

Abstract
We report two female patients with focal segmental glomerulosclerosis and chronic kidney disease. The first patient was found to have a heterozygous, de novo, pathogenic variant in COL4A5 (c.141+1G>A, IVS2+1G>A), which is associated with Alport syndrome. The second patient was found to have a heterozygous, likely pathogenic variant in COL4A4 (c.2842G>T). Both these variants in COL4A5 and COL4A4 are novel, and they were detected using whole exome sequencing and gene panel testing, respectively. Additionally, we discuss the complexities of diagnosis in such cases and the benefits of using the abovementioned diagnostic approaches.
Kidney dysfunction: Identifying mutations linked with focal segmental glomerulosclerosis
Researchers in the USA have identified two mutations linked with unrelated cases of the kidney disease focal segmental glomerulosclerosis (FSGS) and compared different diagnostic techniques. This disease is involved in roughly 40% of adult kidney dysfunction. Using sequencing of all protein-coding genes, Paldeep Atwal’s team at the Mayo Clinic, Jacksonville, identified a novel mutation in an FSGS patient. The mutation was not inherited, indicating that the patient’s siblings are not at increased risk. There were also mutations in other genes which may account for the patient’s vision loss. The team tested a second FSGS patient by screening kidney-related genes specifically, revealing a novel mutation in a different gene. Although the targeted approach is cheaper and quicker, these findings highlight the diagnostic benefits of full sequencing, which can reveal relevant mutations in unexpected genes.
Introduction
Focal segmental glomerulosclerosis (FSGS) is a progressive kidney disease that can be either a primary renal disorder or secondary due to other etiologies, including genetic etiologies1. It is the culprit of approximately 40% of nephrotic syndrome cases in adults and 20% in children2, 3. As the name suggests, FSGS is characterized by focal [less than 50% of glomeruli are affected in light microscopy (LM)] and segmental [less than 50% of a glomerular tuft is affected] glomerular sclerosis and by varying degrees of foot process effacement2. Pathogenic variants in genes that are specific to the function and structure of podocytes, such as TRPC6, ACTN4, WT1, CD2AP, INF2, NPHS2, and PLCE1, have been reported to cause familial FSGS4. Pathogenic variants in collagen type IV genes (COL4A3, COL4A4, and COL4A5) are related to a spectrum of disorders with heterogeneous clinical manifestations.
Alport syndrome (AS), which is caused by glomerular membrane defects, has X-linked and autosomal dominant/recessive modes of inheritance. Classical AS has X-linked inheritance due to pathogenic variants in COL4A5 (86% of cases) and usually presents in early childhood with microscopic or gross hematuria, which progresses to end stage renal disease5.The remaining 15% of AS has autosomal inheritance and is thought to be due to pathogenic variants in the COL4A3 and COL4A4 genes. Thin basement membrane nephropathy (TBMN), which is also characterized by glomerular membrane abnormalities, has been linked to COL4A3 and COL4A4 as well6. Renal disorders causing glomerular basement membrane structural abnormalities are known collectively as collagen IV nephropathies (COL4Ns)7. FSGS typically presents with proteinuria, often in the nephrotic range, which progresses over months to years to a reduction in glomerular filtration rate (GFR). COL4A3 and COL4A4, which are located on chromosome 2, have been associated with FSGS8. Herein, we present two families with novel COL4A4 and COL4A5 variants diagnosed by massively parallel exome sequencing and discuss the complexities of managing these genetic results.
Materials and methods
To identify the molecular bases of the patients’ phenotypes, whole exome sequencing was utilized in the first patient, while the second proband was tested with a nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel.
Results
The exome sequencing revealed a heterozygous, de novo, pathogenic variant in COL4A5 (c.141+1G>A, IVS2+1G>A), a heterozygous, paternally inherited, pathogenic variant in ABCA4 (c.2588G>C, p.G863A) and a heterozygous, maternally inherited, likely pathogenic variant in ABCA4 (c.203C>G, p.P68R). The nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel detected a heterozygous, likely pathogenic variant in COL4A4 (c.2842G>T).
Family 1
Our first proband is a 52-year-old female with stage IV chronic kidney disease (estimated GFR of 28.2 mL/min), microalbuminuria (microalbumin/creatinine ratio of 206 mg/g) and microscopic hematuria. Her kidney disease first started at age 7, when she presented with microscopic hematuria, which was thought to be benign. At that point in time, her kidney function was normal, and she subsequently did well until age 34, when she developed proteinuria (3 grams of protein/24-h urine), hypertension and creatinine clearance of 45 mL/min. She underwent a renal biopsy, which showed FSGS with no foot process effacement, suggestive of a secondary form. She was started on 30 mg prednisone daily, which resulted in remission of her proteinuria. A renal ultrasound scan showed multiple bilateral renal cysts (Fig. 1), though no family history of ADPKD was found. However, her son was also diagnosed with microscopic hematuria at age 7. In addition to the kidney disease, the patient has chronic arthritis, for which she takes low-dose prednisone; hypermobile joints; migraine headaches; and macular degeneration, which was diagnosed at age 40. She underwent a brain MRI to evaluate the headaches, and the result was normal except for a mild Chiari malformation. A genetic etiology to her kidney disease was suspected, and samples from the proband’s mother, father and affected son were submitted for variant segregation analysis by whole exome sequencing (WES). The patient was found to have a heterozygous, de novo, pathogenic variant in COL4A5 (c.141+1G>A, IVS2+1G>A), which is associated with Alport syndrome. As it is de novo, her siblings are unlikely to be at risk for the disorder, although germline mosaicism cannot be excluded. Her son (18 years of age) carries this variant, giving him the genetic diagnosis of Alport syndrome as well. Moreover, the patient was found to be a compound heterozygote for ABCA4 variants. She has a heterozygous, paternally inherited, pathogenic variant in ABCA4 (c.2588G>C, p.G863A) and a heterozygous, maternally inherited, likely pathogenic variant in ABCA4 (c.203 C >G, p.P68R). These variants likely are the cause for the patient’s macular degeneration and vision loss.
Fig. 1.
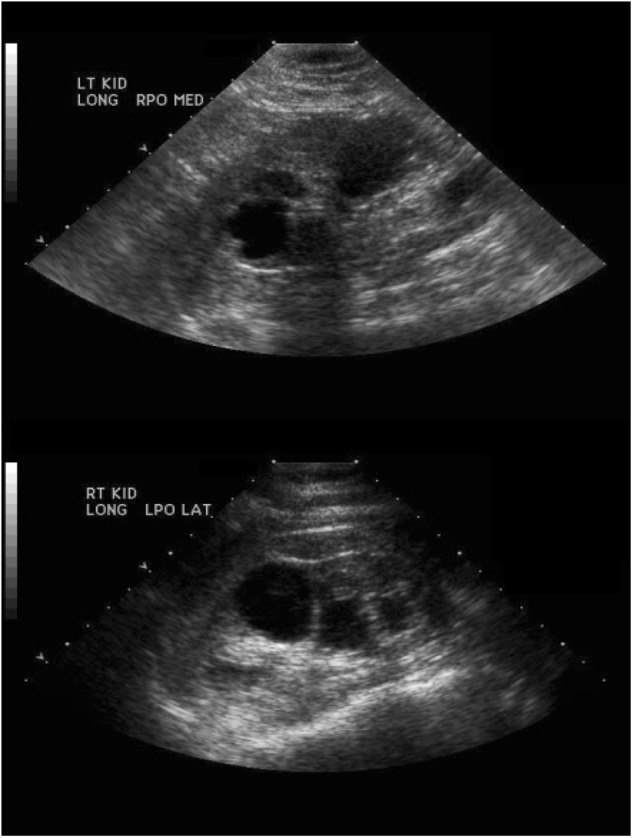
Renal ultrasound showing multiple bilateral cysts
Family 2
Our second proband is a 48-year-old female with stage IV chronic kidney disease (estimated GFR of 22.5 mL/min), nephrotic syndrome (6 g of protein/24-h urine) and hypertension. She underwent a renal biopsy, which revealed FSGS with patchy moderate tubular atrophy, interstitial fibrosis and mild chronic inflammation. She was initially started on prednisone but did not tolerate it due to excessive weight gain of up to 60 pounds and dyspnea. Her renal condition was treated with a combination of ACE inhibitor and ARB medications and excellent blood pressure control, which decreased her proteinuria to 250 mg/24-h urine. Subsequently, her proteinuria started increasing again, and she was started on tacrolimus but showed poor response. She has a history of endometrial adenocarcinoma and endometrioid ovarian carcinoma. The patient also has a history of hypertension and obesity, as well as a family history of cancer (Fig. 2). The proband’s mother and two sons have hematuria. The patient’s sister carried a diagnosis of FSGS and stage IV chronic kidney disease (estimated GFR of 24 mL/min); her brother passed away at age 49 due to cardiac disease and was noted to have had hearing loss at a young age. Our proband was tested with a nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel (Table 1). She was found to have a heterozygous, likely pathogenic variant in COL4A4 (c.2842G>T).
Fig. 2.

Family 2 pedigree. The proband is indicated with an arrow head
Table 1.
Nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel
| Gene | OMIM ID |
|---|---|
| ACTN4 | 604638 |
| ANLN | 616027 |
| ARHGAP24 | 610586 |
| ARHGDIA | 601925 |
| CD2AP | 604241 |
| COL4A3 | 120070 |
| COL4A4 | 120131 |
| COL4A5 | 303630 |
| COL4A6 | 303631 |
| COQ2 | 609825 |
| COQ6 | 614647 |
| COQ8B | 615567 |
| CRB2 | 609720 |
| CUBN | 602997 |
| DGKE | 601440 |
| EMP2 | 602334 |
| FAT1 | 600976 |
| INF2 | 610982 |
| ITGA3 | 605025 |
| ITGB4 | 147557 |
| KANK1 | 607704 |
| KANK2 | 614610 |
| KANK4 | 614612 |
| LAMB2 | 150325 |
| LMX1B | 602575 |
| MYO1E | 601479 |
| NEIL1 | 608844 |
| NEXMIF | 300524 |
| NPHS1 | 602716 |
| NPHS2 | 604766 |
| NUP107 | 607617 |
| NUP205 | 614352 |
| NUP93 | 614351 |
| PAX2 | 167409 |
| PDSS2 | 610564 |
| PLCE1 | 608414 |
| PTPRO | 600579 |
| SCARB2 | 602257 |
| SMARCAL1 | 606622 |
| TRPC6 | 603652 |
| TTC21B | 612014 |
| WDR73 | 616144 |
| WT1 | 607102 |
| XPO5 | 607845 |
Discussion
Type IV collagen-related nephropathy, in which there is disruption of the normal glomerular basement membrane architecture and kidney disease, has been linked to pathogenic variants in COL4A3, COL4A4, and COL4A59. Heterozygous mutations in COL4A3 and COL4A4 result in mild disease with or without isolated microscopic hematuria (MH) characterized by focal or diffuse thinning of the GBM, which is called thin basement membrane nephropathy (TBMN)9. Males hemizygous for COL4A5 pathogenic variants, and individuals of either sex with homozygous or compound heterozygous COL4A3/4 pathogenic variants, are at a greater risk of developing X-linked and autosomal recessive Alport syndrome, respectively10. ESRD develops within the first three decades of life in AS, which is also associated with sensorineural deafness and ocular abnormalities, including asymptomatic dot and fleck retinopathy and lenticonus11,12. Females with heterozygous pathogenic variants in COL4A5 can present a variety of clinical features ranging from asymptomatic hematuria to more severe disease with progression to ESRD13. Moreover, pathogenic mutations in COL4A5 have been identified in families with familial and sporadic FSGS14. The presence of bilateral renal cysts in our patient is interesting, as, to the best of our knowledge, an association between COL4A5 variants and renal cysts has not been reported before.
In our first proband, WES detected a heterozygous variant in COL4A5 (c.141+1G>A, IVS2+1G>A). To the best of our knowledge, this variant has not been reported previously as either a pathogenic or a benign variant. This splice site variant destroys the canonical splice donor site in intron 2 and is predicted to cause abnormal gene splicing. The c.141+1G>A variant has not been not observed in large population cohorts15–17. Based on the ACMG 2015 guidelines, the c c.141+1G>A, IVS2+1G>A variant was classified as a pathogenic variant18. Even though our second proband had diffuse foot process effacement, her poor response to immunosuppression raised suspicion of a genetic etiology19. She was tested with a nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel, which detected a heterozygous and likely pathogenic variant in COL4A4 (c.2842G>T). This variant is predicted to result in a premature protein termination (p.Gly948). This variant alone is probably sufficient to be the primary cause of the renal disease in this patient. Notably, heterozygous missense variants in COL4A4 and COL4A3 have been reported to cause familial or sporadic FSGS18,20.
While the first patient underwent WES, which detected a pathogenic variant in COL4A5, the second patient had a nephrotic syndrome/focal segmental glomerulosclerosis sequencing panel that revealed a likely pathogenic variant in COL4A4. There is an ongoing debate between the advantages and drawbacks of each test, especially in Mendelian disorders. One advantage of WES is variant segregation analysis and the concurrent testing of other family members, which can identify the pattern of Mendelian inheritance and detect other incidental variants that can be of clinical significance. Our first proband, thanks to WES, was found to be compound heterozygous for two ABCA4 variants. This finding helps in explaining her macular degeneration and vision loss and would be of great value when counseling this patient and her family. Gene panels have historically been preferred, especially when looking for genes related to a patient’s phenotype, because of their low cost and short turnaround time, and many clinical geneticists consider them a rapid first-tier test.
Molecular genetic testing is noninvasive, can be highly accurate and is becoming the diagnostic procedure of choice for Alport syndrome since renal pathology findings of FSGS do not necessarily indicate genetic mutation21. Therefore, a multidisciplinary team approach, with expertize in clinical genetics, nephrology, and nephropathology, is important to attain an accurate diagnosis. While the rate of progression of renal disease may be related to the underlying causal mutation, molecular analysis may eventually provide more prognostic data than either renal or skin biopsy. Massively parallel (next generation) sequencing allows simultaneous analysis of the COL4A3, COL4A4, and COL4A5 genes, providing benefits in screening time and cost, and should be considered in similar cases to ours22,23. In summary, we present two families with novel pathogenic variants in COL4A5 and COL4A4 diagnosed by exome sequencing and gene panel testing, respectively, and discuss the merits of these approaches.
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mele C, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. New Engl. J. Med. 2011;365:295–306. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. New Engl. J. Med. 2011;365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 3.Kitiyakara C, Kopp JB, Eggers P. Trends in the epidemiology of focal segmental glomerulosclerosis. Semin. Nephrol. 2003;23:172–182. doi: 10.1053/snep.2003.50025. [DOI] [PubMed] [Google Scholar]
- 4.Buscher AK, et al. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin. Nephrol. 2012;78:47–53. doi: 10.5414/CN107320. [DOI] [PubMed] [Google Scholar]
- 5.Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine. 1999;78:338–360. doi: 10.1097/00005792-199909000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Boye E, et al. Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am. J. Hum. Genet. 1998;63:1329–1340. doi: 10.1086/302106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive alport syndrome with thin basement membrane nephropathy presenting as focal segmental glomerulosclerosis in later life. Nephron. 2015;130:271–280. doi: 10.1159/000435789. [DOI] [PubMed] [Google Scholar]
- 8.Malone AF, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86:1253–1259. doi: 10.1038/ki.2014.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torra R, Tazón-Vega B, Ars E, Ballarín J. Collagen type IV (α3–α4) nephropathy: from isolated haematuria to renal failure. Nephrol. Dial. Transplant. 2004;19:2429–2432. doi: 10.1093/ndt/gfh435. [DOI] [PubMed] [Google Scholar]
- 10.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. New Engl. J. Med. 2003;348:2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 11.Lin F, et al. Whole exome sequencing reveals novel COL4A3 and COL4A4 mutations and resolves diagnosis in Chinese families with kidney disease. BMC Nephrol. 2014;15:175. doi: 10.1186/1471-2369-15-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, et al. A novel heterozygous COL4A4 missense mutation in a Chinese family with focal segmental glomerulosclerosis. J. Cell. Mol. Med. 2016;20:2328–2332. doi: 10.1111/jcmm.12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jais JP, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003;14:2603–2610. doi: 10.1097/01.ASN.0000090034.71205.74. [DOI] [PubMed] [Google Scholar]
- 14.Gast C, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016;31:961–970. doi: 10.1093/ndt/gfv325. [DOI] [PubMed] [Google Scholar]
- 15.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Genomes Project C. A global reference for human genetic variation. Nature. 2015;526:68. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Exome Variant Server NGESPE, Seattle, WA. http://evs.gs.washington.edu/EVS/.
- 18.Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J. Am. Soc. Nephrol. 2018;29:759–774. doi: 10.1681/ASN.2017090958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie J, et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014;6:498–505. doi: 10.1093/jmcb/mju040. [DOI] [PubMed] [Google Scholar]
- 21.Inoue Y, et al. Detection of mutations in the COL4A5 gene in over 90% of male patients with x-linked Alport’s syndrome by RT-PCR and direct sequencing. Am. J. Kidney Dis. 1999;34:854–862. doi: 10.1016/S0272-6386(99)70042-9. [DOI] [PubMed] [Google Scholar]
- 22.Fallerini C, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 2014;86:252–257. doi: 10.1111/cge.12258. [DOI] [PubMed] [Google Scholar]
- 23.Moriniere V, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J. Am. Soc. Nephrol. 2014;25:2740–2751. doi: 10.1681/ASN.2013080912. [DOI] [PMC free article] [PubMed] [Google Scholar]