and wild-type (WT) littermates. Genomic DNA was extracted from tail biopsies for PCR genotyping and sequencing. A T7E1 assay (Viewsolid Biotech, Beijing, People’s Republic of China) and sequence analysis (Sunbio, Beijing, People’s Republic of China) were applied for genotyping.
Measurement of Blood Pressure and Plasma Lipid
A noninvasive computerized CODA tail-cuff blood pressure system (Kent Scientific, Torrington, CT) was used to measure the systolic blood pressure of mice. The equipment was kept clean and free from foreign scents and blood odors. The investigator was blinded with respect to the experimental groups when performing the measurements, and the mice were tested in a randomized order. All the mice underwent training sessions from 1 to 5 pm on 7 consecutive days to become accustomed to the tail-cuff procedure. Sessions of recorded measurements were then made by a single investigator at the same time on 3 consecutive days. Five measurements were performed daily on each mouse, and the average of all 15 measurements was represented as the systolic blood pressure of each mouse. Plasma total cholesterol and triglyceride levels were measured with the kits provided by Zhong Sheng Bio-technology (Beijing, People’s Republic of China). The complete blood count was evaluated by routine blood testing in the experimental animal core facility of Peking University.
Morphometric Analysis and Immunohistochemistry
Mice were euthanatized by CO2 inhalation, followed by perfusion with PBS and fixation with 4% paraformaldehyde. The entire aortas including the segments of the aortic arch, thoracic aorta, and abdominal aorta were dissected to measure the maximal diameters of infrarenal abdominal aortas. Then, the infrarenal abdominal aortas were further embedded in OCT compound and cut into serial frozen sections (7-μm-thick, ≈1 mm apart). Sixteen serial sections for each mouse were analyzed by Verhoeff-Von Gieson staining using a kit from BASO, Inc (Zhuhai, People’s Republic of China). For elastin assessment, elastin degradation was graded on a scale of 1 to 4, where 1 meant <25% degradation, 2 meant 25% to 50% degradation, 3 meant 50% to 75% degradation, and 4 meant >75% degradation.20 To evaluate neutrophil infiltration in the aortic wall, 8 serial sections for each infrarenal abdominal aorta were incubated with rat-originated anti-Ly6G (1:400 dilution, 1.25 μg/mL). An equal amount of rat IgG served as a negative control. Rabbit anti-α-actin (1:500 dilution, 0.4 μg/mL) was used to evaluate the VSMCs content in the aortic wall, an equal amount of rabbit IgG served as a negative control. The data were evaluated blindly by 2 independent investigators and presented as the mean of 8 to 16 serial sections for each mouse.
To evaluate the FAM3D expression in human AAA, we collected AAA tissues from patient undergoing AAA repair operation and fixed them in 4% paraformaldehyde and embedded with paraffin. The procedures for isolation of human-derived tissues were approved by the Ethics Committee of the Peking University Health Science Center. The tissues were sectioned (7-μm-thick, ≈1 mm apart), and 8 serial sections for each infrarenal abdominal aorta were incubated with rabbit anti-FAM3D antibodies (1:200 dilution, 5 μg/mL),13 an equal amount of rabbit IgG served as a negative control. The data were evaluated blindly by 2 independent investigators.
Aortic Single Cell Preparation
The infrarenal abdominal aorta was dissected at different time points during AAA development, and the connective and fat tissues surrounding the vessels were removed completely. The aorta tissues were cut into pieces and digested into single cells in 1 mL of 1× Aorta Dissociation Enzyme Solution as described previously.21 The digested single-cell suspension was used for flow cytometric analysis.
Flow Cytometry Analysis
For analysis of leukocytes from mice after euthanasia by CO2 inhalation, peripheral blood cells collected through cardiac puncture, bone marrow cells obtained from the femur and tibia. and enzyme-digested aortic single-cell suspension were subjected to flow cytometric analysis. In general, bone marrow cells and peripheral blood cells were labeled with FITC-anti-CD45 (0.125 μg/mL), APC-anti-CD11b (0.1 μg/mL), and PE-anti-Ly6G/C (0.1 μg/mL) antibodies to identify monocytes (CD45+CD11b+Ly6ChighLy6G−, CD45+CD11b+Ly6ClowLy6G−) or neutrophils (CD45+CD11b+Ly6G+) following elimination of red blood cells by RBC lysis buffer (Tiangen Biotech Inc, Beijing, China). Aortic single cells were analyzed with FITC-anti-CD45 (0.125 μg/mL), APC-anti-CD11b (0.1 μg/mL), PE-anti-Ly6G (1A8, 0.1 μg/mL), and PerCP/Cy5.5 anti-mouse Ly-6C (1 μg/mL) to identify neutrophils (CD45+CD11b+Ly6G+) or Ly6Chigh (CD45+CD11b+Ly6C+Ly6G−) and Ly6Clow (CD45+CD11b+Ly6C−Ly6G−) monocytes, with FITC-anti-CD45 (0.125 μg/mL), APC-anti-CD11b (0.1 μg/mL), and PE-anti-F4/80 (1 μg/mL) antibodies to identify macrophages (CD45+CD11b+F4/80+) and with FITC-anti-CD45 (0.125 μg/mL), PE-anti-F4/80 (1 μg/mL), and APC-anti-CD86 (1 μg/mL) antibodies to identify proinflammatory activation of macrophages (CD45+F4/80+CD86+). To evaluate the activated VLA-4 and Mac-1 in human neutrophils or monocytes, we applied PE-anti-activated CD29 (0.5 μg/mL) and PE-anti-activated CD11b (0.5 μg/mL) antibodies, respectively, following the isolation of fresh neutrophils or monocytes from peripheral blood of healthy volunteers. Cells were incubated with primary antibodies or IgG on ice for 30 minutes. After the unbound antibodies were washed out, the cells were analyzed using a BD FACS Calibur system (BD Biosciences, San Diego, CA).
In Situ Zymography
MMP (matrix metalloproteinase) activity was detected in infrarenal abdominal aortas using EnzChek Gelatinase/Collagenase Assay Kit (E12055, Invitrogen, Waltham, MA) according to the manufacturer’s instruction. Eleven- to 12-week-old C57BL/6 male mice were treated with elastase for 3 days. After perfusion with PBS and dissection, the tissue was sectioned (7-μm-thick, ≈1 mm apart), and 8 serial sections for each infrarenal abdominal aorta were incubated with DQ gelatin for 1 hour. Images of the tissues were obtained using a confocal microscope (Leica, Wetzlar, Germany). The data were evaluated blindly by 2 independent investigators and presented as the mean of 8 serial sections for each aorta.
Detection of ROS Production
ROS (reactive oxidative species) production was detected by ROS fluorescent probe, dihydroethidine hydrochloride (R001, Vigorous, People’s Republic of China). Following treatment with elastase for 3 days, 11- to 12-week-old C57BL/6 male mice were perfused with PBS and dissected for infrarenal abdominal aorta isolation. Isolated aortic tissue was sectioned (7-µm-thick, ≈1 mm apart), and 8 serial sections for each aorta were incubated with dihydroethidine hydrochloride (5 μmol/L) for 30 minutes. Dihydroethidine hydrochloride–stained ROS was observed using a confocal microscope (Leica, Wetzlar, Germany). The data were evaluated blindly by 2 independent investigators and presented as the mean of 8 serial sections for each aorta.
Apoptosis Assay
The DeadEnd Fluorometric TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) System (G3250, Promega, Madison, WI) was used to detect the vascular cell apoptosis according to the manufacturer’s instructions. In belief, the aortic tissue was sectioned (7-μm-thick, ≈1 mm apart), and 8 serial sections for each infrarenal abdominal aorta were incubated with equilibration buffer for 10 minutes, then TdT (terminal deoxynucleotidyl transferase) reaction mixture was added to the tissues for 1-hour incubation at 37°C followed by 2× SSC buffer used for stopping the reaction. The data were obtained using a confocal microscope (Leica, Wetzlar, Germany) and evaluated blindly by 2 independent investigators.
Cell Culture
The procedures for isolation of human-derived primary cells were approved by the Ethics Committee of the Peking University Health Science Center. Human umbilical vein endothelial cells were isolated from umbilical cords using type I collagenase (100 IU/mL) as previously described.22 Human umbilical vein ECs were maintained in M199 medium (Gibco, San Diego, CA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT), 4.17 mg/L of recombinant human EC growth factor (Sigma-Aldrich, St. Louis, MO), 1.4 IU/mL of heparin sodium (Sigma-Aldrich, St. Louis, MO), 3.0 mg/L of thymidine (Sigma-Aldrich, St. Louis, MO), 5.96 g/L of HEPES, 2.2 g/L of NaHCO3, 200 IU/mL of penicillin, and 100 IU/mL of streptomycin. The cells were digested and passaged with 0.05% trypsin, and cells at passages 5 to 7 were used for experiments. Human neutrophils and monocytes were isolated from the peripheral blood of healthy male volunteers using Polymorphprep (AS1114683, Axis-Shield, Dundee, Scotland) according to the manufacturer’s instructions. Mouse bone marrow neutrophils were separated using a density gradient centrifugation method provided by TBD (Tianjin, People’s Republic of China) according to the manufacturer’s instructions. Neutrophils and monocytes were cultured in RPMI 1640 medium supplemented with 10% FBS. HEK293T cells were purchased from ATCC (Manassas, VA) and were maintained in DMEM supplemented with 10% FBS. Cell cultures were maintained at 37°C in a humidified atmosphere under 5% CO2.
En face Immunofluorescent Staining
Eleven- to 12-week-old C57BL/6 male mice were treated with elastase for 7 days. After perfusion, fixation, and dissection, the infrarenal abdominal aorta was dissected and incubated in 20% goat serum for 15 minutes, then incubated overnight in a rotator with rat anti-CD31 (1:500 dilution, 1 µg/mL) and rabbit anti-FAM3D antibodies (1:200 dilution, 5 µg/mL; 4°C). The secondary antibodies applied for labeling primary antibodies for 1 hour at 37°C were Alexa Fluor 546 goat anti-rat IgG (1:200 dilution, 10 μg/mL) and Alexa Fluor 488 donkey anti-rabbit IgG (1:200 dilution, 10 μg/mL). Aortas were washed 3× with PBS at the intervals between incubations and then stained with Hoechst (1:10 000 dilution, 5 μg/mL, 3 minutes). The infrarenal abdominal aorta was opened by a longitudinal cut through the aortic wall and mounted on slides using anti-fade mounting solution (C1210, APPLYGEN, Beijing, People’s Republic of China), with the intima facing up. Images of the EC monolayer were obtained using a confocal microscope (Leica, Wetzlar, Germany).
Neutrophil Adhesion Assay
Freshly isolated mouse bone marrow neutrophils were fluorescently labeled with 5 μmol/L of DiI in complete RPMI 1640 medium for 30 minutes at 37°C in the dark. Then, confluent human umbilical vein ECs were pretreated with TNF-α (tumor necrosis factor α; 4 ng/mL) for 24 hours in 6-well plates, followed by coculture with labeled neutrophils (1×106/well) for 30 minutes. After nonadherent cells were removed by washing with PBS 3×, the adherent neutrophils were evaluated using a fluorescence microscope (Leica, Wetzlar, Germany); 6 randomly selected fields per well were imaged and subsequently quantified by Image J software (National Institutes of Health, Bethesda, ML).
Neutrophil Transmigration Assay
Human umbilical vein ECs were grown to confluence on collagen I–coated transwell inserts (6.5 mm diameter, 8-µm pore size; Corning Inc., NY). DiI-labeled mouse bone marrow neutrophils (2×105 in 200 μL) were placed in the upper chamber, and mouse FAM3D protein (100, 200, 300, and 400 ng/mL) was added to the lower chamber. After 3 hours of incubation, the neutrophils below the transwell inserts were observed under a fluorescence microscope (Leica, Wetzlar, Germany). The transmigrated neutrophils were quantified by Image J software based on 6 randomly selected fields per transwell insert.
Leukocyte Rolling and Adhesion Under Intravital Microscopy
WT and FAM3D−/− mice (8 weeks old) were intraperitoneally injected with TNF-α (500 ng/mouse). Four hours later, the mice were anesthetized and underwent laparotomy for exposing the mesenteric microvessels. Then, branchless vessels with diameters ranging from 30 to 50 μm and lengths of ≈200 µm were selected for video recording under an inverted intravital microscope (Leica, Wetzlar, Germany). Leukocytes moving more slowly than erythrocytes in the same vessel were defined as rolling, while leukocytes that firmly attached to the same site for >30 seconds were defined as adhesion. The numbers of rolling and adherent leukocytes along microvessels (200-μm in length) were counted and quantified.
Purification of FAM3D Protein
The plasmids pcDNA3.1-human FAM3D-Myc-His or pcDNA3.1-mouse FAM3D-Myc-His were transfected into HEK293T cells using jetPEI (PT-101, Polyplus, Berkeley, CA) according to the manufacturer’s instructions. Sixteen hours after transfection, the culture medium was replaced with OptiMEM (Invitrogen, Waltham, MA), and the cells were cultured for another 5 days. The conditioned medium was harvested, and then FAM3D-Myc-His proteins were purified by the Ni Sepharose High Performance system according to the manufacturer’s instructions (GE Healthcare, Danderyd, Sweden).
Immunofluorescent Staining
Eleven- to 12-week-old C57BL/6 male mice were treated with elastase for 7 days. After perfusion, fixation, and dissection, the tissue was sectioned (7-μm-thick, ≈1 mm apart), and 8 serial sections for each infrarenal abdominal aorta were incubated with rabbit anti-FAM3D antibody (1:200 dilution, 5 μg/mL) overnight at 4°C, The secondary antibodies applied for labeling primary antibodies for 1 hour at 37°C was Alexa Fluor 488 donkey anti-rabbit IgG (1:200 dilution, 10 μg/mL). For double immunofluorescent staining, 8 serial sections for each infrarenal abdominal aorta were incubated with rabbit anti-p-ERK (1:200 dilution, 5 μg/mL), rabbit anti-p-PKC (1:200 dilution, 5 μg/mL), or rabbit anti-p-p38MAPK (1:200 dilution, 5 μg/mL) antibodies together with rat anti-Ly6G (1:400 dilution, 1.25 μg/mL) antibody separately overnight at 4°C. Alexa Fluor 546 goat anti-rat IgG (1:200 dilution, 10 μg/mL) and Alexa Fluor 488 donkey anti-rabbit IgG (1:200 dilution, 10 μg/mL) were applied for labeling corresponding species-originated primary antibodies for 1 hour at 37°C. Sections were washed 3 times with PBS at the intervals between incubations and then stained with Hoechst (1:10 000, 5 μg/mL, 3 minutes). Images were obtained using a confocal microscope (Leica, Wetzlar, Germany). The data were evaluated blindly by 2 independent investigators and presented as the mean of 8 serial sections for each aorta.
Statistical Analysis
All the results are expressed as the mean±SEM. Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA). For statistical comparisons, we first evaluated whether the data were normally distributed. Then, the Brown-Forsythe test was used to check for similar variances among normally distributed data; if the variances were similar, we applied Student t test for 2-group comparisons or analysis of variance for comparisons of >2 groups. Nonparametric tests were used where data were not normally distributed. In all cases, statistical significance was concluded where the 2-tailed probability was <0.05. The details of the statistical analysis applied in each experiment are presented in the corresponding figure legends.
Results
FAM3D Deficiency Attenuates the Development of AAA in Mice
To explore the possible relationship between FAM3D and AAA, we first performed immunohistochemical staining of FAM3D in aneurysmal tissues in AAA patients. Compared with normal vessels, the expression of FAM3D was significantly upregulated in aneurysmal aortas (Figure IA in the online-only Data Supplement). Then we further validated the regulation of FAM3D expression in aortic tissues during the pathogenic process of elastase- or CaPO4-induced AAA by both Western blotting and real-time PCR. We found that FAM3D expression was significantly elevated at 3 and 7 days after elastase induction at both the protein and mRNA levels (Figure 1A; Figure IB in the online-only Data Supplement). Similar results were observed in CaPO4-induced AAA as well (Figure 1B; Figure IC in the online-only Data Supplement). Meanwhile, we also evaluated the regulation of FAM3D expression in Ang II–induced AAA in ApoE−/− mice. Consequently, the protein level of FAM3D was markedly upregulated at early stage (7 days) of AAA formation induced by Ang II (Figure ID in the online-only Data Supplement). Thus, we postulated that FAM3D might be involved in the pathogenesis of AAA.
Figure 1.
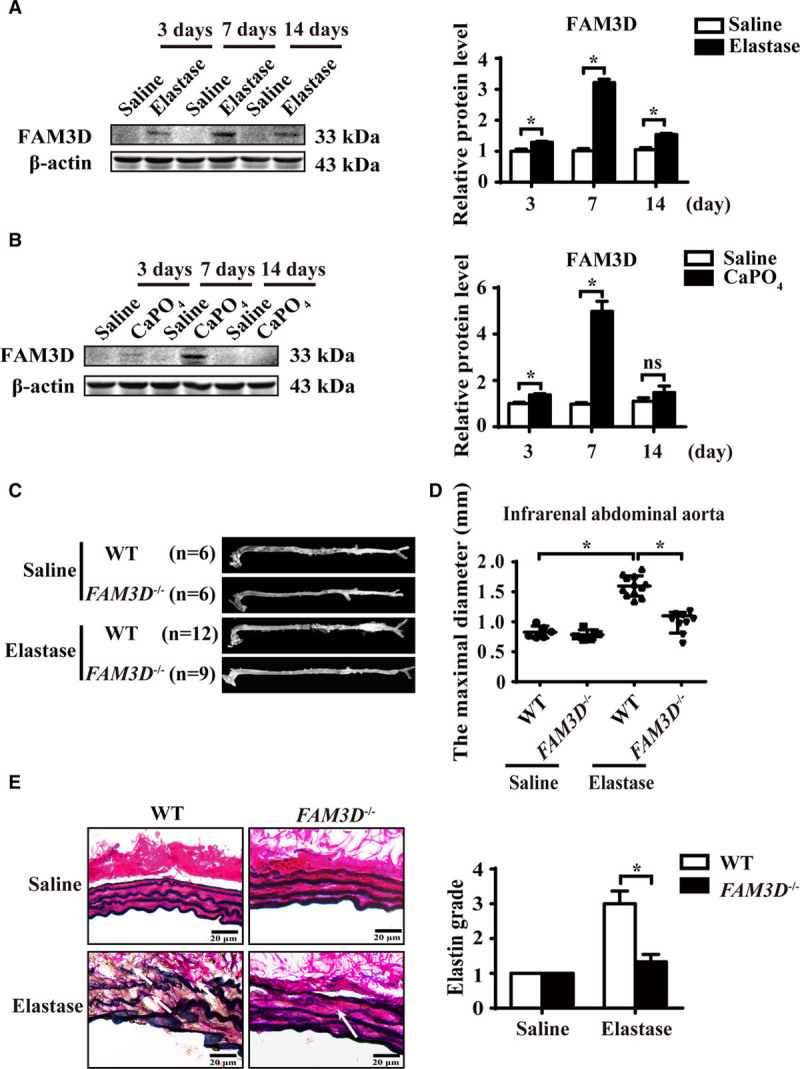
FAM3D (family with sequence similarity 3, member D) deficiency attenuates the development of abdominal aortic aneurysm (AAA) in mice. A and B, Representative Western blot analysis and quantification of FAM3D expression in infrarenal abdominal aortas of C57BL/6 mice treated with elastase (A) or CaPO4 (B) for 3, 7, or 14 days. Saline was applied as a control. n=6. Two-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05. C and D, Representative photographs (C) and quantification of maximal diameters (D) of the infrarenal abdominal aortas in saline- or elastase-induced 11- to 12-week-old wild-type (WT) and FAM3D−/− male mice. Two-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. E, Representative Verhoeff-Van Gieson staining and statistical analysis of elastin degradation in the infrarenal abdominal aortas in saline- or elastase-induced 11- to 12-week-old WT and FAM3D−/− male mice (n=6 for each group). Scale bar, 20 μm. Kruskal-Wallis test, *P<0.05.
To further assess the role of FAM3D in AAA, we generated FAM3D−/− mice by using TALEN technology to delete one cytosine base in exon 3 of the FAM3D gene (Figure IIA through IIC in the online-only Data Supplement). FAM3D was highly expressed in the colon in WT mice, consistent with a previous report.12 Nevertheless, FAM3D was undetectable in various tissues of FAM3D−/− mice, indicating successful knockout (Figure IID in the online-only Data Supplement). The knockout mice were viable, and there were no obvious abnormalities in terms of reproductive capacity, body weight, blood pressure, total cholesterol, or triglyceride levels compared with WT littermates (Table I in the online-only Data Supplement). Interestingly, the baseline number of neutrophils was markedly elevated in the peripheral blood of FAM3D−/− mice compared with that of WT littermates, whereas the proportion of myeloid cells in bone marrow displayed no difference between WT and FAM3D−/− mice (Table II in the online-only Data Supplement). Then, we assessed the formation of AAA in FAM3D−/− mice and their WT littermates. After 14-day elastase induction, the infrarenal abdominal aortas were significantly expanded in WT mice (n=12), whereas FAM3D deficiency markedly abated the elastase-induced increase in the maximal diameters of infrarenal abdominal aortas (1.51±0.07 mm in WT mice versus 1.08±0.07 mm in FAM3D−/− mice; P<0.001; Figure 1C and 1D). Furthermore, Verhoeff-Von Gieson staining, as shown in Figure 1E, demonstrated that FAM3D deficiency also inhibited elastase-induced elastin degradation related to AAA formation. As VSMC apoptosis is one of the major pathologies in AAA development, TUNEL and immunohistochemical staining showed that FAM3D deficiency greatly inhibited VSMC apoptosis and maintained VSMC content in aortic wall during the pathogenesis of elastase-induced AAA (Figure IIIA through IIIC in the online-only Data Supplement), respectively. However, Western blotting displayed that FAM3D did not induce VSMC apoptosis in vitro (Figure IIID in the online-only Data Supplement), implying that FAM3D deficiency may inhibit VSMC apoptosis in elastase-induced AAA through indirect mechanism. Similarly, in CaPO4-induced AAA model, FAM3D−/− mice exhibited attenuated CaPO4-induced infrarenal abdominal aortic enlargement and elastin degradation in the aortic walls compared with their WT littermates (Figure IV in the online-only Data Supplement). Overall, FAM3D deficiency decelerated the development of AAA, implying a pathogenic role of FAM3D in AAA.
FAM3D Deficiency Reduces Neutrophil Recruitment to the Aortic Wall During AAA Formation
Neutrophils and monocytes play critical roles in inflammatory pathology in AAA formation.6,23 We next asked whether FAM3D deficiency–induced reduction in AAA was related to the regulation of neutrophil and monocyte recruitments. We collected elastase-stimulated infrarenal aortic tissues and digested them into single-cell suspensions, which we subjected to flow cytometric analysis. Consistent with a previous study showing that neutrophil recruitment usually occurs in the early stage of AAA,24 the number of neutrophils (CD45+CD11b+Ly6G+) infiltrated into the aortic wall was significantly elevated in WT mice starting at 3 days till 7 days (Figure 2A; Figure VA in the online-only Data Supplement), accompanied with a marked elevation of neutrophils in peripheral blood (Figure 2B; Figure VB in the online-only Data Supplement) and bone marrow at 7 days (Figure VIA in the online-only Data Supplement) and further regressed at 14 days after elastase induction (Figure 2A; Figure VA in the online-only Data Supplement). By contrast, FAM3D deficiency significantly inhibited the accumulation of neutrophils in the infrarenal abdominal aortic wall at 3 and 7 days but increased the number of neutrophils in peripheral blood (Figure 2A and 2B; Figure VA and VB in the online-only Data Supplement). However, there was no difference in the number of neutrophils in bone marrow between WT and FAM3D−/− mice, excluding a possible role of FAM3D in the modulation of neutrophils in bone marrow (Figure VIA in the online-only Data Supplement). Thus, these results indicated that FAM3D may play a critical role in neutrophil recruitment during AAA formation. Additionally, Ly6Chigh (CD45+CD11b+Ly6ChighLy6G−, proinflammatory) and Ly6Clow (CD45+CD11b+Ly6ClowLy6G−, patrolling) monocyte recruitments in aortic wall were significantly elevated from 3 days after elastase induction, whereas infiltrated macrophage (CD45+CD11b+F4/80+) in the aortic wall was significantly enhanced at 14 days after elastase treatment (Figure 2C and 2D; Figure VE and VF in the online-only Data Supplement). Especially, the number of Ly6Chigh monocytes was declined, but macrophage infiltration was elevated in aortic wall during 7 to 14 days, implying the differentiation of Ly6Chigh monocytes to macrophages may contribute to this alteration of cell population. By contrast, FAM3D deficiency greatly decreased the degree of Ly6Chigh monocyte recruitment and macrophage infiltration but exhibited no effect on the recruitment of Ly6Clow monocytes. Moreover, the numbers of monocytes in either peripheral blood or bone marrow (Figures VB through VD and VIB in the online-only Data Supplement) as well as the inflammatory activation of macrophages (Figure VIC in the online-only Data Supplement) exhibited no significant difference between WT and FAM3D−/− mice induced by elastase. Interestingly, although both neutrophil and monocyte recruitments were significantly elevated at 3 days in the process of elastase-induced AAA, FAM3D deficiency exhibited its inhibitive effects on neutrophil recruitment (3 days) earlier than Ly6Chigh monocyte recruitment (7 days) and macrophage infiltration (14 days; Figure 2A, 2C, and 2D). Because neutrophils are able to further regulate monocyte/macrophage accumulation at inflammatory sites,25,26 it was plausible that FAM3D deficiency primarily reduced neutrophil recruitment from the blood into the aortic wall. Further immunohistochemical staining confirmed that FAM3D−/− mice displayed a great decrease in neutrophil infiltration in the infrarenal abdominal aortic wall compared with WT littermates in the early stage of AAA formation (7 days; Figure 2E).
Figure 2.
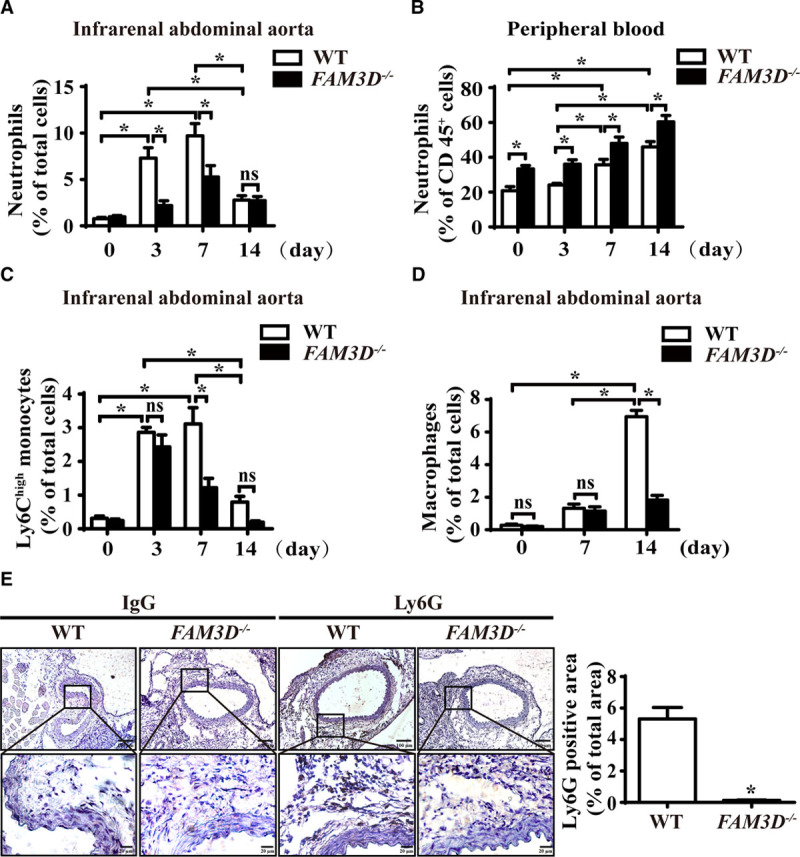
FAM3D (family with sequence similarity 3, member D) deficiency reduces neutrophil recruitment to the aortic wall during abdominal aortic aneurysm (AAA) formation. A, Quantification of neutrophils in the infrarenal abdominal aortas of elastase-treated wild-type (WT) and FAM3D−/− mice at 0 day (WT, n=6; FAM3D−/−, n=6), 3 days (WT, n=7; FAM3D−/−, n=7), 7 days (WT, n=7; FAM3D−/−, n=6), and 14 days (WT, n=9; FAM3D−/−, n=7). Two-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance. B, Quantification of neutrophils in the peripheral blood of elastase-induced WT and FAM3D−/− mice at 0 day (WT, n=6; FAM3D−/−, n=6), 3 days (WT, n=7; FAM3D−/−, n=7), 7 days (WT, n=7; FAM3D−/−, n=6), and 14 days (WT, n=9; FAM3D−/−, n=7). Two-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. C, Quantification of Ly6Chigh monocytes in the infrarenal abdominal aortas of elastase-treated WT and FAM3D−/− mice at 0 day (WT, n=6; FAM3D−/−, n=6), 3 days (WT, n=7; FAM3D−/−, n=7), 7 days (WT, n=7; FAM3D−/−, n=7), and 14 days (WT, n=7; FAM3D−/−, n=7). Two-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance. D, Quantification of macrophages in infrarenal abdominal aortas of elastase-treated WT and FAM3D−/− mice at 0 day (WT, n=6; FAM3D−/−, n=6), 7 days (WT, n=7; FAM3D−/−, n=6), and 14 days (WT, n=9; FAM3D-/-, n=7). Two-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance. E, Representative immunohistochemical staining and quantification of neutrophil infiltration in infrarenal abdominal aortas of WT and FAM3D−/− mice 7 days after elastase induction, using the neutrophil-specific antibody Ly6G. Rat IgG was applied as a negative control. WT, n=8; FAM3D−/−, n=7. Scale bar, 20 μm. Mann-Whitney test, *P<0.05.
As neutrophil-derived neutrophil elastase, proteinase-3, MMP, and ROS have been all reported to mediate AAA development,6,27 we next evaluated whether FAM3D deficiency–decreased neutrophil infiltration affected the production and activity of these pathogenic mediators during AAA formation. As shown in Figure VIIA and VIIB in the online-only Data Supplement, the protein expression of neutrophil elastase and proteinase-3 were significantly downregulated in FAM3D−/− aneurysmal tissues compared with those of WT mice. Moreover, at 3 days of elastase induction, when FAM3D only modulated the infiltration of neutrophils rather than monocytes/macrophages, in situ zymography and dihydroethidine hydrochloride staining exhibited that FAM3D deficiency markedly attenuated MMP activity and ROS production in aneurysmal tissue, respectively (Figure VIIC and VIID in the online-only Data Supplement).
Blockage of FAM3D by Neutralizing Antibody Ameliorates AAA Formation and Neutrophil Recruitment
To further verify the pathogenic role of FAM3D in AAA formation and neutrophil recruitment, we applied a validated FAM3D neutralizing antibody 6D713 through intraperitoneal injection into 11- to 12-week-old C57BL/6 male mice during the elastase induction (Figure 3A). Then, we assessed the formation of AAA in elastase-induced mice. As shown in Figure 3B and 3C, 6D7 inhibited elastase-induced expansion of infrarenal aortas in mice (1.59±0.09 mm in IgG-treated mice versus 0.92±0.05 mm in FAM3D nAb-treated mice; P<0.001). Furthermore, Verhoeff-Von Gieson staining demonstrated that 6D7 also attenuated elastin degradation related to AAA formation (Figure 3D). As well, flow cytometric analysis displayed that 6D7 inhibited the neutrophil recruitment in aneurysmal aortic wall (Figure 3E). Overall, these results indicated that FAM3D may be a potential therapeutic target for AAA formation.
Figure 3.
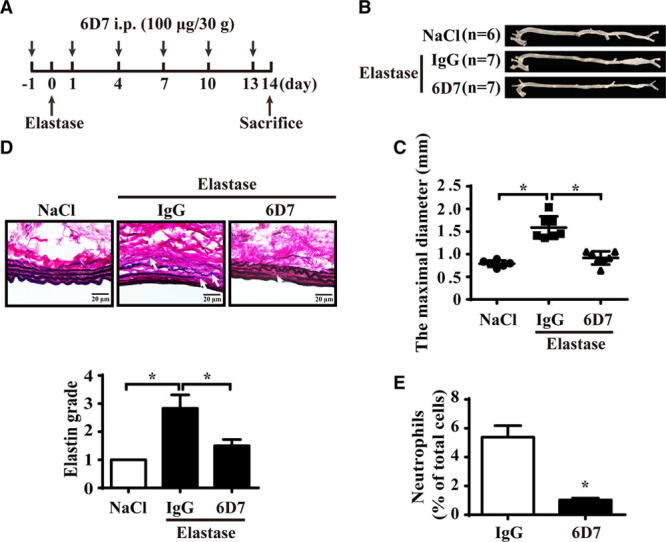
FAM3D (family with sequence similarity 3, member D) neutralization antibody 6D7 attenuates the development of abdominal aortic aneurysm (AAA) in mice. A, Schematic illustration of the time points of intraperitoneal injection of 6D7 or mouse IgG (100 μg/30 g body weight). B and C, Representative photographs (B) and quantification of maximal diameters (C) of the infrarenal abdominal aortas of 11- to 12-week-old C57BL/6 male mice treated with elastase for 14 days along with mouse IgG or 6D7 injection (NaCl, n=6; elastase+IgG, n=7; elastase+6D7, n=7). One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. D, Representative Verhoeff-Van Gieson staining and statistical analysis of elastin degradation in the infrarenal abdominal aortas of 11- to 12-week-old C57BL/6 male mice treated with elastase for 14 days along with mouse IgG or 6D7 injection (NaCl, n=6; elastase+IgG, n=7; elastase+6D7, n=7). Scale bar, 20 μm. Kruskal-Wallis test, *P<0.05. E, Quantification of neutrophils in the infrarenal abdominal aortas of wild-type (WT) mice treated with elastase for 7 days along with mouse IgG or 6D7 injection (IgG, n=7; 6D7, n=7). Unpaired Student t test, *P<0.05.
FAM3D is Upregulated in ECs by Vascular Pathogenic Stimuli
As FAM3D was upregulated in aneurysmal tissues, we next explored the cellular origins of FAM3D in AAA development. First, the immunofluorescent staining displayed the upregulated FAM3D mainly located in intima but not in media at 7 days after elastase induction (Figure VIIIA in the online-only Data Supplement), indicating EC-derived FAM3D may play a critical role in AAA formation. Furthermore, en face immunofluorescent staining demonstrated that EC-colocalized FAM3D was upregulated at early stage (7 days) of elastase-induced AAA formation (Figure 4A). Because neutrophil-derived chemokines are also involved in neutrophil recruitment, we measured the expression of FAM3D in neutrophils as well. As a result, FAM3D was not expressed in mouse or human neutrophils with or without TNF-α treatment but was expressed in the colon (Figure VIIIB in the online-only Data Supplement). To further study the regulation of FAM3D expression, we measured the expression of FAM3D in ECs and VSMCs stimulated by TNF-α. Similarly, the expression of FAM3D did not exhibit a significant alteration in VSMCs (Figure VIIIC in the online-only Data Supplement). By contrast, ECs did express and secrete FAM3D. After TNF-α stimulation, FAM3D was markedly and dose-dependently upregulated at both the protein and mRNA levels, while secreted FAM3D was also increased in EC-conditioned medium (Figure 4B through 4D). Moreover, we detected FAM3D protein level in ECs treated with TNF-α (4 ng/mL) for various time points and demonstrated that TNF-α upregulated FAM3D expression in ECs as early as 4 hours of stimulation (Figure 4E). Similarly, other inflammatory stimuli, such as lipopolysaccharide and oscillatory shear stress (0.5±4 dyne/cm2), enhanced FAM3D expression in ECs compared with vehicle and pulsatile shear stress (12±4 dyne/cm2), respectively (Figure VIIID through VIIIF in the online-only Data Supplement).
Figure 4.
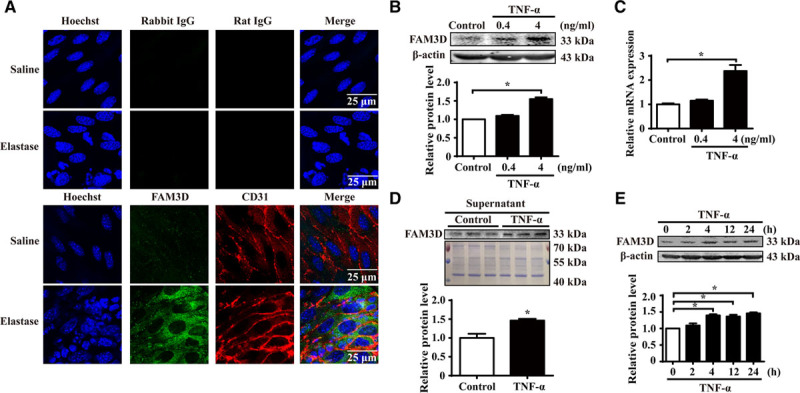
FAM3D (family with sequence similarity 3, member D) is expressed and upregulated in endothelial cells. A, Representative immunofluorescent en face staining of FAM3D expression in the endothelial layer of infrarenal abdominal aortas from 11- to 12-week-old male C57BL/6 mice 7 days after saline (n=6) or elastase (n=6) treatment. Rabbit IgG and rat IgG were used as the negative controls of FAM3D and CD31 antibodies, respectively. B, Representative Western blot analysis and quantification of FAM3D expression in human umbilical vein endothelial cells (HUVECs) treated with TNF-α (tumor necrosis factor α) for 24 hours. n=3. One-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05. C, Real-time polymerase chain reaction (PCR) analysis of FAM3D expression in HUVECs treated with TNF-α for 24 hours. β-Actin was applied as an internal control. n=6. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. D, Representative Western blot analysis and quantification of FAM3D in the supernatant of HUVECs stimulated by TNF-α (4 ng/mL) for 24 hours. Coomassie blue staining of the gel loaded with cell lysates of HUVECs was applied as the internal control. n=3. Unpaired Student t test, *P<0.05. E, Representative Western blot analysis and quantification of FAM3D expression in HUVECs treated with TNF-α (4 ng/mL) for 0, 2, 4, 12, and 24 hours. n=3. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05.
Endothelial Cell–Derived FAM3D Contributes to Neutrophil Recruitment
Neutrophil recruitment involves interaction between ECs and neutrophils, including neutrophil tethering, rolling, and adhesion to ECs, as well as neutrophil transendothelial migration (transmigration).28 Then, we verified whether EC-derived FAM3D mediates these processes of neutrophil recruitment. As shown by in vitro neutrophil and EC coculture experiments, the FAM3D-neutrolizing antibody 6D7 (20 μmol/L) significantly inhibited neutrophil adhesion to TNF-α-stimulated ECs (Figure 5A), indicating that EC-derived FAM3D was involved in neutrophil adhesion. Furthermore, we intraperitoneally injected TNF-α into WT and FAM3D−/− mice, respectively. Four hours later, as TNF-α sufficiently upregulated FAM3D expression in ECs during the period (Figure 4E), we observed the rolling and adhesion of leukocytes, most of which were neutrophils in 4 hours after TNF-α treatment,29 in mesentery microvessels by intravital microscopy. As demonstrated by images (Figure 5B) and videos (Movies I–IV in the online-only Data Supplement), intraperitoneal injection of TNF-α led to neutrophil rolling and adhesion, whereas FAM3D deficiency greatly inhibited TNF-α-induced neutrophil adhesion (28.75±5.020 in WT mice versus 7±0.9636 in FAM3D−/− mice; P<0.001) but not rolling in vivo. To further evaluate neutrophil transmigration, we applied a transwell system and found that FAM3D dose-dependently induced neutrophil transmigration across EC layers (Figure 5C).
Figure 5.
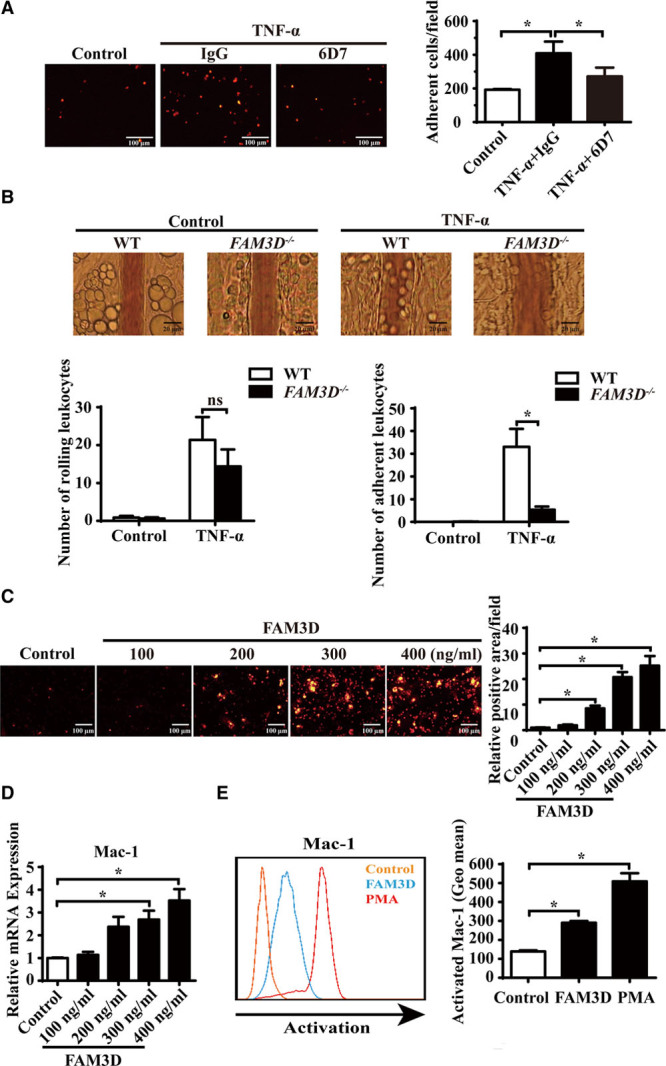
FAM3D (family with sequence similarity 3, member D) induces neutrophil adhesion and transmigration and activates Mac-1 (macrophage-1 antigen) in neutrophils. A, Representative photographs and quantification of the adhesion of DiI-labeled neutrophils to human umbilical vein endothelial cells (HUVECs) after coculture in the absence or presence of FAM3D neutralization antibody 6D7 (20 μmol/L) or an equal amount of mouse IgG for 2 hours. HUVEC monolayers were stimulated with 4 ng/mL TNF-α (tumor necrosis factor α) for 24 hours prior to coculture. n=12. One-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05. B, Representative intravital microscopic photographs and quantification of leukocyte rolling and adhesion to mesentery microvessels of wild-type (WT; n=8) and FAM3D−/− (n=8) mice 4 hours after intraperitoneal injection with or without TNF-α (500 ng for each mice). Two-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance. C, Representative photographs and quantification of DiI-stained mouse neutrophils transmigrated through a confluent layer of HUVECs in response to 4 hours of treatment with mouse FAM3D. n=12. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. D, Real-time polymerase chain reaction (PCR) quantification of mRNA levels of Mac-1 (CD11b) in mouse neutrophils stimulated by FAM3D for 4 hours. β-Actin was applied as an internal control. n=6. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. E, Representative flow cytometric analysis and quantification of activated Mac-1 (CD11b) induced by FAM3D (10 nmol/L) or phorbol-12-myristate-13-acetate (PMA; 10 nmol/L) in human neutrophils. n=6. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance.
FAM3D Specifically Activates Mac-1 in Neutrophils
Neutrophil tethering and rolling are dependent on the selectin ligands PSGL-1 (P-selectin glycoprotein ligand-1) and CD44, as well as L-selectin, while neutrophil adhesion and transmigration are mediated by integrins, such as VLA-4 and Mac-1.30 We therefore applied real-time PCR to examine whether FAM3D regulated the expression of these molecules. We found that FAM3D dose-dependently upregulated the mRNA expression of Mac-1 but not the expression of other molecules (Figure 5D; Figure IXA through IXD in the online-only Data Supplement). In addition to upregulated expression, the activation of the integrins VLA-4 and Mac-1 also contributes to the interaction between neutrophils and ECs and further mediates neutrophil recruitment.31,32 Consequently, we specifically measured activated VLA-4 and Mac-1 by flow cytometry. Consistent with the expression results, FAM3D activated Mac-1 but not VLA-4 (Figure 5E; Figure IXE in the online-only Data Supplement). In short, FAM3D specifically upregulated and activated Mac-1 in neutrophils.
FAM3D Induces Neutrophil Recruitment via FPR-Gi Protein/β-Arrestin-Mac-1 Signaling
FPR1 and FPR2 have been identified as the functional receptors of FAM3D.13 We next explored whether FPR1/FPR2 mediated FAM3D-induced Mac-1 activation and neutrophil adhesion. When neutrophils were preincubated with cyclosporin H (5 μmol/L) or WRW4 (5 μmol/L), which are antagonists of FPR1 and FPR2, respectively, FAM3D-induced Mac-1 activation and neutrophil adhesion were significantly inhibited. Meanwhile, the dual antagonist Boc-PLPLP (10 ng/mL), which inhibits both FPR1 and FPR2, blocked FAM3D-induced neutrophil activation and adhesion to ECs (Figure 6A; Figure XA and XB in the online-only Data Supplement). Furthermore, intraperitoneal injection of FAM3D induced neutrophil recruitment in peritoneal cavity, whereas the pretreatment of cyclosporin H or WRW4 markedly inhibited FAM3D-induced neutrophil recruitment in mice (Figure 6B). Thus, both FPR1 and FPR2 mediated FAM3D-induced Mac-1 activation and neutrophil adhesion and recruitment.
Figure 6.
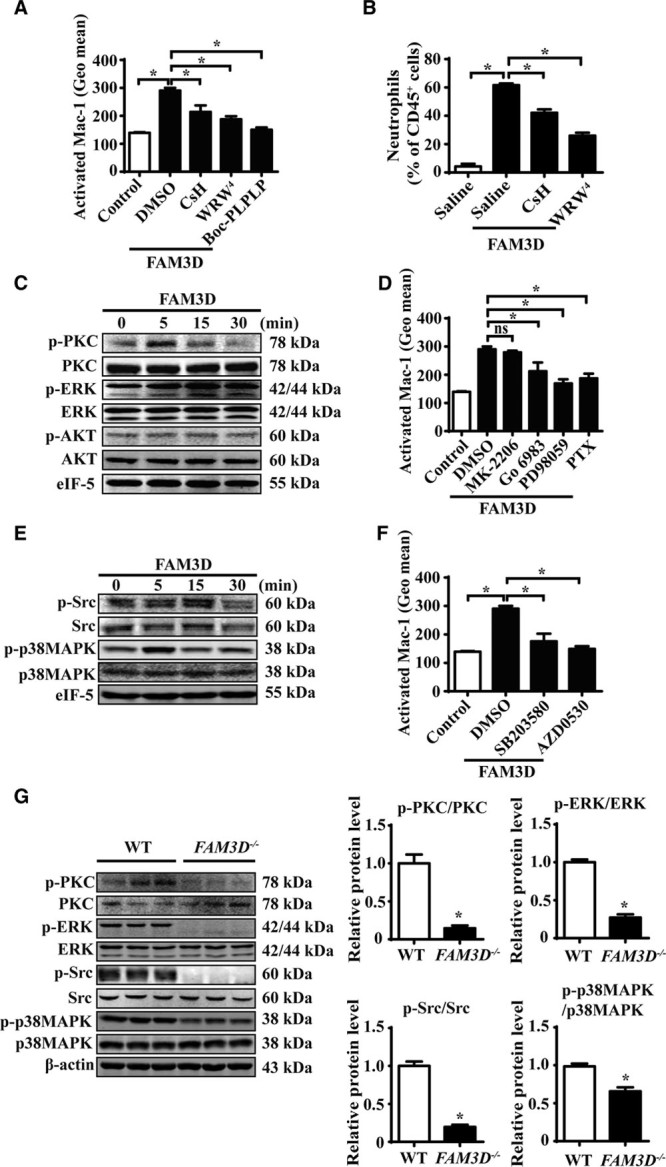
FAM3D (family with sequence similarity 3, member D) induces Mac-1 (macrophage-1 antigen) activation via FPR (formyl peptide receptor)-Gi protein/β-arrestin signaling. A, Flow cytometric quantification of activated Mac-1 (CD11b) in human neutrophils pretreated with 10 ng/mL of Boc-PLPLP, 5 μmol/L of cyclosporine H (CsH), or 5 μmol/L of WRW4 for 2 hours followed by FAM3D (400 ng/mL) treatment for another 15 minutes. n=6. One-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05. B, C57BL/6 mice (11–12 weeks old) underwent the intraperitoneal injection of CsH (10 μg/30 g body weight) or WRW4 (10 μg/30 g body weight) at 1 hour prior to FAM3D induction. Then FAM3D (10 μg/30 g body weight) was injected into peritoneal cavity for neutrophil recruitment in 6 hours. The percentage of neutrophils in CD45+ peritoneal cells was analyzed as CD45+CD11b+Ly6G+ by flow cytometry. n=7. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. C, Representative Western blot analysis of PKC (protein kinase C), ERK (extracellular regulated MAP kinase), and AKT (PKB, protein kinase B) activation in human neutrophils treated with 400 ng/mL FAM3D for various periods. D, Human neutrophils were pretreated with 20 μmol/L of MK-2206, 100 nmol/L of Go6983, 10 μmol/L of PD98059 or 10 ng/mL of PTX for 2 hours, followed by FAM3D (400 ng/mL) treatment for another 15 minutes, and the activation of Mac-1 (CD11b) was analyzed by flow cytometric assay. n=6. One-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons, *P<0.05, ns, no significance. E, Representative Western blot analysis of Src and p38MAPK (mitogen-activated kinase-like protein) activation in human neutrophils treated with 400 ng/mL FAM3D for various periods. F, Human neutrophils were pretreated with 10 μmol/L of SB203580 or 5 μmol/L of AZD0530, respectively, for 2 hours followed by FAM3D (400 ng/mL) treatment for another 15 minutes; the activation of Mac-1 was analyzed by flow cytometric assay. n=6. One-way ANOVA followed by Tukey’s test for multiple comparisons, *P<0.05. G, Representative Western blot analysis and quantification of PKC, ERK, Src, and p38MAPK in peripheral blood neutrophils isolated from wild-type (WT; n=6) and FAM3D−/− (n=6) mice treated with elastase for 7 days. Unpaired Student t test, *P<0.05. HUVECs indicates human umbilical vein endothelial cells; PTX, pertussis toxin; and TNF-α, tumor necrosis factor α.
FPR1 and FPR2 are the GPCRs (G-protein-coupled receptors that transduce Gi-protein- and β-arrestin-dependent signals.33 Gi-protein-dependent signals through neutrophil FPRs trigger the dissociation of the GPCR-specific Gi subunit from the shared Gβγ dimer and subsequently activate the PKC, AKT, and ERK signal transduction pathways and further facilitate neutrophil activation.34 On the other hand, FPR downstream of both β-arrestin-1- and β-arrestin-2-related signals mediate neutrophil chemotaxis and scaffold Src and p38MAPK.35–37 Although ERK1/2 is also reported as the downstream target of β-arrestin transduction, FPR-mediated ERK1/2 activation occurs through Gi protein but not dependent on β-arrestin-1/2.38 To measure Gi-protein-dependent signaling, we evaluated the phosphorylation of PKC, ERK1/2, and AKT in FAM3D-treated neutrophils by Western blotting (Figure 6C; Figure XC through XE in the online-only Data Supplement). We found that FAM3D transiently induced PKC phosphorylation at 5 minutes and persistently led to ERK1/2 phosphorylation from 5 minutes until 30 minutes. Nevertheless, FAM3D did not activate AKT in neutrophils. Furthermore, we separately applied specific inhibitors of Gi protein, PKC, ERK1/2, and AKT to evaluate Mac-1 activation. As shown in Figure 6D, PTX (pertussis toxin, a Gi protein inhibitor), Go6983 (a PKC inhibitor), and PD98059 (an ERK1/2 inhibitor), but not MK-2206 (an AKT inhibitor), blocked FAM3D-induced Mac-1 activation, indicating the involvement of PKC and ERK1/2 in Gi-protein-dependent signaling that mediates the effects of FAM3D in neutrophils. To further detect β-arrestin-dependent signaling of FPR1/FPR2, we measured Src and p38MAPK activation by Western blotting (Figure 6E; Figure XF and XG in the online-only Data Supplement). FAM3D transiently induced Src phosphorylation at 15 minutes, as well as p38MAPK activation at 5 minutes, in neutrophils. Moreover, both the p38MAPK inhibitor SB203580 and the Src inhibitor AZD0530 significantly blocked FAM3D-activated Mac-1, implying that FAM3D also activated p38MAPK- and Src-related β-arrestin-dependent signaling to induce Mac-1 activation (Figure 6F).
To further verify the occurrence of FAM3D-activated FPR1/FPR2 signaling in vivo, we compared both Gi-protein- and β-arrestin-dependent signals in neutrophils from elastase-treated WT and FAM3D−/− mice. As the process of EC-derived chemokine-activated neutrophil signaling and recruitment mainly happened in peripheral blood, we first isolated peripheral blood neutrophils and found that FAM3D deficiency inhibited Gi-protein-dependent PKC and ERK1/2 phosphorylation, as well as β-arrestin-dependent Src and p38MAPK activation (Figure 6G). Moreover, we also evaluated the signals of neutrophils in aortic wall by immunofluorescent staining. As the result, FAM3D deficiency significantly decreased the neutrophil infiltration, as well as PKC, ERK1/2, and p38MAPK phosphorylation (Figure XI in the online-only Data Supplement). Thus, FAM3D led to neutrophil recruitment via FPR1/2-Gi protein/β-arrestin-Mac-1 signaling during AAA formation.
Discussion
In the current study, we established that FAM3D, a novel chemokine, mediated the interaction between ECs and neutrophils by activating neutrophil Mac-1. Mechanistically, FAM3D, as an agonist for both FPR1 and FPR2, activated their downstream signaling cascades, including Gi-protein- and β-arrestin-related pathways (Figure 7). FAM3D would be a suitable therapeutic target, as FAM3D deficiency significantly attenuates neutrophil recruitment and ameliorates AAA development in vivo.
Figure 7.

Schematic illustration of Mac-1 (macrophage-1 antigen) activation induced by FAM3D (family with sequence similarity 3, member D). Pathological stimuli including TNF-α, LPS, and OSS promote the expression and secretion of endothelial cell (EC)–generated FAM3D. FAM3D activates Mac-1 on neutrophils through FPR (formyl peptide receptor) 1 and FPR2, dependent on Gi-protein- and β-arrestin-related signals. The activated Mac-1 interacts with ICAM-1 on the endothelial surface, mediating neutrophil recruitment and abdominal aortic aneurysm (AAA) development. ICAM-1 indicates intercellular adhesion molecule 1; LPS, lipopolysaccharide; OSS, oscillatory shear stress; and TNF-α, tumor necrosis factor α.
Previous studies indicated that neutrophils are the first leukocytes recruited into the aortic wall and may exhibit pathogenic effects in AAA development.5,6 Chemokine-mediated interaction between ECs and neutrophils facilitates neutrophil recruitment and exaggerates vascular inflammation.39 Although application of neutralizing antibodies against chemokine receptors CXCR1 and CXCR2 attenuates neutrophil recruitment and ameliorates acute aortic dissection rupture and elastase-induced AAA,9,24,40 whether corresponding chemokines, the ligands of these receptors, could be a therapeutic target for AAA is still a lack of evidence. Here, we identified the pathogenic role of FAM3D, a novel chemokine, in neutrophil recruitment and AAA formation, and we found that deficiency of FAM3D significantly inhibited the development of AAA in mouse models. FAM3D may serve as a potential therapeutic target for aortic aneurysmal diseases. CXCL1, CXCL2, and CXCL8 are the chemotactic agonists to neutrophil recruitment and are upregulated in aortic aneurysm/dissection. Likewise, these chemokines may also contribute to AAA formation, which should be confirmed by further investigation.
Neutrophil recruitment is a process of interaction between ECs and neutrophils consisting of tethering, rolling, adhesion, and transmigration. When expressed on neutrophils, the selectin ligands PSGL-1 and CD44, as well as L-selectin, interact with endothelial E-/P-selectin and PSGL-1, respectively, mediating neutrophil tethering and rolling on the EC layer.28 L-selectin−/− mice with impaired neutrophil tethering and rolling displayed attenuated aortic neutrophil recruitment and reduced aortic diameters after elastase induction compared with WT littermates.41 On the other hand, neutrophil-derived VLA-4 and Mac-1 specifically recognize VCAM-1 (vascular cell adhesion molecular 1) and ICAM-1 (intercellular adhesion molecule 1) on ECs, helping neutrophils adhere to ECs and further transmigrate into the subendothelial space.30 Here, we found that FAM3D induced neutrophil recruitment and aggravated AAA development by uniquely activating Mac-1-mediated neutrophil adhesion and transmigration but not PSGL-1-, CD44-, or L-selectin-related neutrophil rolling. Our study is in accordance with previous report that application of Mac-1 subunit CD18-blocking antibodies significantly decreased neutrophil adhesion and transmigration, thereby ameliorating AAA development in rats.42
Herein we also identified that FPR1/2 signals including Gi-protein- and β-arrestin-dependent pathways mediated FAM3D-induced Mac-1 activation and neutrophil adhesion/transmigration. FPRs are GPCRs that recognize classic chemoattractants derived from pathogens, damaged host tissues, and tumors. The human FPR family has 3 members, FPR1, FPR2, and FPR3, of which neutrophils mainly express the former 2 subtypes. Several FPR ligands have been verified including bacteria-derived N-formyl peptides as well as endogenous peptides/proteins produced by host. Among the endogenous ligands, some selectively ignite either FPR1 or FPR2.43 In contrast, annexin A1 interacts with both FPR1 and FPR2 and exhibits a protective role by resolving inflammation.44,45 In current study, we established that FAM3D-induced activation of Mac-1 was dependent on both FPR1 and FPR2 in neutrophils. Our observation was in line with previous report that FAM3D was a dual agonist of both FPR1 and FPR2.13 Of note, FPRs, a group of GPCRs coupled with Gi/o proteins, transduce both Gi-protein- and β-arrestin-related signals.34,46 Although some of their downstream signals have been reported to be involved in AAA formation,47,48 the role of FPRs in the pathogenesis of AAA is still elusive. In the current study, we additionally found that Gi-protein-dependent PKC and ERK and β-arrestin-dependent Src and p38MAPK synergistically mediated FAM3D-FPR-induced Mac-1 activation in neutrophils. Our study is consistent with previous reports that FPR downstream targets of Gi protein signaling, including PKC and ERK, are significantly activated in aneurysmal tissues.47,49 In particular, ERK deficiency attenuates elastase-induced AAA in mice.47 In addition, β-arrestin-2 contributes to Ang II–induced AAA formation in mice,48 and p38MAPK signaling, the downstream of β-arrestin, is also markedly activated after AAA formation.50
FAM3D deficiency repressed Ly6Chigh monocyte recruitment and macrophage infiltration in infrarenal abdominal aortas after elastase induction. As Mac-1 also plays a critical role in monocyte recruitment, as well as FPRs are markedly expressed in monocytes/macrophages,51,52 we evaluated the activation of Mac-1 in human monocytes after FAM3D treatment. Indeed, FAM3D could activate Mac-1 in monocytes as well (Figure XII in the online-only Data Supplement). However, FAM3D deficiency exhibited its inhibitive effects on neutrophil recruitment (3 days) earlier than the Ly6Chigh monocyte (7 days)/macrophage (14 days) infiltration. Because neutrophils are able to further regulate monocyte/macrophage accumulation at inflammatory sites, it was plausible that FAM3D deficiency–inhibited monocytes/macrophages recruitment may also partially depend on the inhibition of neutrophil infiltration.
Regarding to the cellular origins of FAM3D involved in AAA formation, we identified that FAM3D was mainly upregulated in ECs rather than VSMCs during the pathogenesis of AAA. As the same family members with FAM3D, the role of FAM3A-C in ECs still remain elusive, although FAM3A has been reported to be predominantly expressed in ECs.11 Hence, we detected the regulation of other FAM3 family members, including FAM3A-C. Interestingly, distinct with FAM3D, FAM3A-C mRNA levels were not regulated in ECs by TNFα stimulation (Figure XIII in the online-only Data Supplement). Thus, to explore the effects of FAM3A-C on regulation of EC function requires further investigation.
In conclusion, FAM3D, a dual agonist of FPR1 and FPR2, activates downstream signals, including Gi-protein- and β-arrestin-dependent pathways, and further activates Mac-1 in neutrophils. FAM3D deficiency attenuates neutrophil recruitment and rescues AAA development in vivo, thereby indicating that FAM3D may hold promise as a therapeutic target for AAA.
Sources of Funding
This research was supported by funding from the National Natural Science Foundation of the People’s Republic of China (NSFC; 81730010, 81670436, 81220108004, 91539203, 91739116, 31770940), the National Key R&D Program of China (2016YFC0903000), and the 111 Project of the Chinese Ministry of Education (B07001).
Disclosures
None.
Nonstandard Abbreviations and Acronyms
- AAA
- abdominal aortic aneurysm
- ECs
- endothelial cells
- ERK
- extracellular regulated MAP kinase
- FAM3D
- family with sequence similarity 3, member D
- FPR
- formyl peptide receptor
- GPCR
- G-protein-coupled receptors
- ICAM-1
- intercellular adhesion molecule 1
- Mac-1
- macrophage-1 antigen
- MAPK
- mitogen-activated kinase-like protein
- PKC
- protein kinase C
- PTX
- pertussis toxin
- TNF-α
- tumor necrosis factor α
- VCAM-1
- vascular cell adhesion molecular 1
- VLA-4
- very late antigen-4
- VSMC
- vascular smooth muscle cell
- WT
- wild-type
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.118.311289/-/DC1.
Highlights
FAM3D (family with sequence similarity 3, member D) activates Gi-protein- and β-arrestin-related FPR (formyl peptide receptor) signalings.
FAM3D activates Mac-1 (macrophage-1 antigen) in neutrophils through a mechanism dependent on FPR signaling.
FAM3D deficiency inhibits neutrophil recruitment and ameliorates abdominal aortic aneurysm development.
References
- 1.Johnston KW, Rutherford RB, Tilson MD, Shah DM, Hollier L, Stanley JC. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J Vasc Surg. 1991;13:452–458. doi: 10.1067/mva.1991.26737. [DOI] [PubMed] [Google Scholar]
- 2.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8:92–102. doi: 10.1038/nrcardio.2010.180. doi: 10.1038/nrcardio.2010.180. [DOI] [PubMed] [Google Scholar]
- 3.Rizas KD, Ippagunta N, Tilson MD., III Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol Rev. 2009;17:201–210. doi: 10.1097/CRD.0b013e3181b04698. doi: 10.1097/CRD.0b013e3181b04698. [DOI] [PubMed] [Google Scholar]
- 4.Buck DB, van Herwaarden JA, Schermerhorn ML, Moll FL. Endovascular treatment of abdominal aortic aneurysms. Nat Rev Cardiol. 2014;11:112–123. doi: 10.1038/nrcardio.2013.196. doi: 10.1038/nrcardio.2013.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 6.Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR., Jr Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112:232–240. doi: 10.1161/CIRCULATIONAHA.104.517391. doi: 10.1161/CIRCULATIONAHA.104.517391. [DOI] [PubMed] [Google Scholar]
- 7.Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014;10:593–619. doi: 10.1586/1744666X.2014.894886. doi: 10.1586/1744666X.2014.894886. [DOI] [PubMed] [Google Scholar]
- 8.Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest. 2013;93:768–778. doi: 10.1038/labinvest.2013.71. doi: 10.1038/labinvest.2013.71. [DOI] [PubMed] [Google Scholar]
- 9.Anzai A, Shimoda M, Endo J, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116:612–623. doi: 10.1161/CIRCRESAHA.116.304918. doi: 10.1161/CIRCRESAHA.116.304918. [DOI] [PubMed] [Google Scholar]
- 10.Shi GP. Role of cathepsin C in elastase-induced mouse abdominal aortic aneurysms. Future Cardiol. 2007;3:591–593. doi: 10.2217/14796678.3.6.591. doi: 10.2217/14796678.3.6.591. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Xu G, Patel A, et al. Cloning, expression, and initial characterization of a novel cytokine-like gene family. Genomics. 2002;80:144–150. doi: 10.1006/geno.2002.6816. [DOI] [PubMed] [Google Scholar]
- 12.de Wit NJ, IJssennagger N, Oosterink E, Keshtkar S, Hooiveld GJ, Mensink RP, Hammer S, Smit JW, Müller M, van der Meer R. Oit1/Fam3D, a gut-secreted protein displaying nutritional status-dependent regulation. J Nutr Biochem. 2012;23:1425–1433. doi: 10.1016/j.jnutbio.2011.09.003. doi: 10.1016/j.jnutbio.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Peng X, Xu E, Liang W, et al. Identification of FAM3D as a new endogenous chemotaxis agonist for the formyl peptide receptors. J Cell Sci. 2016;129:1831–1842. doi: 10.1242/jcs.183053. doi: 10.1242/jcs.183053. [DOI] [PubMed] [Google Scholar]
- 14.Alsiraj Y, Thatcher SE, Charnigo R, Chen K, Blalock E, Daugherty A, Cassis LA. Female mice with an XY sex chromosome complement develop severe angiotensin II-induced abdominal aortic aneurysms. Circulation. 2017;135:379–391. doi: 10.1161/CIRCULATIONAHA.116.023789. doi: 10.1161/CIRCULATIONAHA.116.023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilmink AB, Quick CR. Epidemiology and potential for prevention of abdominal aortic aneurysm. Br J Surg. 1998;85:155–162. doi: 10.1046/j.1365-2168.1998.00714.x. doi: 10.1046/j.1365-2168.1998.00714.x. [DOI] [PubMed] [Google Scholar]
- 16.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB Council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. doi: 10.1161/ATVBAHA.117.309524. doi: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Yu B, Liu Z, Li J, Ma M, Wang Y, Zhu M, Yin H, Wang X, Fu Y, Yu F, Wang X, Fang X, Sun J, Kong W. Homocysteine directly interacts and activates the angiotensin II type I receptor to aggravate vascular injury. Nat Commun. 2018;9:11. doi: 10.1038/s41467-017-02401-7. doi: 10.1038/s41467-017-02401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamanouchi D, Morgan S, Stair C, Seedial S, Lengfeld J, Kent KC, Liu B. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg. 2012;56:455–461. doi: 10.1016/j.jvs.2012.01.038. doi: 10.1016/j.jvs.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao G, Fu Y, Cai Z, Yu F, Gong Z, Dai R, Hu Y, Zeng L, Xu Q, Kong W. Unspliced XBP1 confers VSMC homeostasis and prevents aortic aneurysm formation via FoxO4 interaction. Circ Res. 2017;121:1331–1345. doi: 10.1161/CIRCRESAHA.117.311450. doi: 10.1161/CIRCRESAHA.117.311450. [DOI] [PubMed] [Google Scholar]
- 20.Satoh K, Nigro P, Matoba T, O’Dell MR, Cui Z, Shi X, Mohan A, Yan C, Abe J, Illig KA, Berk BC. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II-induced aortic aneurysms. Nat Med. 2009;15:649–656. doi: 10.1038/nm.1958. doi: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butcher MJ, Herre M, Ley K, Galkina E. Flow cytometry analysis of immune cells within murine aortas. J Vis Exp. 2011;53:pii: 2848. doi: 10.3791/2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng S, Li W, Xu M, Bai X, Zhou Z, Han J, Shyy JY, Wang X. Calcitonin gene-related peptide promotes angiogenesis via AMP-activated protein kinase. Am J Physiol Cell Physiol. 2010;299:C1485–C1492. doi: 10.1152/ajpcell.00173.2010. doi: 10.1152/ajpcell.00173.2010. [DOI] [PubMed] [Google Scholar]
- 23.Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. doi: 10.1038/nrcardio.2017.52. doi: 10.1038/nrcardio.2017.52. [DOI] [PubMed] [Google Scholar]
- 24.Pagano MB, Bartoli MA, Ennis TL, Mao D, Simmons PM, Thompson RW, Pham CT. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A. 2007;104:2855–2860. doi: 10.1073/pnas.0606091104. doi: 10.1073/pnas.0606091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wantha S, Alard JE, Megens RT, van der Does AM, Döring Y, Drechsler M, Pham CT, Wang MW, Wang JM, Gallo RL, von Hundelshausen P, Lindbom L, Hackeng T, Weber C, Soehnlein O. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. 2013;112:792–801. doi: 10.1161/CIRCRESAHA.112.300666. doi: 10.1161/CIRCRESAHA.112.300666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. 2009;114:4613–4623. doi: 10.1182/blood-2009-06-221630. doi: 10.1182/blood-2009-06-221630. [DOI] [PubMed] [Google Scholar]
- 27.Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, Pham CTN. Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler Thromb Vasc Biol. 2016;36:1660–1669. doi: 10.1161/ATVBAHA.116.307786. doi: 10.1161/ATVBAHA.116.307786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 29.Jung U, Norman KE, Scharffetter-Kochanek K, Beaudet AL, Ley K. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J Clin Invest. 1998;102:1526–1533. doi: 10.1172/JCI119893. doi: 10.1172/JCI119893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan SY, Shen Q, Rigor RR, Wu MH. Neutrophil transmigration, focal adhesion kinase and endothelial barrier function. Microvasc Res. 2012;83:82–88. doi: 10.1016/j.mvr.2011.06.015. doi: 10.1016/j.mvr.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herter JM, Margraf A, Volmering S, Correia BE, Bradshaw JM, Bisconte A, Hill RJ, Langrish CL, Lowell CA, Zarbock A. PRN473, an inhibitor of Bruton’s tyrosine kinase, inhibits neutrophil recruitment via inhibition of macrophage antigen-1 signalling. Br J Pharmacol. 2018;175:429–439. doi: 10.1111/bph.14090. doi: 10.1111/bph.14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luissint AC, Lutz PG, Calderwood DA, Couraud PO, Bourdoulous S. JAM-L-mediated leukocyte adhesion to endothelial cells is regulated in cis by alpha4beta1 integrin activation. J Cell Biol. 2008;183:1159–1173. doi: 10.1083/jcb.200805061. doi: 10.1083/jcb.200805061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17:638–650. doi: 10.1016/j.intimp.2013.06.034. doi: 10.1016/j.intimp.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selvatici R, Falzarano S, Mollica A, Spisani S. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur J Pharmacol. 2006;534:1–11. doi: 10.1016/j.ejphar.2006.01.034. doi: 10.1016/j.ejphar.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 35.Gera N, Swanson KD, Jin T. β-Arrestin 1-dependent regulation of Rap2 is required for fMLP-stimulated chemotaxis in neutrophil-like HL-60 cells. J Leukoc Biol. 2017;101:239–251. doi: 10.1189/jlb.2A1215-572R. doi: 10.1189/jlb.2A1215-572R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagener BM, Marjon NA, Prossnitz ER. Regulation of N-formyl peptide receptor signaling and trafficking by arrestin-Src kinase interaction. PLoS One. 2016;11:e0147442. doi: 10.1371/journal.pone.0147442. doi: 10.1371/journal.pone.0147442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Y, Zhang Y, Guo Y, Zhang Y, Xu M, He B. β2-Adrenoceptor involved in smoking-induced airway mucus hypersecretion through β-arrestin-dependent signaling. PLoS One. 2014;9:e97788. doi: 10.1371/journal.pone.0097788. doi: 10.1371/journal.pone.0097788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gripentrog JM, Miettinen HM. Formyl peptide receptor-mediated ERK1/2 activation occurs through G(i) and is not dependent on beta-arrestin1/2. Cell Signal. 2008;20:424–431. doi: 10.1016/j.cellsig.2007.11.002. doi: 10.1016/j.cellsig.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. 2011;17:1381–1390. doi: 10.1038/nm.2514. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houard X, Touat Z, Ollivier V, Louedec L, Philippe M, Sebbag U, Meilhac O, Rossignol P, Michel JB. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res. 2009;82:532–541. doi: 10.1093/cvr/cvp048. doi: 10.1093/cvr/cvp048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hannawa KK, Eliason JL, Woodrum DT, Pearce CG, Roelofs KJ, Grigoryants V, Eagleton MJ, Henke PK, Wakefield TW, Myers DD, Stanley JC, Upchurch GR., Jr L-selectin-mediated neutrophil recruitment in experimental rodent aneurysm formation. Circulation. 2005;112:241–247. doi: 10.1161/CIRCULATIONAHA.105.535625. doi: 10.1161/CIRCULATIONAHA.105.535625. [DOI] [PubMed] [Google Scholar]
- 42.Strindberg G, Ricci MA, Guibord RS, Mercier M, Nichols P, Bednar MM. CD-18 monoclonal antibody blocks the early events of aneurysm expansion. Ann N Y Acad Sci. 1996;800:266–267. doi: 10.1111/j.1749-6632.1996.tb33327.x. [DOI] [PubMed] [Google Scholar]
- 43.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drechsler M, de Jong R, Rossaint J, Viola JR, Leoni G, Wang JM, Grommes J, Hinkel R, Kupatt C, Weber C, Döring Y, Zarbock A, Soehnlein O. Annexin A1 counteracts chemokine-induced arterial myeloid cell recruitment. Circ Res. 2015;116:827–835. doi: 10.1161/CIRCRESAHA.116.305825. doi: 10.1161/CIRCRESAHA.116.305825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walther A, Riehemann K, Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5:831–840. doi: 10.1016/s1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- 46.Gabl M, Holdfeldt A, Sundqvist M, Lomei J, Dahlgren C, Forsman H. FPR2 signaling without β-arrestin recruitment alters the functional repertoire of neutrophils. Biochem Pharmacol. 2017;145:114–122. doi: 10.1016/j.bcp.2017.08.018. doi: 10.1016/j.bcp.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 47.Ghosh A, DiMusto PD, Ehrlichman LK, Sadiq O, McEvoy B, Futchko JS, Henke PK, Eliason JL, Upchurch GR., Jr The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J Am Coll Surg. 2012;215:668–680.e1. doi: 10.1016/j.jamcollsurg.2012.06.414. doi: 10.1016/j.jamcollsurg.2012.06.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trivedi DB, Loftin CD, Clark J, Myers P, DeGraff LM, Cheng J, Zeldin DC, Langenbach R. β-Arrestin-2 deficiency attenuates abdominal aortic aneurysm formation in mice. Circ Res. 2013;112:1219–1229. doi: 10.1161/CIRCRESAHA.112.280399. doi: 10.1161/CIRCRESAHA.112.280399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones JA, Stroud RE, Kaplan BS, Leone AM, Bavaria JE, Gorman JH, III, Gorman RC, Ikonomidis JS. Differential protein kinase C isoform abundance in ascending aortic aneurysms from patients with bicuspid versus tricuspid aortic valves. Circulation. 2007;116(11 s)(uppl):I144–I149. doi: 10.1161/CIRCULATIONAHA.106.681361. doi: 10.1161/CIRCULATIONAHA.106.681361. [DOI] [PubMed] [Google Scholar]
- 50.Martorell S, Hueso L, Gonzalez-Navarro H, Collado A, Sanz MJ, Piqueras L. Vitamin D receptor activation reduces angiotensin-II-induced dissecting abdominal aortic aneurysm in apolipoprotein E-knockout mice. Arterioscler Thromb Vasc Biol. 2016;36:1587–1597. doi: 10.1161/ATVBAHA.116.307530. doi: 10.1161/ATVBAHA.116.307530. [DOI] [PubMed] [Google Scholar]
- 51.El Zein N, D’Hondt S, Sariban E. Crosstalks between the receptors tyrosine kinase EGFR and TrkA and the GPCR, FPR, in human monocytes are essential for receptors-mediated cell activation. Cell Signal. 2010;22:1437–1447. doi: 10.1016/j.cellsig.2010.05.012. doi: 10.1016/j.cellsig.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Yang X, Chordia MD, Du X, Graves JL, Zhang Y, Park YS, Guo Y, Pan D, Cui Q. Targeting formyl peptide receptor 1 of activated macrophages to monitor inflammation of experimental osteoarthritis in rat. J Orthop Res. 2016;34:1529–1538. doi: 10.1002/jor.23148. doi: 10.1002/jor.23148. [DOI] [PubMed] [Google Scholar]