

Abstract
The EU-protected slug Geomalacus maculosus Allman occurs only in the West of Ireland and in northern Spain and Portugal. We explored the microbial community found within the faeces of Irish specimens with a view to determining whether a core microbiome existed among geographically isolated slugs which could give insight into the adaptations of G. maculosus to the available food resources within its habitat. Faecal samples of 30 wild specimens were collected throughout its Irish range and the V3 region of the bacterial 16S rRNA gene was sequenced using Illumina MiSeq. To investigate the influence of diet on the microbial composition, faecal samples were taken and sequenced from six laboratory reared slugs which were raised on two different foods. We found a widely diverse microbiome dominated by Enterobacteriales with three core OTUs shared between all specimens. While the reared specimens appeared clearly separated by diet in NMDS plots, no significant difference between the slugs fed on the two different diets was found. Our results indicate that while the majority of the faecal microbiome of G. maculosus is probably dependent on the microhabitat of the individual slugs, parts of it are likely selected for by the host.
Introduction
While the study of gut microbial communities is becoming increasingly popular, there is still a dearth of research focusing on those of wild animal populations1. This is despite the large influence that factors such as habitat and food availability are likely to have on the gut microbial composition. In fact, it has been shown that captive animals have a distinctly different gut microbiome than those from the wild2,3 which is hardly surprising, as a major mode of colonisation of the intestinal tract with microbes is through the environment4,5. Hence, the gut microbiome of a species should reflect, at least to an extent, the bacteria which can be found associated with the food or water it ingests in its habitat. Food availability within habitats is, among others, dependent on abiotic factors as well as seasonality and it has been shown that the composition of the gut microbiome of some animals differs between sites and season6–8. Additionally, geographical patterns of enteric microbial communities have been discovered in Galapagos iguanas with the microbiota being more distinct the further the islands are separated from each other9. While the authors suggest that the dominant drivers of the observed differentiation are host-bacterial interactions and differences in diet, historical and contemporary processes of ecological drift could also be a factor. In Drosophila, diet was found to have such a large effect on the gut microbiome that samples clustered by food rather than by host species10. Apart from habitat and diet specific microbes the gut harbours a “core microbiome”, members of which have likely co-evolved with their hosts and fulfil important functions including nutrient extraction such as cellulose degradation in termites11 or aid with the breakdown of toxins which have been ingested with the diet12,13. These bacteria are often specialized gut symbionts and are transmitted vertically from the eggs, through coprophagy or social interactions and it was found that gut communities of social insects were usually more distinctive and consistent than those of non-social invertebrates4. There are also indications that some species are deliberately choosing food items which contain byproducts of desirable bacteria to shape their own gut microbiota (e.g. Drosophila melanogaster14).
Studies of the gut and faecal microbiome of gastropods show that these contain microbes that possess cellulolytic activity15–17 and facilitate digestion of lignocellulose by the host16,17. This could account for the remarkable efficiency of terrestrial slugs and snails in breaking down plant fibre18,19. Cardoso et al.15 show that a change in diet causes a shift in the gut microbial community of Achatina fulica, similar to that observed in humans and other animals, and the authors suggest that the snail gut microbiota might be able to influence the energy balance equation and affect how much energy is extracted from the diet15. With only a handful of studies investigating the gut microbiome of slugs, more research is needed to determine the influence of environment and diet on the microbial community of these terrestrial molluscs. The first step is to complete a detailed inventory of the microroganisms associated with the gut of slug. To this end this study focuses on Geomalacus maculosus Allman, an EU protected slug species which is found only in the West of Ireland and the North of Iberia. Recent research has shown that the Irish population was probably introduced from Iberia sometime after the last glacial maximum (LGM) and that specimens from different locations within Ireland could not be distinguished using the mitochondrial markers 16 S rRNA and COI20. In Ireland G. maculosus inhabits deciduous and coniferous forests as well as a range of open habitats including blanket bogs and wet grasslands where it feeds on lichens, liverworts, bryophytes and fungi which it grazes from rocks or the bark of trees21–23. Hence its gut microbiota might be highly adapted to aid the digestion of non-vascular plants which are staples of its diet.
This study is the first to assess the diversity of bacteria found within faeces of the protected slug G. maculosus. We employed a two-pronged approach, utilising faecal samples from slugs that were collected from the wild as well as from laboratory hatched specimens to address our aims:
To determine whether the slug is a major selector of its microbiome or whether their gut microbes are more reflective of their environment, we collected faecal samples from slugs which were sampled from eleven different sites/seven different habitats. If the former is the case we would expect a substantial ‘core microbiome’ shared by all specimens, if the latter is the case, we hypothesize that the microbial signatures of slugs collected from the same site/habitat will be more similar than those collected from different sites/habitats.
To explore the impact of diet on the microbial community composition, we fed laboratory reared slugs from the same egg clutch on two different foods but under the same environmental conditions (e.g. substrate, moisture, temperature). We hypothesize that if diet was the major determinant of the gut microbiome composition, there would be a high degree of separation between the two groups and a high degree of similarity/shared phylotypes within them.
In the light of the protected status of the species, the identification of beneficial microbes could enable predictions about the adaptations of the slug to its habitat and thus help explain its limited distribution. This study also contributes to the further understanding of general invertebrate host-microbe interactions.
Results
Microbial diversity
Excluding the negative control, a total of 3,126 Operational Taxonomic Units (OTUs) belonging to 31 phyla and 76 associated classes of bacteria were observed within our samples. The most frequently observed phylum which was dominant in nearly all faecal samples was the Proteobacteria (73.1%), followed by Bacteroidetes (7.5%). All other phyla apart from Planctomycetes, Acidobacteria, Verrucomicrobia, Firmicutes and Actinobacteria had an abundance of less than 1%, 6.3% of OTUs were unassigned. The most abundant orders (>2.5%) were Enterobacteriales (41.2%), Rhodospirillales (10.9%), Rhizobiales (7.9%) Burkholderiales (5.3%), Sphingobacteriales (4.8%), Planctomycetales (3.6%) and Flavobacteriales (2.7%), however, their abundance between the faecal samples was found to be very variable (Fig. 1). The LCBD values which are shown for each sample (Fig. 1) are a comparative index of uniqueness with large values indicating the samples that have strongly different species compositions compared to the other ones, these include the negative control, most reared specimens and a range of other samples even from within one sample site (Fig. 1). An average of 277 OTUs (±92 standard deviation (SD)) were observed per sample. Many of these were low abundance OTUs: an average of 29.3% (±6.1 SD) were observed as singletons in a sample and an average of 16.6% (±2.7 SD) were observed as doubletons within a sample (Supplementary Table S1).
Figure 1.

Taxaplot showing all orders with >2.5% abundance within the faecal samples and the LCBD of each sample. They are sorted by sample site (see Fig. 4; L = fed on lichen, O = fed on oats, X = negative control) and grouped by habitat (ES = Exposed Siliceous).
The negative control contained 234 OTUs, the majority of which belonged to the Proteobacteria and Actinobacteria, both accounting for more than 40% of the sample (Fig. 1). The most abundant OTU was Lapillicoccus (Actinobacteria: Micrococcales; 20.2%), other abundant OTUs were Rubrobacter (Actinobacteria: Rubrobacterales; 12.4%) and Acinetobacter (γ-Proteobacteria: Pseudomonadales; 11.1%). The dominant OTUs of the negative control were found only in trace abundances in all other samples (≤0.1% of sequences per sample), and 44 of the 234 OTUs of the negative control were not found in any other sample.
Alpha and Beta Diversity
Significant differences in species richness, Pielou’s Evenness and Shannon’s Diversity Index were found between sites. Samples from Crookhaven and Glengarriff Woods had the highest species richness, which was significantly greater (at P < 0.05) than that at Glanteenassig Forest, Derreen Forest and Cloosh Forest as well as that from the reared specimens. The lowest species richness was recorded from the oat-fed reared specimens (significantly lower than samples collected from all sites except Ballycarbery, Lough Currane, Raferigeen and Derrycunnihy Woods) and from Raferigeen (significantly lower than samples collected from Ballaghbeama Gap and Derreen Forest). Samples from Ballaghbeama Gap had the highest evenness/Shannon Index which was significantly higher than that at Raferigeen, Cloosh Forest, Lough Currane and of the reared specimens. Lough Currane had the lowest evenness/Shannon index which was significantly lower than that at Ballaghbeama Gap and Derreen Forest, (Supplementary Fig. S1).
No clear separation of samples by either sample site or habitat could be observed in the NMDS plots (Supplementary Fig. S2). However, the spread of the samples from the conifer plantation habitat was consistently found to be less than those of the other habitats. No separation of clusters was observed by either the substrate from which the slugs were collected (rock or tree) or by the environment type (forest or open habitat) (Supplementary Fig. S2). While the PERMANOVA found groups to be significantly different when the faecal samples were grouped by sample site (R2 = 0.45, P = 0.004 (Bray-Curtis); R2 = 0.45, P = 0.001 (unweighted UniFrac) and R2 = 0.42, P = 0.03 (weighted UniFrac)), unequal variances could be (partially) responsible for the differences observed between the centroids of the groups which is supported by the absence of clearly separated groups in the NMDS plots (Supplementary Fig. S2).
A separation of the reared specimens samples by diet could be observed in the NMDS plots (not shown), however, the sample number was too small (N = 3 for each grouping) that the difference within the groups was not statistically significant despite relatively high R2 values (Bray-Curtis: R2 = 0.4, P = 0.1, unweighted UniFrac R2 = 0.36, P = 0.1, weighted UniFrac R2 = 0.4, P = 0.2). When displayed together on an NMDS plot, the reared specimens appeared separated from the wild specimens when using the Bray-Curtis (Fig. 2; R2 = 0.52, P = 0.001) and unweighted UniFrac (R2 = 0.49, P = 0.001) distances but not with the weighted UniFrac distances (R2 = 0.47, P = 0.001; Supplementary Fig. S2).
Figure 2.
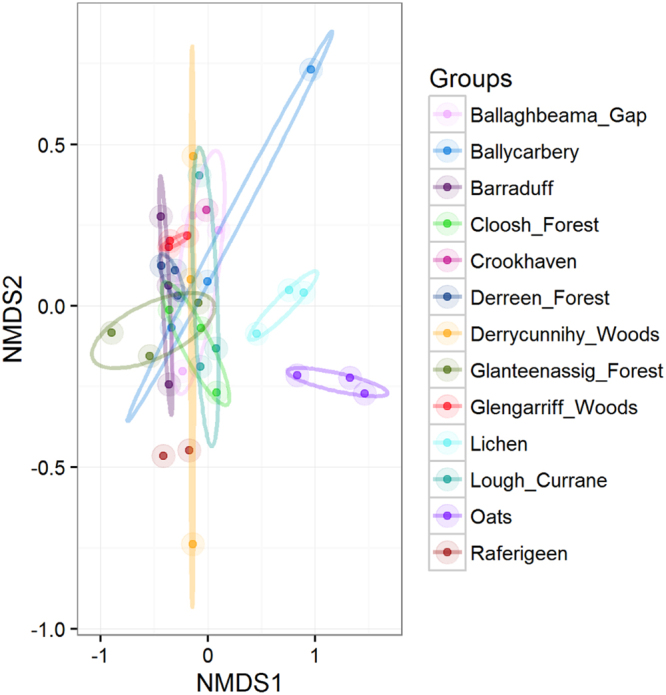
NMDS plot based on Bray-Curtis distances showing the reared and the wild specimens colour coded by sample site (R2 = 0.52, P = 0.001).
Core microbiome
Three OTUs (at ≥97% similarity) were found in all faecal samples of the wild and reared slugs, one was an unassigned short read (53 bp) and two belonged to the Enterobacteriaceae (γ-Proteobacteria: Enterobacteriales). One of these was further classified by using blastn and the NCBI database to Citrobacter freundii (100% similarity) the other one to Buttiauxella noackiae (99% similarity). They accounted for approximately 14% (B. noackiae) and 9% (C. freundii) of all sequences respectively and were the most abundant OTUs alongside Rahnella sp. (γ-Proteobacteria: Enterobacteriales 7.2%), Microvirga sp. (γ-Proteobacteria: Rhizobiales; 4.3%) and Acidiphilium sp. (α-Proteobacteria: Rhodospirillales; 3.4%), which occurred in 89, 94 and 89% of samples respectively. While these five OTUs were dominant in some of the samples, in other samples they accounted for less than 10% of all OTUs. The abundance of the two core OTUs within the samples ranged from <0.1% to 70% with a mean of 13.2% ± 17 SD (B. noackiae) and 8.3% ±16.3 SD (C. freundii) respectively. Apart from the three OTUs that were found in all samples, ten further OTUs were observed in at least 90% (=33 specimens) of samples (Fig. 3), with Microvirga sp. and an OTU from the family Comamonadaceae being significantly more abundant in the wild specimens (Padj < 0.001; Supplementary Fig. S3) and Enterobacter aerogenes UCI 45 being significantly more abundant in the reared specimens (Padj < 0.001; Supplementary Fig. S3). The primary source of the bacterial phylotypes from the NCBI database which were identified as matches to our sequences was soil (Supplementary Fig. S4), followed by sea and sea water, sediment and rhizosphere. In total, 245 biomes (e.g. forest), environmental features (e.g. plantation) or materials (e.g. soil) were assigned to the microbes, indicating that they occur ubiquitously in the environment.
Figure 3.

OTUs found in at least 90% of samples (33 of 36) in the order of overall abundance.
Discussion
The faecal samples of G. maculosus were found to harbour a diverse microbial community which differed greatly between individuals. In fact, 40% (1,220) of OTUs were found exclusively in single specimens. Interestingly, the highest number of unique OTUs was from the Crookhaven sample (107 OTUs) where only one slug was sampled indicating that certain OTUs might be associated with specific sites or individual slugs. However, even samples collected from slugs found at the same site were found to differ considerably. This could be explained by the rather sedentary nature of G. maculosus, which generally does not cover large distances within its habitat24,25. As the lichen and bryophyte species differ between habitats and even between trees and rocks within one site, so do the associated microbes which the slugs ingest from the environment through feeding. The observed differences between the faecal bacterial communities of specimens collected from the same site as well as the large amount of low abundance OTUs can hence likely be attributed to micro-structuring within the habitat of the slug.
The lower microbial species richness that we found in the faeces of the reared specimens when compared to the wild slugs has also been observed in a range of other studies26. This seems plausible, as they were brought up under more controlled conditions, however, further investigations into biotic and abiotic factors at the sample sites would be necessary to determine why microbial species richness might differ between them.
We found a great overlap of bacterial phylotypes and genera between our study and a range of other studies investigating the gut and/or faecal microbiome of terrestrial slugs16,17,27 and snails15,28–30, particularly among the Enterobacteriales (Supplementary Fig. S5). More than 40% of sequences that were found in the faecal samples of this study belonged to this order, including the two identified core OTUs. These bacteria are part of the gut flora of many animals as well as humans and are also frequently found in water and soil31. Genera that were observed in at least four of the eight compared studies (including this work) and could be considered part of a ‘typical’ terrestrial gastropod microbiome include Aeromonas (γ-Proteobacteria: Aeromonadales), Buttiauxella (γ-Proteobacteria: Enterobacteriales), Citrobacter (γ-Proteobacteria: Enterobacteriales), Kluyvera (γ-Proteobacteria: Enterobacteriales) and Pseudomonas (γ-Proteobacteria: Pseudomonadales) (Supplementary Fig. S5). Buttiauxella and Kluyvera are frequently isolated from slugs and snails32 and the authors even consider molluscs to be the natural source and ecological niche of these bacteria32. Additionally, several phylotypes belonging to these genera including A. hydrophilia, B. agrestis, C. freundii, K. intermedia and P. fluorescens have been linked with cellulolytic or xylanolytic and pectinolytic activity in the terrestrial slug Arion ater16,17 and in the silkworm Bombyx mori33. The likely importance of these bacteria for G. maculosus is clearly indicated by the presence of C. freundii and B. noackiae in all specimens which might serve a key role in the digestion of lichens and bryophytes.
As can be seen by the vast differences in the microbial community composition of our samples (Figs 1 and 3), the environment from which the slugs were collected has most certainly a major impact on the bacteria found in their faeces. However, as discussed above, our sample sites were too complex in structure and too large for a rather sedentary animal to detect site- or even habitat-specific microbial signatures. This is also obvious from the NMDS plots, where there was no clear separation between samples belonging to the same category (Supplementary Fig. S2). Interestingly, conifer plantation was the habitat which was clustering most closely together in the NMDS plots (Supplementary Fig. S2). This could be because conifer plantations in Ireland are predominantly monocultures with a lower species richness of lichens and bryophytes compared to semi-natural woodlands34,35 resulting in less diverse food sources for G. maculosus and hence a less variable microbial community.
Although a separation of the oat and lichen reared slugs was noticeable in the NMDS plots, the sample size was too small to determine whether this was statistically significanant. A total of 702 OTUs were observed in the faeces of the reared slugs, 25 (3.6%) of which were found in all six hatchlings. Out of 386 OTUs observed in the faeces of the oat-fed slugs, 60 (15.5%) were found in all three hatchlings, while 92 out of an observed 516 OTUs (17.8%) were shared by all lichen-fed slugs. This is surprisingly low, considering these slugs were from the same parent and were reared on identical substrate before the samples were taken. Due to the very small size of juvenile faecal pellets, we were forced to wait three weeks before we could collect enough faecal material for DNA extraction. This may have biased the end results, as the DNA extraction was from a composite sample. While the samples were not subjected to changing environmental conditions or shifts in temperatures, a recent study36 describes that facultatively aerobic and aerobic bacteria increase while anaerobic bacteria decrease within faecal samples over time, thus affecting the final proportions of taxa. A more precise way of describing the microbial communities found within hatchlings would be the dissection of the juvenile slugs and the examination of the bacteria associated with their gut rather than their faeces.
Three OTUs were shared between all reared and wild specimens and a further 13 were found in at least 90% of samples. This finding indicates that G. maculosus is a selector of at least part of its microbiome; an assumption which is further supported by the presence of several genera which have the proven ability to aid the digestion in slugs and are common gut bacteria in other terrestrial gastropods15,16,27–30. It was mentioned earlier that some Buttiauxella and Kluyvera strains might in fact be specifically adapted to live inside the gut of molluscs32. The ubiquitousness of Citrobacter in terrestrial gastropod guts could suggest a similar scenario for certain strains of this genus, however, many of the observed phylotypes (including Citrobacter) are rather commonly found in the environment and especially within soil and water, where they can be taken up through feeding. A vertical transfer of some bacteria, which is seen, in particular, among social insects that possess distinctive and consistent gut microbial communities4, could also be considered. While G. maculosus lacks parental care and sociality, a transmission of microbes could occur via the egg. Hatching G. maculosus slugs do not consume their eggs, even if these are left within the same container for a few days (pers. obs.), however, they do eat a tiny hole in their egg shell before emerging which might be sufficient for microbial transfer.
In conclusion we showed that the microbial communities found within the faecal samples of G. maculosus are highly variable even between slugs collected from the same site. We hypothesize that this reflects the significant influence of the microhabitat on the composition of the microbial gut community of G. maculosus. While diet may influence the gut microbiome of the reared specimens, a larger sample size and a different experimental design are required to further test this hypothesis. To determine the impact of local habitat and feed in shaping the gut microflora of G. maculosus the microbiome of local food sources should also be considered. The core microbiome consisted of three OTUs which were found within the faecal samples of all wild and reared slugs, at least two of which have likely beneficial functions for their slug host. As similar bacterial phylotypes, many of which have been linked with cellulolytic, xylanolytic or pectinolytic activity, were observed in several other gut microbiota studies on terrestrial molluscs, there is a possibility that these might be selected for by the hosts.
Methods
Sampling
Wild specimens
In June and July 2012, 50 G. maculosus specimens were collected under licence from eleven different locations (between 15 and 200 km apart) within Ireland (Fig. 4). Slugs were sampled from tree trunks or rocks from seven different habitats37: blanket bog, heath, exposed siliceous rock, wet grassland, deciduous woodland, mixed woodland and coniferous plantations. They were transferred into sterile petri dishes and observed until they defecated. Freshly collected faeces were transferred into sterile Eppendorf tubes which were initially stored in a mobile freezer compartment at −6 °C before being moved to a −80 °C freezer in the laboratory two days later.
Figure 4.

(a) The Irish distribution area of G. maculosus (shaded), the arrow indicates the localised population of the species in County Galway; (b) Sites sampled during this study, different habitats are encircled in a different colour. The number of faecal samples used in the following analyses from each site is given in brackets. The maps were generated using ArcGIS 10.2 http://resources.arcgis.com/en/help/install-guides/arcgis-server/10.2/; (a) was created by Dr Gesche Kinderman and modified with permission for this publication.
Laboratory reared specimens
In June 2014, a clutch of ten eggs was laid by a slug captured two weeks beforehand from the mixed woodland site in Glanteenassig (site A, Fig. 4). The eggs were removed from the parent slug and put into a petri dish containing moist tissue paper and kept at room temperature. After hatching, each slug was transferred into a single petri dish where four slugs were fed with porridge oats, while three slugs were fed with different lichens collected from Cloosh Forest (site T, Fig. 4); three eggs did not hatch. As faecal amounts of the juveniles were small, they were collected over a period of three weeks in the same manner as described above and immediately stored at −80 °C.
DNA extraction, PCR and sequencing
DNA was extracted from the faecal samples collected from the fifty sampled and six reared slugs using the PowerSoil DNA Isolation Kit (MoBIO, Carlsbad, CA, USA). The V3 region of the 16 S rRNA was amplified with the universal bacterial primers 341 F (5′-CTACGGGAGGCAGCAG-3′) and 518 R (5′-ATTACCGCGGCTGCTGG-3′) using the following conditions: two minutes initial denaturation at 98 °C followed by 30 cycles of 20 seconds at 98 °C, 30 seconds at 58 °C and 30 seconds at 72 °C. The final extension step was for five minutes at 72 °C. One µl of purified DNA was added to a 24 µl PCR mixture containing one unit of Q5 High-Fidelity DNA Polymerase (New England BioLabs, Ipswich, MA, USA) and 0.25 µM of each primer. Each sample was amplified three times and the combined PCR products were run on a 2% agarose gel and subsequently excised and gel extracted using the MinElute Gel Extraction Kit (QIAGEN, Hilden, Germany). As not all 50 samples amplified satisfactorily, the purified PCR products of 36 samples (30 from wild specimens, six from reared specimens) were sent to Research and Testing Laboratory, Texas, USA for sequencing on Illumina MiSeq (sample weight and DNA amount of PCR products are shown in Supplementary Table S2).
The contamination of samples with foreign DNA can pose a problem38, especially when working with low microbial biomass samples as in this study. Therefore, a negative control (blank extraction), followed by the same PCR protocol as that of the faecal samples, was also included for sequencing. Additionally, the risk of skewing our results was prevented by using the same extraction kit for all samples38.
Sequence analyses
Quality control and pairing
The paired-end reads were filtered and trimmed using Sickle v1.20039 by applying a sliding window approach and trimming regions with an average base quality less than 20. A 10 bp length threshold was subsequently applied to discard reads that fall below this length. BayesHammer40 from the Spades v2.5.0 assembler was used to error correct the paired-end reads followed by pandaseq v2.4 with a minimum overlap of 50 bp to assemble the forward and reverse reads into a single sequence. This approach was chosen as it has resulted in a reduction of substitution errors by 77–98% with an average of 93.2% for MiSeq datasets in a previous study41.
Construction of OTU table and phylogenetic tree
After obtaining the consensus sequences from each sample, the UPARSE v7.0.1001 pipeline (https://bitbucket.org/umerijaz/amplimock/src) was used for OTU construction. The reads were barcoded according to sample and pooled together before being dereplicated and sorted by decreasing abundance, singletons were discarded. They were then clustered based on 97% similarity discarding reads that were shorter than 32 bp. Chimeras were filtered using the “Gold” database (http://drive5.com/uchime/ uchime_download.html) that is derived from the ChimeraSlayer reference database in the Broad Microbiome Utilities (http://microbiomeutil.sourceforge.net/). To generate OTU tables for different samples, the original barcoded reads were matched against clean OTUs with 97% similarity. The representative OTUs were then taxonomically classified against the RDP database using the standalone RDP classifier v2.642 with the default–minWords option of 5. OTUs assigned to ‘Chloroplast’, ‘Mitochondria’ and ‘Eukaryota’ were filtered from the OTU table prior to further analysis. To obtain the phylogenetic distances between OTUs, they were multisequence aligned against each other using mafft v7.04043. FastTree v2.1.744 was used on these alignments to generate an approximately-maximum-likelihood phylogenetic tree.
Statistical analysis
All statistical analyses were performed in R45. Alpha diversity (Pielou’s evenness, species richness and Shannon index) was calculated using the package vegan46, the aov function was used to calculate pairwise ANOVA p-values which were then drawn on top of the alpha diversity figures (see Fig. 1 and Supplementary Fig. S1). Beta diversity was calculated using the packages vegan (Bray-Curtis) and phyloseq.47 (weighted and unweighted UniFrac48). While Bray-Curtis considers the species abundance count, UniFrac also includes the phylogenetic distance between the branch lengths of OTUs observed in different samples. Weighted UniFrac accounts for the abundance of OTUs and unweighted UniFrac considers their presence or absence. To visualise the similarity of the samples, vegan’s metaMDS function was used to produce a non-metric multidimensional scaling (NMDS) plot of community data (OTUs at 3% divergence) based on the Bray-Curtis, weighted and unweighted UniFrac distances. The samples were grouped for different metadata categories (sample site, habitat, substrate and environment type) and standard deviations of the (weighted) averages were drawn as ellipses onto the plot using vegan’s ordiellipse function (see Supplementary Fig. S2).
To test whether the centroids and dispersion of the groups differ, vegan’s adonis function was used for a Permutational Multivariate Analysis of Variance (PERMANOVA) by partitioning distance matrices among sources of variation (both qualitative and quantitative information). This function fits linear models (e.g., factors, polynomial regression) to distance matrices and uses a permutation test with pseudo-F ratios.
To find OTUs that are significantly different between metadata categories, the function DESeqDataSetFromMatrix() from the DESeq2 package49 was used, with a significance value cut-off of P < 0.001. This function allows negative binomial GLM fitting (as abundance data from metagenomic sequencing is overdispersed) and Wald statistics for abundance data. After correcting for multiple comparisons, it reports OTUs that have log-fold changes between selected metadata categories, in this case between wild and reared specimens (see Supplementary Fig. S3).
We performed Local Contribution to Beta Diversity (LCBD) analysis50 by using Hellinger transformation to compute the total sum of squares of the species composition for all samples from which the sample-wise local contributions to beta diversity could be derived as a proportion of the total beta diversity. These values were then plotted as bubbles under stacked bar plots to indicate samples that differ markedly in their species composition (see Fig. 1).
Seqenv51 was used to search OTU sequences against the blastn nucleotide database (NCBI), and the textual information on isolation sources of the reference genomes for the OTUs were collated on which we ran a text mining algorithm to identify and parse words associated with the Environmental Ontology (EnvO). The normalized frequencies of EnvO terms for each OTU were then multiplied with the sequence counts from the OTU table to generate a sample-wise EnvO abundance table on which we performed the same differential analysis as we did for OTUs (see Supplementary Fig. S4).
Statistical scripts, workflows, and software used in this manuscript can be found at http://userweb.eng.gla.ac.uk/umer.ijaz#bioinformatics.
Data availability
Sequencing results are available in the Sequence Read Archive (SRA) database at NCBI under BioProject ID PRJNA386253, accession number SUB2570565.
Electronic supplementary material
Acknowledgements
We are grateful to Tim Collins for help with collecting G. maculosus faeces and to Erin Johnston for supplying the slug eggs. Many thanks to Aoife Duff, Ciara Keating, Agata Lisik and Enrico Tatti for advice on laboratory work. We thank the Wildlife Licensing Unit of the National Park and Wildlife Service for supplying us with license number C145/2011. This study was partly funded by a School of Natural Sciences Project Award from the National University of Ireland Galway and by NERC NE/L011956/1.
Author Contributions
I.R., M.G. and C.J.S. designed the study; I.R. carried out the sampling and laboratory work and drafted the manuscript; I.R. and U.Z.I. analysed the data; C.J.S., M.G. and U.Z.I. provided critical revisions of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-28720-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Inga Reich, Email: reichi@oregonstate.edu.
Umer Zeeshan Ijaz, Email: Umer.Ijaz@glasgow.ac.uk.
Cindy J. Smith, Email: Cindy.Smith@glasgow.ac.uk
References
- 1.Amato KR. Co-evolution in context: The importance of studying gut microbiomes in wild animals. Microbiome Science and Medicine. 2013;1:10–29. doi: 10.2478/micsm-2013-0002. [DOI] [Google Scholar]
- 2.Nakamura N, et al. Analysis of the hydrogenotrophic microbiota of wild and captive black howler monkeys (Alouattapigra) in Palenque National Park, Mexico. Am. J. Primatol. 2011;73:909–919. doi: 10.1002/ajp.20961. [DOI] [PubMed] [Google Scholar]
- 3.Nelson TM, Rogers TL, Carlini AR, Brown MV. Diet and phylogeny shape the gut microbiota of Antarctic seals: A comparison of wild and captive animals. Environ. Microbiol. 2012;15:1132–1145. doi: 10.1111/1462-2920.12022. [DOI] [PubMed] [Google Scholar]
- 4.Engel P, Moran NA. The gut microbiota of insects – diversity in structure and function. FEMS Microbiol. Rev. 2016;37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 5.Newton ILG, Sheehan KB, Lee FJ, Horton MA, Hicks RD. Invertebrate systems for hypothesis-driven microbiome research. Microbiome Science and Medicine. 2013;1:1–9. doi: 10.2478/micsm-2013-0001. [DOI] [Google Scholar]
- 6.Kobayashi Y, Koike S, Miyaji M, Hata H, Tanaka K. Hindgut microbes, fermentation and their seasonal variations in Hokkaido native horses compared to light horses. Ecol. Res. 2016;21:285–291. doi: 10.1007/s11284-005-0118-x. [DOI] [Google Scholar]
- 7.King GM, Judd C, Kuske CR, Smith C. Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from coastal Louisiana, USA. PLoS One. 2012;7:e51475. doi: 10.1371/journal.pone.0051475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One. 2012;7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lankau EW, Hong PY, Mackie RI. Ecological drift and local exposures drive enteric bacterial community differences within species of Galapagos iguanas. Mol. Ecol. 2012;21:1779–1788. doi: 10.1111/j.1365-294X.2012.05502.x. [DOI] [PubMed] [Google Scholar]
- 10.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 2011;7:e1002272. doi: 10.1371/journal.pgen.1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 12.Ping L, et al. A novel Dps-type protein from insect gut bacteria catalyses hydrolysis and synthesis of N-acyl amino acids. Environ. Microbiol. 2007;9:1572–1583. doi: 10.1111/j.1462-2920.2007.01279.x. [DOI] [PubMed] [Google Scholar]
- 13.Kikuchi Y, et al. T. Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. USA. 2012;10:8618–8622. doi: 10.1073/pnas.1200231109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broderick NA, Lemaitre B. Gut-associated microbes of Drosophila melanogaster. Gut Microbes. 2012;3:307–321. doi: 10.4161/gmic.19896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardoso AM, et al. Gut bacterial communities in the giant land snail Achatina fulica and their modification by sugarcane-based diet. PLoS One. 2012;7:e33440. doi: 10.1371/journal.pone.0033440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joynson R, Swamy A, Bou PA, Chapuis A, Ferry N. Characterization of cellulolytic activity in the gut of the terrestrial land slug Arion ater: Biochemical identification of targets for intensive study. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014;177–178:29–35. doi: 10.1016/j.cbpb.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Joynson, R., Pritchard, L., Osemwekha, E. & Ferry, N. Metagenomic analysis of the gut microbiome of the common black slug Arion ater in search of novel lignocellulose degrading enzymes. Front. Microbiol. 8, 10.3389/fmicb.2017.02181 (2017). [DOI] [PMC free article] [PubMed]
- 18.Davidson DH. Assimilation efficiencies of slugs on different food materials. Oecologia. 1976;26:267–273. doi: 10.1007/BF00345295. [DOI] [PubMed] [Google Scholar]
- 19.Charrier M, Daguzan J. Food consumption: production and energy budget in Helix aspersa Müller (Gastropoda Pulmonata) Ann. Nutr. Aliment. 1980;34:147–166. [PubMed] [Google Scholar]
- 20.Reich I, et al. Genetic study reveals close link between Irish and Northern Spanish specimens of the protected Lusitanian slug Geomalacus maculosus. Biol. J. Linn. Soc. Lond. 2015;116:156–168. doi: 10.1111/bij.12568. [DOI] [Google Scholar]
- 21.Reich, I., O’Meara, K., Mc Donnell R.J. & Gormally, M.J. An assessment of the use of conifer plantations by the Kerry Slug (Geomalacus maculosus) with reference to the impact of forestry operations. Irish Wildlife Manuals, No. 64. National Parks and Wildlife Service, Department of Arts, Heritage and the Gaeltacht, Dublin, https://www.npws.ie/sites/default/files/publications/pdf/IWM64.pdf (2012).
- 22.Platts EA, Speight MCD. The taxonomy and distribution of the Kerry Slug Geomalacus maculosus Allman, 1843 (Mollusca: Arionidae) with a discussion of its status as a threatened species. Ir. Nat. J. 1988;22:417–430. [Google Scholar]
- 23.Mc Donnell R, O’Meara K, Nelson B, Marnell F, Gormally M. Revised distribution and habitat associations for the protected slug Geomalacus maculosus (Gastropoda, Arionidae) in Ireland. Basteria. 2013;77:33–37. [Google Scholar]
- 24.Reich I, Mc Donnell RJ, Mc Inerney C, Callanan S, Gormally MJ. EU-protected slug Geomalacus maculosus and sympatric Lehmannia marginata in conifer plantations: what does mark-recapture method reveal about population densities? J. Moll. Stud. 2017;83:27–35. doi: 10.1093/mollus/eyw039. [DOI] [Google Scholar]
- 25.Mc Donnell, R. J. & Gormally, M. J. Distribution and Population Dynamics of the Kerry Slug, Geomalacus maculosus (Arionidae). Irish Wildlife Manual No. 54. National Parks and Wildlife Service, Department of Arts, Heritage and the Gaeltacht, Dublin, https://www.npws.ie/sites/default/files/publications/pdf/IWM%2054.pdf (2011).
- 26.Bahrndorff S, Alemu T, Alemneh T, Nielsen JL. The microbiome of animals: Implications for conservation biology. Int. J. Genomics. 2016;2016:5304028. doi: 10.1155/2016/5304028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson, P. G. Characterisation of the bacterial flora associated with the grey field slug Deroceras reticulatum and assessment of its suitability as a target for biological control. Edinburgh Research Archive, https://www.era.lib.ed.ac.uk/handle/1842/5276 (2010).
- 28.Charrier M, Fonty G, Gaillard-Martinie B, Ainouche K, Andant G. Isolation and characterization of cultivable fermentative bacteria from the intestine of two edible snails, Helix pomatia and Cornu aspersum (Gastropoda: Pulmonata) Biol. Res. 2006;39:669–681. doi: 10.4067/S0716-97602006000500010. [DOI] [PubMed] [Google Scholar]
- 29.Stalder GL, Lonaric I, Walzer C. Diversity of Enterobacteria including β-lactamase producing isolates associated with the Spanish slug (Arion vulgaris) Sci. Total Environ. 2014;479–80:11–16. doi: 10.1016/j.scitotenv.2014.01.103. [DOI] [PubMed] [Google Scholar]
- 30.Rossing, J. S. & Hietpas, N. J. Isolation of anaerobic microbiota of slug intestinal tract. UW-L Journal of Undergraduate Research Xhttps://www.uwlax.edu/urc/jur-online/PDF/2007/rossing-hietpas.pdf (2007).
- 31.Brenner, D. & Farmer, J. Enterobacteriaceae in Bergey’s Manual of Systematics of Archaea and Bacteria (ed. Whitman, W. B.) 1–24 (John Wiley and Sons, 2015).
- 32.Müller HE, Brenner DJ, Fanning GR, Grimont PAD, Kämpfer P. Emended description of Buttiauxella agrestis with recognition of six new species of Buttiauxella and two new species of Kluyvera: Buttiauxella ferragutiae sp. nov., Buttiauxella gaviniae sp. nov., Buttiauxella brennerae sp. nov., Buttiauxella izardii sp. nov., Buttiauxella noackiae sp. nov., Buttiauxella warmboldiae sp. nov., Kluyvera cochleae sp. nov. and Kluyvera georgiana sp. nov. Int. J. Syst. Bacteriol. 1996;46:50–63. doi: 10.1099/00207713-46-1-50. [DOI] [PubMed] [Google Scholar]
- 33.Anand AAP, et al. Isolation and characterization of bacteria from the gut of Bombyx mori that degrade cellulose, xylan, pectin and starch and their impact on digestion. J. Insect Sci. 2010;10:107. doi: 10.1673/031.010.10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey JW, Davey S, Peace AJ, Ferris R, Harding K. Lichens and bryophyte communities of planted and semi-natural forests in Britain: the influence of site type, stand structure and deadwood. Biol. Conserv. 2002;107:165–180. doi: 10.1016/S0006-3207(02)00057-5. [DOI] [Google Scholar]
- 35.Coote L, et al. Epiphytes of Sitka spruce (Picea sitchensis) plantations in Ireland and the effects of open spaces. Biodivers. Conserv. 2007;16:4009–4024. doi: 10.1007/s10531-007-9203-5. [DOI] [Google Scholar]
- 36.Menke S, Meier M, Sommer S. Shifts in the gut microbiome observed in wildlife faecal samples exposed to natural weather conditions: lessons from time-series analyses using next-generation sequencing for application in field studies. Methods Ecol. Evol. 2015;6:1080–1087. doi: 10.1111/2041-210X.12394. [DOI] [Google Scholar]
- 37.Fossitt, J. A. A Guide To Habitats In Ireland. (The Heritage Council of Ireland, Dublin, 2000).
- 38.Salter SJ, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87–87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi, N. A. & Fass, J. N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files. Version 1.21: Available at https://github.com/najoshi/sickle (2011).
- 40.Nikolenko SI, Korobeynikov AI, Alekseyev MA. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14:S7. doi: 10.1186/1471-2164-14-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schirmer M, et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015;43:e37–e37. doi: 10.1093/nar/gku1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price MN, Dehal PS, Arkin AP. FastTree 2 - Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2013)
- 46.Oksanen, J. et al. vegan: Community Ecology Package, R Package version 2.2-1, http://CRAN.R-project.org/package=vegan (2015).
- 47.Mc Murdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Legendre P, De Cáceres M. Beta diversity as the variance of community data: dissimilarity coefficients and partitioning. Ecol. Lett. 2013;16:951–963. doi: 10.1111/ele.12141. [DOI] [PubMed] [Google Scholar]
- 51.Sinclair L, et al. Seqenv: linking microbes to environments through text mining. PeerJ Preprints. 2016;4:e2317v1. doi: 10.7717/peerj.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing results are available in the Sequence Read Archive (SRA) database at NCBI under BioProject ID PRJNA386253, accession number SUB2570565.