

Supplemental Digital Content is available in the text
Keywords: airway immunity, microbiota profiling, obstructive sleep apnea
Abstract
Lung microbiota may affect innate immunity and treatment consequence in the obstructive sleep apnea (OSA) patients. Bronchoalveolar lavage fluid (BALF) was obtained from 11 OSA patients and 8 patients with other lung diseases as control, and used for lung microbiota profiling by PCR amplification and sequencing of the microbial samples. It was demonstrated that phyla of Firmicutes, Fusobacteria, and Bacteriodetes were relatively abundant in the lung microbiota. Alpha-diversity comparison between OSA and control group revealed that Proteobacteria and Fusobacteria were significantly higher in OSA patients (0.3863 ± 0.0631 and 0.0682 ± 0.0159, respectively) than that in control group (0.119 ± 0.074 and 0.0006 ± 0.0187, respectively, P < .05 for both phyla). In contrast, Firmicutes was significantly less in OSA patients (0.1371 ± 0.0394) compared with that in the control group (0.384 ± 0.046, P < .05). Comparison within a group (ß-diversity) indicated that the top 5 phyla in the OSA lung were Proteobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Acidobacteria, while the top 5 phyla in the control group were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Acidobacteria. These findings indicated that lung microbiota in OSA is distinct from that of non-OSA patients. Manipulation of the microbiota may be an alternative strategy to augment airway immunity and to reduce susceptibility to airway infection.
1. Introduction
In human body, there are various kinds of microbial communities, which are also called microbiota. Commensal bacteria such as Streptococcus, Veillonella, Prevotella, and Actinomyces are the predominant microbiota in healthy human lungs.[1–3] These microbiota are important for preventing respiratory infection[2,4,5] in that they can regulate innate and acquired immunity.
It has been reported that probiotic commensal bacteria of gut microbiota has substantial and continuous effects on human health and physiological development, including in maturing the immune system and in preventing pathogen invasion.[6] Similarly, manipulation of lung microbiota by regulating pathogen activity and enhancing the natural immune system using probiotic supplementary therapies may become an alternative treatment for variety kinds of lung diseases, including chronic obstructive pulmonary disease (COPD), asthma, cystic fibrosis, and cancer. In this regard, association between lung microbiota and lung diseases such as COPD, asthma, and cystic fibrosis has been studied.[7–13] However, the microbiome profiling in obstructive sleep apnea (OSA) patients has not been studied.
OSA is a chronic, highly prevalent sleep disorder, with an estimated prevalence of approximately 22% in men and 17% in women.[14] Studies in the understanding of OSA pathogenesis have shown that impaired narrow upper airway anatomy and pharyngeal dilator muscle activity are considered as key contributors to OSA pathogenesis.[15] In addition, some nonanatomical factors, such as microaspiration and alteration of lung microbiota, may also affect the severity of OSA in a substantial proportion of OSA patients. The role of microbiota in the pathogenesis of OSA has not been investigated. The current study was, therefore, designed to investigate microbiota in the lower airways of OSA patients.
2. Materials and methods
2.1. Patient recruitment
Patients, who were hospitalized with OSA into the Department of Respiratory Disease and Critical Care Medicine, Xinjiang People's Hospital (Urumqi, Xinjiang) from June 2015 to February 2016, were enrolled into this study. These OSA patients were diagnosed following the Guideline of Adult Obstructive Sleep Apnea by the Chinese Association of Respiratory Diseases and had not been treated before the enrollment. A written consent form was obtained from each patient and the Study Protocol was approved by the Ethic Committees of Xinjiang People's Hospital.
Inclusion criteria included following the guideline on diagnosis of OSA in the adult by The Chinese Medical Association, Branch of Sleep Respiratory Disease, 2011; Patients had never received any treatment for OSA; Patients, who had unknown cough but normal chest X-ray, would undergo fibreoptic bronchoscopy for further diagnosis and treatment; Patients had no smoking history; and Patients signed the informed consent, and agreed to participate in this clinical study.
Exclusion criteria included smokers, or patients with neuromuscular disorders, infectious disease, rheumatoid and autoimmune diseases, cancer, peripheral vascular diseases, coagulation disorders, liver or kidney diseases, severe mental illness, acute renal failure, severe heart or brain diseases (cardiac infarction or stroke within last 6 months, or heart failure), central or mixed sleep apnea, injury or surgery within last 3 months, treated with steroid or immunosuppression drug, or cytotoxic drugs, treated with free radical scavenger, and patients with chronic hypoxia were excluded from the study.
As shown in Fig. 1, 11 of 35 OSA patients were enrolled into the current study who met the inclusion criteria described above; for the subjects of control group, 8 of 22 non-OSA patients were selected. These patients were hospitalized during the same period as the OSA patients, but had no abnormal chest imaging appearance.
Figure 1.
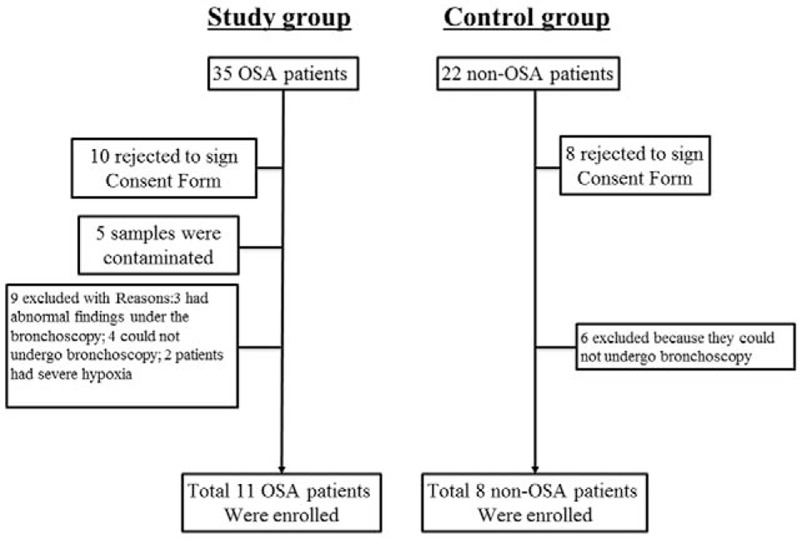
Flow chart of patients’ enrollment.
2.2. Sleep apnea test
Sleep apnea was examined with a sleep diagnostic device (Compumedics, Compumedics E-series, Compumedics Inc; Abbotsford, Victory, Australia) and data were analyzed with Profusion PSG software. All participants were restricted from tea, coffee, or any sedatives on the day of testing. Following parameters were recorded for at least 7 hours during the sleep at night: electroencephalogram, submandibular EMG, electrooculogram, nose flow, chest and abdomen movement, blood oxygen saturation, electrocardiogram, leg movement, and body position.
2.3. Laboratory test in BALF samples
Bronchoalveolar lavage (BAL) was then performed in all participants by instilling pre-warmed sterile saline (120 mL each time and 2 times) and aspirating (60 mL each time and 2 times). After centrifugation of the BALF, the supernatant was saved at −80°C for laboratory test and the pellet was used for microbiome profiling.
2.4. Bacterial DNA extraction, amplification, and examination
Bacterial DNA was extracted from the sediment of alveolar lavage fluid, amplified by PCR, sequenced by Illumina, and the bioinformatics of the sequence results were analyzed (Supplement Figure S1). To accomplish this, DNA amount was quantified by Nano Drop 2000 and quality of DNA was examined by electrophoresis (Supplement Figure S2). Briefly, V4-V5 region of bacterial 16S rRNA was amplified by PCR by using 357F and 926R primers (Supplement Figure S3). Sequencing was performed with QIIE1.7.0 and ChimeraSlayer. Sequences were polymerized and 97% consistency between PCR products and 16S rRNA was classified, and SILWA database was referred for labeling.
2.4.1. Illumina high throughput sequencing
As wrong information might exist in the raw data of primary sequencing, quality control and data filtering on these raw data were performed in order to obtain clean data. Operational Taxonomic Units (OUT) clustering was then performed on the basis of sequence similarity, and information of OUT distribution in each sample and species were obtained through analysis.
Following primer sequences were used for PCR amplification: 515F: 5’-GTGCCAGCMGCCGCGGTAA-3’; 806R: 5’-GGACTACHVGGGTWTCTAAT-3’. Products of PCR amplification were examined by electrophoresis as shown in Supplement Figure S4.
2.5. Statistical analysis
Data were analyzed using SAS jmp pro10.0 software (SAS Institute Inc., Cary, NC. Data were expressed as mean ± SD. Comparison of Study group and Control group was performed by homogeneity test of variance followed by t test or Wilcoxon test.
Sequencing raw data were filtered using Trimmomatic software (http://www.usadellab.org/cms/?page=trimmomatic) for quality control and data stitching, which was performed with Mothur (http://www.mothur.org) and Flash. The number of distinct OTUs was calculated with the clean and stitched data. Clean contigs were generated using barcode and primer information, using Qiime (http://qiime.org/index.html). Quality scores across the bases was shown in the Supplement Figure S5.
Relative abundance of microbiota was analyzed on the basis of the OTU distribution of each sample at phylum level. OTU rarefaction curve was obtained and used to show variety of microbiota and to compare abundances of varying kinds of microbiota (Supplement Figure S6).
Shannon–Wiener curve was obtained by the Shannon Index formula:
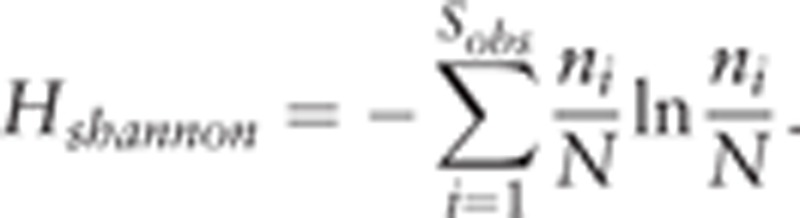 |
This curve was used to indicate different kinds of microbiomes in the patients’ samples, as shown in Supplement Figure S7.
Simpson curve was also used to indicate the number of regional microbiomes (Supplement Figure S8). Simpson curve was calculated by the following formula:
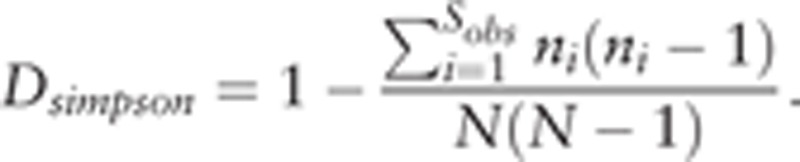 |
3. Results
3.1. Characteristics of the OSA and control groups enrolled into the current study
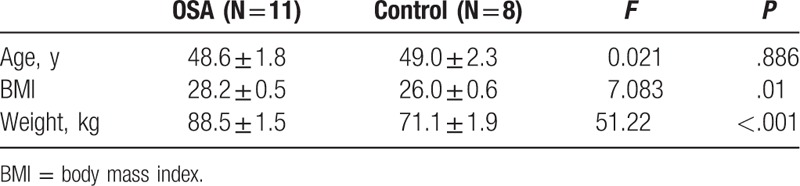
The characteristics of the study and control subjects are compared and presented in Table 1. As summarized in Table 1, OSA patients had significantly higher body mass index and body weight than that of control subjects (BMI: 28.2 ± 0.5 vs 26.0 ± 0.6, P = .01; body weight: 88.5 ± 1.5 kg vs 71.1 ± 1.9 kg, P < .001).
Table 1.
Characteristics of the study and control subjects.
3.2. Relative abundance of microbiomes at phylum level in the OSA and control groups
Figure 2 shows microbiota at phylum level, which were larger or equals 1% in relative abundance. Less than 1% in relative abundance was classified as “Others,” which were either “unclassified” or “unidentified.” Analysis of the overall relative abundance of microbiota in all 19 subjects revealed that phyla of Firmicutes, Fusobacteria, and Bacteriodetes were relatively abundant (Fig. 2).
Figure 2.
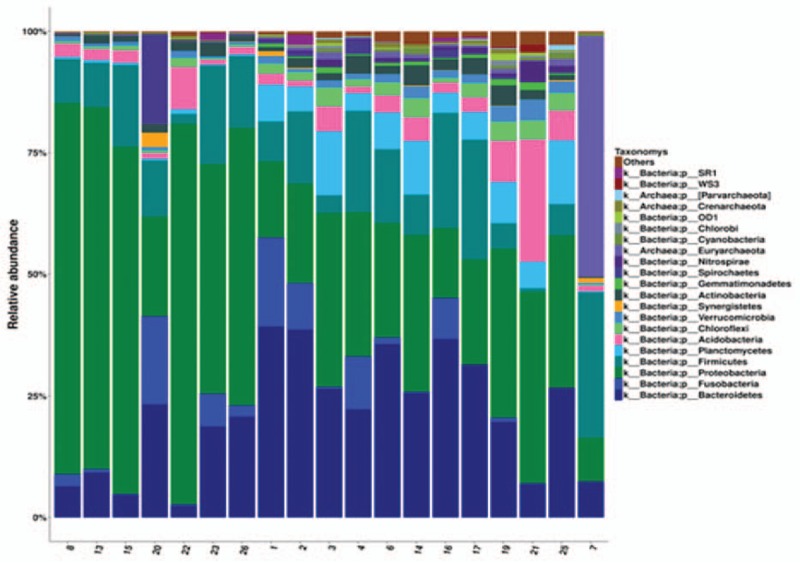
Relative abundance of microbiota at phylum level. Relative abundance of microbiota at phylum level in all samples from 11 OSA and 8 control subjects were plotted. Horizontal axis: sample ID; vertical axis: relative abundance of larger than 1% at phylum level. If the relative abundance was less than 1%, they were classified as “Others,” which included “unclassified” and “unidentified” bacteria. Each color bar represents 1 phylum of bacteria.
Comparison of microbiota from 11 OSA patients’ BALF to that from 8 control subjects (diversity between groups) is performed and presented as Fig. 3. It was found that Proteobacteria and Fusobacteria were significantly higher in OSA patients (0.3863 ± 0.0631 and 0.0682 ± 0.0159, respectively) than that in control group (0.119 ± 0.074 and 0.0006 ± 0.0187, respectively, P < .05 for both phyla). In contrast, Firmicutes was significantly less in OSA patients (0.1371 ± 0.0394) compared with that in the control group (0.384 ± 0.046, P < .05). There was no significant difference in comparison of other microbiota in OSA patients from control group (P > .05).
Figure 3.
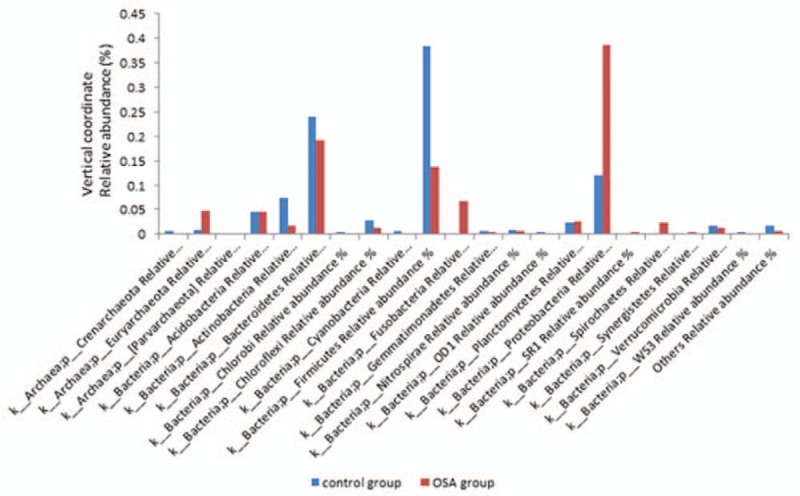
Phylum level composition of microbiota in OSA and control group. Bacterial DNA was extracted, amplified by PCR, and sequenced as described in the Materials and methods. Relative abundance of microbiota was analyzed and comparison between the 2 groups was performed. Horizontal axis: bacterial phyla; vertical axis: relative abundance of each microbiota. Blue bar: OSA group; red bar: control group.
Comparison of microbiota within OSA group or control group (ß-diversity within a group) is presented as Fig. 4A (OSA group) and B (control group). As shown in Fig. 4A, the top 5 phyla in the OSA patients’ lower airway were Proteobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Acidobacteria. Of them, Proteobacteria, Bacteroidetes, and Firmicutes accounted for 90% of the microbiota in OSA patients. In contrast, the top 5 phyla in the control group were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Acidobacteria. Of them, the top 3 of Firmicutes, Bacteroidetes, Proteobacteria accounted over 90% of the microbiota in the control subjects (Fig. 4B).
Figure 4.
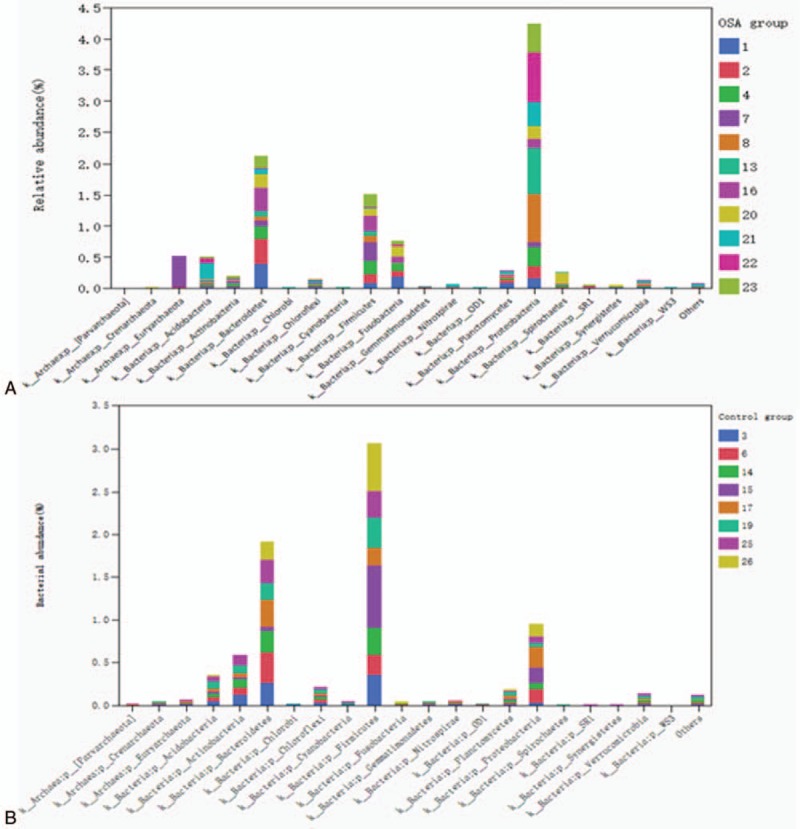
Relative abundance of microbiota at phylum level in the OSA group (A) and control group (B). Bacterial DNA was extracted, amplified by PCR, and sequenced as described in the Materials and methods. Relative abundance of microbiota was analyzed and ß-comparison within OS or control group was performed. Horizontal axis: bacterial phyla; vertical axis: relative abundance of each microbiota. Each color of the bar represents patients’ identification number.
3.3. Heat map of relative abundance by samples
Figure 5 revealed the clustering of average relative phyla abundances of lower airway microbiota in both OSA patients and control subjects. As shown by the heat map, relative phyla abundances of microbiota were crossly distributed among the OSA samples and control samples.
Figure 5.
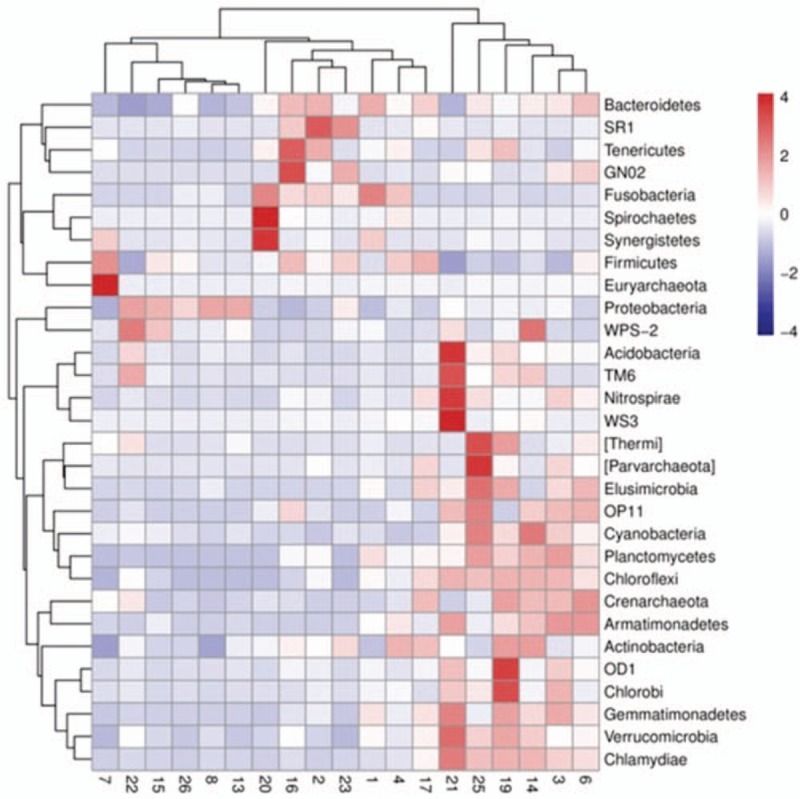
Heat-map of the top 30 bacteria at phylum level. Abundance of microbiota from both 11 OSA patients and normal subjects BALF samples was analyzed by Z-score as described in the Materials and methods. Vertical axis: sample clustering; horizontal axis: phylum clustering.
Next, genus level relative abundances of 40 bacteria were analyzed. It was found that abundance of Fusobacteria was significantly increased in OSA group compared with that in control group (P < .05). In contrast, genus level relative abundances of Clostridium, Acinetobacter, Planctomycetes, New Sphingomonas, Ciliate genus, Ancient genus, and Silk sulfur bacteria genus were significantly lower in the OSA patients’ samples than that from control subjects (P < .05).
3.4. Analysis on evolution of the lung microbiota
An evolution tree was established using the relative abundance data at genus level lung microbiota. To accomplish this, the top 50 bacteria with highest relative abundance and specific OTU were selected to establish the evolution tree. As shown in Fig. 6, the top 5 common bacteria in lung microbiota were Prevotella, Porphyromonas, Acinetobacter, Fusobacteria, and Bacteroidetes.
Figure 6.
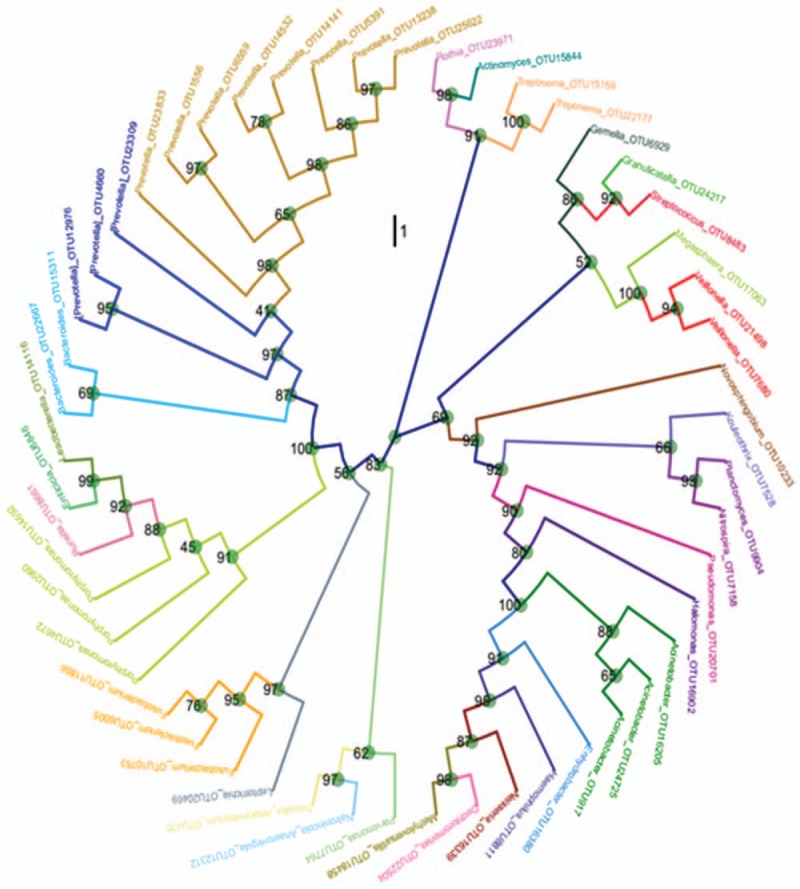
Evolutionary tree of the top 50 OTU bacteria. An evolutionary tree was established using relative abundances at genus level and the top 50 operational taxonomic unit (OTU). Name of the bacteria at genus level was corresponded to OUT identification number. Bootstrap number, which was an indicator of accuracy, was shown at each bifurcation point.
4. Discussion
OSA patients are predisposed to increased microaspiration and gastric reflux,[15] and it has been reported that OSA patients had an increased risk of community-acquired pneumonia.[16] Therefore, the current study was designed to investigate lower airway microbiota in OSA patients in comparison to that of control subjects with respiratory diseases other than OSA. We demonstrated that there was diverse microbiome in the airways of OSA patients and control subjects. Specifically, by diversity comparison (between the groups), we found that Proteobacteria and Fusobacteria were significantly higher in OSA patients than that in control group, while Firmicutes was significantly less in OSA patients compared with that in the control group. By ß-diversity (within the group) comparison, we found that the top 5 phyla in the OSA patients’ lower airway were Proteobacteria, Bacteroidetes, Firmicutes, Fusobacteria, and Acidobacteria. Of them, Proteobacteria, Bacteroidetes, and Firmicutes accounted for 90% of the microbiota in OSA patients. In contrast, the top 5 phyla in the control group were Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Acidobacteria. Of them, the top 3 of Firmicutes, Bacteroidetes, Proteobacteria accounted for over 90% of the microbiota in the control group lower airways.
Healthy airway microbiota is essential to properly maintain the airway immunity and to reduce the opportunity of respiratory tract infection. Alteration of this healthy microbiota may lead to increased susceptibility of airway mucosa to pathogens, and consequently results in lung injury or chronic airway inflammation.[17,18] The lung microbiome, which is distinct from that of other body site microbiome,[9] may be manipulated to restore “healthy” microbial communities via use of probiotics or antibiotics. Altered airway microbiota may contribute to the development of pneumonia or airway infection in OSA patients.[4,16] Therefore, further understanding of the role of the complex airway microbiome in OSA may have beneficial effect to the patients and it requires investigation of the interactions among genes of the microbiota and host. In this regard, clustering of relative phyla abundances of lower airway microbiota in both OSA patients and control groups in the current study revealed that relative phyla abundances of microbiota were crossly distributed among the OSA and control samples, suggesting microbiome may have beneficial cross-talk with the host lung and altered relative abundance and diversity of lung microbiota may result in increased susceptibility to inflammation and injury.
Similar to the gut microbial alterations, alteration of microbial profiles and their metabolites may happen in the lungs.[19] Thus, monitoring the dynamic changes of lung microbiota is crucial in understanding the response of the polymicrobial ecosystem to intense antibiotic treatments. In this regard, many studies have compared the differences of airway microbiota in variety kinds of lung diseases and studied the role of lung microbiota in the pathogenesis or severity of lung diseases, including COPD, asthma, and cystic fibrosis.[7–13,20,21] For instance, in a study of the taxonomic and functional profiles of lung microbiota on human nonmalignant lung tissue, Yu et al[9] reported that lung tissue microbiota was clearly distinct from the microbiotas reported at other body sites (oral cavity, nasal cavity, gut, skin, and vagina). However, to our knowledge, a study on the correlation between OSA and lung microbiota has not been reported. Here, we report the characteristics of lower airway microbiota in OSA and found the top 5 common bacteria in the OSA lung microbiota are Prevotella, Porphyromonas, Acinetobacter, Fusobacteria, and Bacteroidetes.
The current study had several limitations. First, this investigation was limited by the small sample size. Second, the control group consisted of patients with other lung diseases in that it was unethical to recruit normal subjects for bronchoscopy and BAL. Third, the BALF samples were utilized in this study. There is still controversy regarding whether BAFL samples might be contaminated with upper airway or oral residual bacteria.[22–24] Thus, longitudinal studies and studies evaluating different lung segments will be needed in the future study to confirm this issue. Fourth, smokers and patients with comorbidities of other organs or system disorders were excluded from this study, and thus, it may not be representative of the general population. In this regard, studies on the direct effect of smoking on the lower airway microbiome are controversial,[4,9,25] and it remains to be further investigated.
Taken together, findings of the current study indicated that indigenous lung microbiota was different between OSA and patients with other lung diseases. This might be due to increased chance of microaspiration[26,27] or inefficient microbial clearance in OSA patients. Hence, future in-depth studies of the lower airway microbiome in a variety of lung diseases including OSA are necessary to further explore the gut-lung axis and the potential role of extrapulmonary microbes in the development of respiratory disease as well as in the treatment of airway inflammation.
Author contributions
Nanfang Li designed the study, Dongmei Lu prepared the manuscript, Ayinigeer Abulimiti and Li Cai collected the data, Ling Zhou and Jing Hong analyzed the data, Xiaoguang Yao searched the references. All the authors made the final approval.
Conceptualization: Nanfang Li.
Data curation: Xiaoguang Yao.
Formal analysis: Ayinigeer Abulimiti.
Investigation: Jing Hong.
Methodology: Ling Zhou.
Project administration: Li Cai.
Writing – original draft: Dongmei Lu.
Supplementary Material
Footnotes
Abbreviations: BAL = bronchoalveolar lavage, BALF = bronchoalveolar lavage fluid, COPD = chronic obstructive pulmonary disease, OSA = obstructive sleep apnea, OUT = operational taxonomic unit, OUT = operational taxonomic units.
Funding/support: The project was supported by The Xinjiang Uygur Autonomous Region Natural Science Foundation, projector number 2016D01C114.
The study protocol was approved by the Ethic Committees of Xinjiang People's Hospital.
A written consent form was obtained from each patient.
The authors have no conflict of interests.
Supplemental Digital Content is available for this article.
References
- [1].Cernadas M. It takes a microbiome: commensals, immune regulation, and allergy. Am J Respir Crit Care Med 2011;184:149–50. [DOI] [PubMed] [Google Scholar]
- [2].Beck JM, Young VB, Huffnagle GB. The microbiome of the lung. Transl Res 2012;160:258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dickson RP, Erb-Downward JR, Prescott HC, et al. Cell-associated bacteria in the human lung microbiome. Microbiome 2014;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Morris A, Beck JM, Schloss PD, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 2013;187:1067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rabin HR, Surette MG. The cystic fibrosis airway microbiome. Curr Opin Pulm Med 2012;18:622–7. [DOI] [PubMed] [Google Scholar]
- [6].Kosiewicz MM, Zirnheld AL, Alard P. Gut microbiota, immunity, and disease: a complex relationship. Front Microbiol 2011;2:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sze MA, Dimitriu PA, Hayashi S, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012;185:1073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet 2014;384:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu G, Gail MH, Consonni D, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol 2016;17:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Segal LN, Clemente JC, Wu BG, et al. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax 2017;72:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sze MA, Morris A. Launching into the deep: does the pulmonary microbiota promote chronic lung inflammation and chronic obstructive pulmonary disease pathogenesis? Am J Respir Crit Care Med 2016;193:938–40. [DOI] [PubMed] [Google Scholar]
- [12].Hauptmann M, Schaible UE. Linking microbiota and respiratory disease. FEBS Lett 2016;590:3721–38. [DOI] [PubMed] [Google Scholar]
- [13].Depner M, Ege MJ, Cox MJ, et al. Bacterial microbiota of the upper respiratory tract and childhood asthma. J Allergy Clin Immunol 2017;139:826–34.e13. [DOI] [PubMed] [Google Scholar]
- [14].Franklin KA, Lindberg E. Obstructive sleep apnea is a common disorder in the population: a review on the epidemiology of sleep apnea. J Thorac Dis 2015;7:1311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schindler A, Mozzanica F, Sonzini G, et al. Oropharyngeal dysphagia in patients with obstructive sleep apnea syndrome. Dysphagia 2014;29:44–51. [DOI] [PubMed] [Google Scholar]
- [16].Chiner E, Llombart M, Valls J, et al. Association between obstructive sleep apnea and community-acquired pneumonia. PLoS One 2016;11:e0152749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Segal LN, Rom WN, Weiden MD. Lung microbiome for clinicians. New discoveries about bugs in healthy and diseased lungs. Ann Am Thorac Soc 2014;11:108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lemon KP, Armitage GC, Relman DA, et al. Microbiota-targeted therapies: an ecological perspective. Sci Transl Med 2012;4:137rv5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 2014;20:159–66. [DOI] [PubMed] [Google Scholar]
- [20].Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One 2010;5:e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li J, Hao C, Ren L, et al. Data mining of lung microbiota in cystic fibrosis patients. PLoS One 2016;11:e0164510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Charlson ES, Bittinger K, Chen J, et al. Assessing bacterial populations in the lung by replicate analysis of samples from the upper and lower respiratory tracts. PLoS One 2012;7:e42786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med 2011;184:957–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio 2015;6:e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 2011;6:e16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Morse CA, Quan SF, Mays MZ, et al. Is there a relationship between obstructive sleep apnea and gastroesophageal reflux disease? Clin Gastroenterol Hepatol 2004;2:761–8. [DOI] [PubMed] [Google Scholar]
- [27].Teramoto S, Ohga E, Matsui H, et al. Obstructive sleep apnea syndrome may be a significant cause of gastroesophageal reflux disease in older people. J Am Geriatr Soc 1999;47:1273–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.