

Key Points
TL in LSCs is significantly shortened at diagnosis of CML and correlates with LSC burden.
TL in nonleukemic myeloid cells in deep molecular remission is unaffected by long-term TKI treatment.
Abstract
Telomere length (TL) in peripheral blood (PB) cells of patients with chronic myeloid leukemia (CML) has been shown to correlate with disease stage, prognostic scores, response to therapy, and disease progression. However, due to considerable genetic interindividual variability, TL varies substantially between individuals, limiting its use as a robust prognostic marker in individual patients. Here, we compared TL of BCR-ABL−, nonleukemic CD34+CD38− hematopoietic stem cells (HSC) in the bone marrow of CML patients at diagnosis to their individual BCR-ABL+ leukemic stem cell (LSC) counterparts. We observed significantly accelerated telomere shortening in LSC compared with nonleukemic HSC. Interestingly, the degree of LSC telomere shortening was found to correlate significantly with the leukemic clone size. To validate the diagnostic value of nonleukemic cells as internal controls and to rule out effects of tyrosine kinase inhibitor (TKI) treatment on these nontarget cells, we prospectively assessed TL in 134 PB samples collected in deep molecular remission after TKI treatment within the EURO-SKI study (NCT01596114). Here, no significant telomere shortening was observed in granulocytes compared with an age-adjusted control cohort. In conclusion, this study provides proof of principle for accelerated telomere shortening in LSC as opposed to HSC in CML patients at diagnosis. The fact that the degree of telomere shortening correlates with leukemic clone’s size supports the use of TL in leukemic cells as a prognostic parameter pending prospective validation. TL in nonleukemic myeloid cells seems unaffected even by long-term TKI treatment arguing against a reduction of telomere-mediated replicative reserve in normal hematopoiesis under TKI treatment.
Visual Abstract
Introduction
Chronic myeloid leukemia (CML) is a clonal hematopoietic stem cell (HSC) disease caused by an acquired reciprocal translocation between chromosomes 9 and 22 (the so-called Philadelphia translocation, Ph) resulting in the BCR-ABL fusion gene. BCR-ABL, a constitutively active tyrosine kinase, promotes cell survival and proliferation through several intracellular signal transduction pathways eventually leading to malignant transformation.1 It is the primitive CML leukemic stem cell (LSC) compartment that harbors the BCR-ABL translocation and typically expands with disease progression.2
It is well established that the pathobiology of CML differs between chronic phase (CP) and advanced stages, such as accelerated phase (AP) and blast crisis (BC). Whereas CP is characterized primarily by an increased cellular turnover and expansion of the leukemic committed progenitor compartment, late stages of CML typically acquire additional molecular aberrations, eventually leading to a differentiation block resulting in increasing blast counts, and secondarily, signs of hematopoietic insufficiency.3 At the stem cell compartment level, LSCs typically represent a minority of HSC at diagnosis. However, their contribution to the SC pool continuously increases from early CP to late CP (reviewed by Eaves and Eaves2 and Alvarez et al4). Recently, it has been shown that the degree of leukemic involvement in the HSC pool at diagnosis measured as the degree of Ph-positivity in the CD34+CD38− stem cell by fluorescent in situ hybridization (FISH) is correlated with prognosis and response to nilotinib,5 dasatinib,6 and imatinib first-line therapy.7
Telomeres shorten with each cell division, and telomere length (TL) reflects the replicative history of a cell. Previous studies in peripheral blood (PB) cells of CML patients showed dramatically reduced TL of leukemic cells as opposed to nonleukemic T cells8 and revealed a correlation of age-adapted TL with disease stage, response to treatment, and remaining duration of CP8-13 (reviewed by Brümmendorf and Balabanov14). Apart from variation related to the methodology used for TL measurement, telomere biology in healthy individuals in vivo is substantially influenced by 2 main parameters, age and genetic interindividual variability.15 Although age adjustment of TL can now be easily achieved by adaptation of individual results to healthy control cohorts, the high and mostly genetic interindividual variability in TL15 has so far still somewhat limited the prognostic and predictive value of routine TL assessment in individual CML patients, as no patient-specific BCR-ABL–negative internal control cells have been readily available.
Here, we first investigated if telomere shortening can be detected in LSC as opposed to nonclonal HSC, and, if so, the degree of intraindividual telomere shortening in LSC at diagnosis was correlated with leukemic involvement of the HSC compartment and could thus potentially serve as a discriminator between early and late CP CML. For this purpose, we analyzed TL in the LSC and nonleukemic CD34+CD38− HSC (ΔTLLSC-HSC) of CP-CML patients at diagnosis using a modified confocal quantitative FISH (Q-FISH) technique. Indeed, we were able to establish a correlation between accelerated telomere shortening in LSC and the degree of leukemic involvement of the CD34+CD38− HSC compartment that (pending prospective validation in clinical trials) could potentially become the first epigenetic biomarker in newly diagnosed CML.
Given the rather limited accessibility of LSC and nonclonal HSC for clinical routine prognostication of CML, we alternatively considered to compare the previously established TL measurement in leukemic myeloid PB cells obtained from CML patients at diagnosis8,13 to their nonclonal myeloid counterparts later obtained in deep molecular remission. In order to validate this control population, we first had to rule out a potential preexisting telomere deficit in nonleukemic cells already present at the time of malignant transformation (as it had been recently observed in AML patients).16 Furthermore, given the fact that long-lasting tyrosine kinase inhibitor (TKI) treatment is typically required to achieve stable deep molecular remission in CML patients before treatment discontinuation, an effect of this treatment on TL in nonclonal myeloid cells had to be included. These questions were prospectively addressed by analyzing TL in PB of patients in deep molecular remission eligible for treatment discontinuation as part of a German-Dutch scientific substudy within the pan-European EURO-SKI trial (NCT01596114).
Methods and patients
TL analysis of the BM slides
Single-blinded bone marrow (BM) samples were obtained from 15 patients of the first-line CML study (NCT00852566) of the Nordic CML Study Group (NCMLSG).6 Inclusion criteria for the NCMLSG trial were CML at first diagnosis in CP and no previous TKI treatment (see Hjorth-Hansen et al6 and Mustjoki et al7). Detailed patient characteristics are shown in Table 1. Written informed consent was obtained from all patients, and the study was conducted in accordance with the Declaration of Helsinki. Approval by the ethical committee was given for all analyses. Initial cell preparation was carried out as previously reported.7 Briefly, BM samples from initial diagnosis were subjected to Ficoll separation; next, CD34+ cells were obtained by immune-magnetic bead separation and subsequently stained with CD34 and CD38 antibodies. Next, CD34+ cells were sorted by fluorescence-activated cell sorting (80% being CD34+CD38+; 5% being CD34+CD38−) and spun onto cytoslides. The percentage of BCR-ABL+ and BCR-ABL− cells was first determined by FISH, using dual-fusion dual-color BCR-ABL1 probe (Vysis; Abbott Laboratories, Abbott Park, IL), and clone size was calculated according to standard procedures.7
Table 1.
Clinical baseline data of the 15 patients enrolled in the Nordic first-line CML study (NCT00852566)
| Patient number | Age, y | Sex | Sokal score | Euro score | Clone size, % | Leukocytes, × 109/L | Hb, g/dL | Platelets, × 109/L |
|---|---|---|---|---|---|---|---|---|
| 1 | 72 | M | 0.93 | 1106 | 89 | 37 | 123 | 275 |
| 2 | 63 | M | 1.60 | 1902 | 91 | 119 | 99 | 264 |
| 3 | 51 | F | 0.82 | 1242 | 87 | 40 | 118 | 847 |
| 4 | 55 | M | 0.75 | 749 | 99 | 65.1 | 141 | 462 |
| 5 | 61 | M | 0.74 | 871 | 57 | 19.6 | 126 | 362 |
| 6 | 60 | F | 0.8 | 1002 | 75 | 97 | 111 | 401 |
| 7 | 61 | M | 1.39 | 1567 | 87 | 288 | 82 | 395 |
| 8 | 46 | F | 0.75 | 287 | 35 | 38.6 | 114 | 425 |
| 9 | 64 | M | 0.84 | 928 | 16 | 22 | 125 | 390 |
| 10 | n.a. | n.a. | n.a. | n.a. | 30 | n.a. | n.a. | n.a. |
| 11 | 55 | F | 0.73 | 1053 | 79 | 75 | 95 | 137 |
| 12 | 61 | F | 0.90 | 1242 | 32 | 32 | 129 | 796 |
| 13 | 58 | F | 0.69 | 791 | 37 | 38 | 138 | 188 |
| 14 | 51 | F | 0.76 | 1177 | 48 | 73.9 | 129 | 503 |
| 15 | 41 | F | 0.67 | 204 | 83 | 11.4 | 137 | 706 |
F, female; Hb, hemoglobin; M, male; n.a., not available.
In the next step, BCR-ABL FISH images of the sorted cell populations (Figure 1A-B) were captured using a laser scanning confocal microscope (LSCM; LSM710; Zeiss, Oberkochen, Germany) running Zen 2009 software (Zeiss). Standard software tools were used to save designated space coordinates (eg, x1, y1, z1) for every image of the conditions. Image magnification was ×63. Digital zoom was ×2.5. Fifty to 100 cells per condition were captured. The same cytoslides were then washed and reprocessed for telomere Q-FISH (Figure 1B) using our previously published protocol.17-20 Slides were dehydrated and air dried before telomeres were hybridized with a fluorescent Cy3-(C3TA2)-PNA probe (Daejeon, Panagene, South Korea) for 2 hours. Slides were washed in formamide buffer for 30 minutes before cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole solution (Sigma, Darmstadt, Germany). Cell relocalization at the LSCM was done using an indirect approach based on a system of precoordinates that allowed for a more precise tracking of the saved spots previously captured. Multitracking mode of 0.5-μm steps was then used to acquire the 4′,6-diamidino-2-phenylindole and telomere signal. Maximum intensity projections of 3 to 5 single consecutive steps were carried out. To quantify the fluorescence intensity of the telomere signal, images were analyzed using Definiens software (Definiens, Munich, Germany). Quantification of BCR-ABL FISH staining and telomere Q-FISH analysis were carried out separately in a single blinded fashion. Intensity of telomeres was given in arbitrary units. Mean TL of BCR-ABL− cells was subtracted from BCR-ABL+ cells for each patient and calculated as ΔTLLSC-HSC in order to minimize intraindividual TL variation.
Figure 1.

Representative laser scanning confocal microscopy images obtained after applying a 2-phase combined FISH technique based on coordinate assembly protocol for assessment of TL within the same CP CML patient’s CD34+38− compartment. (A) In phase 1, BCR-ABL+ and BCR-ABL− cells were identified by BCR-ABL FISH staining. (B) In phase 2, PNA-Tel Q-FISH is used to restain the samples allowing telomere quantification in the BCR-ABL+ and BCR-ABL− cells within the same individual’s CD34+38− compartment. Image magnification ×630. Digital zoom ×2.5. DAPI, 4′,6-diamidino-2-phenylindole.
TL analysis of the PB
PB samples for TL analysis of 134 CML patients were prospectively obtained within a scientific substudy of the pan-European EURO-SKI trial (NCT01596114). Inclusion criteria of the EURO-SKI trial were treatment by the TKI imatinib, dasatinib, or nilotinib for at least 3 years and confirmed deep molecular response (MR4) (see Saussele et al21). The primary aim of the study was to evaluate the molecular relapse-free survival after TKI cessation. Written consent was obtained from all patients, and the study was conducted in accordance with the Declaration of Helsinki. Samples were acquired at study inclusion, and TL analysis was carried out using flow cytometry and FISH (flow-FISH) as described previously.18,19 Age adaption was carried out on the basis of 356 healthy controls as described previously, and TL is given in kilobases.18,19 Detailed patient characteristics are shown in supplemental Table 1.
Statistical analysis
SPSS (version 16) was used for statistical analysis. Wilcoxon matched pairs test, Pearson`s correlation, and 1-sample Student t test were used for sample analysis. Log-rank test was used for analysis of time to MR loss. Statistical significance was set at P < .05.
Results
TL in CD34+38− LSC as a marker to discriminate early from late CP CML
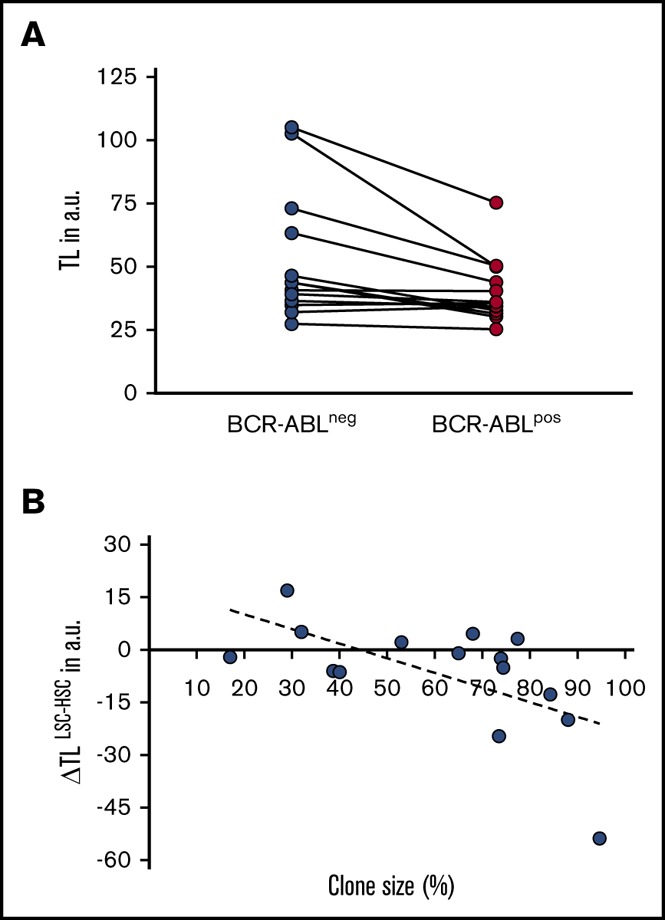
The purpose of the study was to test whether BCR-ABL–negative HSC can be used as an internal intraindividual control for genetic variability of TL in LSC. The aim was to gain deeper insights into the clonal expansion of the LSC compartment during CP CML as a potential reflection of whether an individual patient at diagnosis is biologically still in early vs already in late CP.8 For this purpose, we analyzed TL in individual BCR-ABL+ CD34+CD38− sorted BM cells of CML patients at first diagnosis and compared with their phenotypically identical nonleukemic BCR-ABL− CD34+CD38− counterparts.7,22-24 Sequential LSCM for BCR-ABL FISH and telomere Q-FISH was used (Figure 1A-B). In analogy to previous studies in PB,8,13 we found a statistically significant telomere deficit (−4.9 arbitrary fluorescent units [a.u.] range: −53.7 to 16.9 a.u., P = .04) in BCR-ABL+ LSC compared with BCR-ABL− nonleukemic HSC at diagnosis (Figure 2A).
Figure 2.

TL in BCR-ABL− vs BCR-ABL+cells of CP CML patients at first diagnosis. (A) TL (in a.u.) in CD34+CD38− BCR-ABL+ LSC is significantly shortened compared with BCR-ABL− HSC (P = .040). (B) Difference in TL between BCR-ABL+ LSC and BCR-ABL− HSC (ΔTLLSC-HSC, in a.u.) as a function of clone size (ie, the percentage of Ph+ cells) in the BM CD34+CD38− population of CP CML patients at diagnosis (r2 = 0.367, P = .016).
We next assessed whether size of the leukemic clone in CML patients at diagnosis was indeed paralleled by accelerated telomere shortening in the respective LSC fraction. The mean clone size (ie, proportion of BCR-ABL+) in the CD34+CD38− fraction of the study cohort of BCR-ABL+ patients was 59.9% ± 32% (mean ± standard deviation; Table 1). We then calculated the difference in TL between BCR-ABL+ and BCR-ABL− cells (ΔTLLSC-HSC) and correlated the difference with the clone size. CML LSC burden was found to be directly correlated with accelerated telomere shortening (expressed as ΔTLLSC-HSC) probably due to the substantial replicative expansion of the BCR-ABL+ stem cell pool already before diagnosis of CML (R2 = 0.367; P = .016; n = 15; Figure 2B).
TL in nonclonal cells assessed in deep molecular remission
Based on previous results describing a preexisting TL deficit in nonleukemic cells from patients with AML in remission after induction therapy,16 we wanted to analyze TL in nonleukemic cells from CML patients in MR.4 In addition, we aimed to investigate whether we could detect a correlation between TL and previous TKI treatment duration and/or a potential use of TL in nonclonal cells as a predictive marker for molecular relapse after TKI cessation. Therefore, we prospectively analyzed 134 CML patients enrolled into the telomere substudy of the EURO-SKI trial. In order to correct for the well-described age dependency of TL,15 we age-adapted TL of granulocytes (aaTLgran) and lymphocytes (aaTLlymph) on the basis of a preexisting large control cohort described previously.18,19 We observed that in lymphocytes mean aaTLlymph (−0.22 ± 1.6 kb, n = 133; P = .02) of CML patients was slightly but significantly shortened (Figure 3A-C). In granulocytes, however, we did not detect a statistically significant difference (0.16 ± 1.13 kb, n = 133; P = .10) compared with age-adapted healthy donors (Figure 3B-C).
Figure 3.

TL of 134 CML patients within the EURO-SKI study. (A) TL (in kb) vs age (in years) in PB lymphocytes of CML patients with loss of (orange squares; n = 58) or maintained (blue circles; n = 75) molecular residual disease. (B) TL (in kb) vs age (in years) in PB granulocytes of CML patients with loss of (green squares; n = 58) or maintained (yellow circles; n = 75) molecular residual disease. (C) Age-adjusted TL (aaTL) of PB lymphocytes (shown in blue; P = .020) and granulocytes (shown in yellow; P = .100) from the EURO-SKI study patients. n.s., not significant.
We next tested whether TL at time of TKI cessation correlates with the risk of molecular relapse expressed as duration of molecular remission after TKI cessation. Interestingly, we found a trend toward a correlation between age-adjusted TL shortening and loss of major molecular response (MR3) over time for the granulocyte (P = .08; supplemental Figure 1A) but not for the lymphocyte (P = .12; supplemental Figure 1B) compartment. We further wanted to analyze the possible impact of previous duration of TKI treatment before TKI cessation on TL in deep molecular remission. We did not observe any significant correlation of aaTLlymph neither in the subgroup with loss of MR3 (R2 = 0.02, P = .35) nor in the subgroup with sustained MR3 (R2 = 0.03, P = .18; supplemental Figure 2A) in the lymphocyte nor in the granulocyte subpopulation (R2 = 0.04, P = .13 for loss of MR3 and R2 = 0.01, P = .46 for sustained MR3, respectively) (supplemental Figure 2B).
Taken together and in line with previous pilot studies,10 TL in nonleukemic granulocytes obtained in major molecular response did not differ from healthy individuals. We conclude that durable treatment (median duration of TKI treatment: 7.2 years, range 3.0 to 14.0 years) with TKIs did not seem to affect TL in nonleukemic myelopoiesis. Whether TKI treatment might play a role for the rather slight but significant shortening of TL observed in lymphocytes will need to be investigated in follow-up studies.
Discussion
Despite its model character for molecular targeted therapy of cancer,25 risk stratification and prognostication of CML have not changed very much since the first development of the Sokal26 and later Hasford Score27 in 1984 and 1998, respectively. Although clinical scores became clinically more refined and adapted to the TKI era, the modification and improvement toward the EUTOS28 or ELTS29 score were mostly achieved by regrouping or different weighing of clinical routine parameters. However, surprisingly and different from most other hematological malignancies, only few additional molecular or cytogenetic abnormalities have so far been identified to gain clinical impact on prognostication or prediction of treatment response in CML at diagnosis.30 This results in our sustained inability to clinically predict phase transition from CP to AP/BC, a clinical endpoint still of utmost importance because outside allogeneic stem cell transplantation, prognosis in advanced stage CML is still very poor with overall survival in the range of 6 to 12 months.
We have previously shown that TL measured in PB of patients with CML can discriminate leukemic myeloid from nonleukemic T cells and correlates with disease stage, clinical prognostic scores,11 and response to treatment13,31 (reviewed in Keller et al32). In addition, we could discriminate early vs late CP by correlating TL with remaining duration of CP in retrospective analysis.8 Furthermore, TL was found to correlate with Ph-positivity of long-term culture initiating cells, LTC-IC measured by conventional cytogenetics (Tim H. Brümmendorf, T. L. Holyoake, P. M. Lansdorp, C. Eaves, unpublished results). Moreover, accelerated telomere shortening (typically observed in late CP CML) in myeloid neoplasia was found to be associated with genetic instability (reviewed in Ohyashiki et al33 and Vajen et al34) as well as with a characteristic inflammatory signature (termed telomere-associated secretory phenotype) potentially contributing to disease progression in a murine model of BCR-ABL+, telomerase knock-out cells.35 Finally, effective execution of telomerase inhibition in a CML cell-line model was shown to be dependent of intact p53 signaling.36 Taken together, impaired telomere maintenance in leukemic as opposed to non-LSCs is suggested to mirror disease progression within CML CP and to be functionally involved in phase transition toward AP/BC.
Until 20 years ago, prognostication of CML and other hematological disorders (reviewed by Brümmendorf and Balabanov14) on the basis of TL measurement has significantly been impaired by suboptimal methodology, age dependency of the parameter, and even more so, by substantial interindividual variability that (on the basis of twin studies) can mostly be attributed to genetic causes.15 Substantial improvements in the methodology to study TL in eukaryotes have been introduced with the development of Q-FISH–based methodologies, particularly with flow-FISH to measure the average length of telomeres in cells37 (also allowing to study large control cohorts used for age-adaptation15,18,19,38). Nevertheless, genetic individual variability can only significantly be reduced by expressing TL in leukemic (target) cells in relation to nonleukemic (control) cells with presumably normal TL. We therefore decided to analyze intraindividual TL specifically in LSC in relation to HSC of selected patients with CML and to compare these results with the degree of Ph-positivity in the CD34+38− stem cell compartment at diagnosis. For this purpose, we use a 2-step combination of routine FISH methodology used for the identification of BCR-ABL+ cells5,7 with a recently developed confocal Q-FISH for TL measurement.17 Thereby, we could demonstrate for the first time that immature LSCs indeed have significantly shortened TL compared with phenotypically identical nonleukemic HSC at diagnosis, thus validating conclusions drawn from previous TL measurements of PB cells and extending these results to the HSC compartment.8,11,13,31 Interestingly and despite the relatively small sample size available for analysis, accelerated telomere shortening was found to be correlated with an increasing leukemic clone size in the HSC compartment, a novel parameter that was previously shown to be correlated with prognosis and response to TKI treatment.5,7 It is important to note that other factors than the replicative history of LSC alone are being reflected in the clone size measured at diagnosis. However, together with previous studies,8,13 the current data support the hypothesis that progressive telomere shortening represents an epigenetic biomarker of CML LSC, correlates with disease progression in CP, and (via increased genetic instability)39 contributes to phase transition of CML.
The assumption that nonleukemic myeloid cells in patients with CML at diagnosis have unaltered TL (and could thereby be used as controls) has not been formally proven to date. In theory and in analogy to AML,16 nonleukemic cells could conceivably have harbored a preexisting, preleukemic telomere deficit potentially contributing to disease onset and evolution. Furthermore, CML cells could potentially alter normal HSC via extrinsic factors that might impact not only on their differentiation and self-renewal capacity but eventually also on TL.40 Consequently, we decided to study TL in nonleukemic cells of CML patients by analyzing the PB obtained in deep molecular remission under TKI treatment within the EURO-SKI study. We found that TL in nonleukemic cells from CML patients was not different between myeloid and lymphoid cells (Figure 2C) and, as opposed to previous studies in AML,16 not meaningfully shortened in myeloid cells compared with age-adjusted controls. Furthermore, no correlation between TKI treatment duration and telomere shortening was observed arguing against sustained replicative telomere-mediated damage to the nonleukemic HSC pool as a consequence of long-lasting TKI treatment. The rather discrete but significant shortening of TL in the lymphoid compartment (that can be translated into 2 to 3 additional population doublings) could potentially be attributed to an increased replicative demand in the lymphoid compartment under TKI treatment and/or a shift in lymphocyte subfractions (with known differences in TL38,41,42) observed to a different degree under the individual TKI treatments.
In conclusion, we propose that despite the presence of quiescent HSC subfractions, overall BCR-ABL+ LSC are characterized by an increased cellular turnover leading to accelerated telomere shortening. At CML-CP diagnosis, telomeres in BCR-ABL+ myeloid cells and as shown here, also in CD34+38− LSC, are already significantly shortened compared with their respective nonleukemic HSC counterparts, and this difference is increasing progressively with decreasing remaining duration of CP.8 Potentially, replication-independent mechanisms such as increased oxidative stress due to BCR-ABL–mediated increased reactive oxygen species production43 on the 1 hand and telomerase activity and/or alternative lengthening of telomeres44 on the other hand might additionally impact on TL (reviewed by Lansdorp45 and by Vasko et al46). However, based on the overall continuous telomere shortening observed, we hypothesized that, by measuring the degree of effective telomere shortening in each individual patient, we might be able to overcome the problem of interindividual variability in TL assessment, thus allowing a better prediction of the remaining duration of CP, and as a consequence, progression toward AP/BC in the future.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
A special thank you to the centers participating in the EURO-SKI and NordCML006 clinical trials. The authors thank Lucia Vankann for the excellent technical support, and the Confocal Microscopy Unit, a core facility of the Interdisciplinary Center for Clinical Research Aachen within the Faculty of Medicine at the RWTH Aachen University.
This work was in part supported by a grant from the Stiftung “Lichterzellen” (T.H.B.) as well as by grants from Finnish Cancer Organizations, Finnish Cancer Institute, and Sigrid Juselius Foundation (M.S.).
Authorship
Contribution: A.-S.B. wrote the manuscript, performed experiments collected, and analyzed and interpreted the data; M.S.V.F. performed experiments and analyzed and interpreted the data; S.A.A., J.R., H.H.-H., A.H., F.-X.M., and S.S. planned the study design, provided clinical data and samples, and analyzed the data; V.K., J.D., J.J., G.O., P.E.W., P.A.W.t.B., and T.F. provided substantial clinical data and samples; S.W., S.H., S.I., M.S., and S.K. collected and analyzed data; S.M. and F.B. planned study design and analyzed and interpreted the data; T.H.B. conceived and planned the study design, interpreted the data, and provided financial support; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: T.H.B. received research funding from Novartis and Pfizer, gave presentations, and participated in advisory boards for Novartis, Pfizer, Ariad, and Janssen (no personal honoraria). H.H.-H. leads the NCMLSG, which conducted the NordCML006 trial with support from BMS. J.J. receives research support from Novartis and BMS, received speaker’s fees from Incyte, Pfizer, Celgene, Roche, and BMS, and participated in advisory boards of BMS, Pfizer, and Novartis. S.K. received research funding from Novartis, BMS, and Janssen (none related to this study), gave presentations, and participated in advisory boards for Novartis, Pfizer, BMS, Incyte/Ariad, and Janssen. S.M. has received honoraria and research funding from Novartis, Pfizer, and BMS (not related to this study). G.O. received honoraria and/or research funding form Novartis, ARIAD, BMS, ROCHE, J&J, and Celgene. J.R. has received honoraria and research funding from Novartis, Pfizer, and BMS (not related to this study). S.S. received honoraria from Novartis, BMS, Incyte, Pfizer, and research funding from Novartis and BMS. The remaining authors declare no competing financial interests.
Correspondence: Tim H. Brümmendorf, Department of Hematology, Oncology, and Stem Cell Transplantation, University Hospital Aachen, Pauwelsstr 30, 52074 Aachen, Germany; e-mail: tbruemmendorf@ukaachen.de.
References
- 1.Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer. 2008;8(5):341-350. [DOI] [PubMed] [Google Scholar]
- 2.Eaves CJ, Eaves AC In: Carella AMDG, Eaves CJ, Goldman JM, Hehlmann R, eds. Chronic myeloid leukaemia: biology and treatment. London: Martin Dunitz; 2001:73-100 [Google Scholar]
- 3.Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94(suppl 2):S107-S121. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez RH, Kantarjian H, Cortes JE. The biology of chronic myelogenous leukemia: implications for imatinib therapy. Semin Hematol. 2007;44(1 suppl 1):S4-S14. [DOI] [PubMed] [Google Scholar]
- 5.Thielen N, Richter J, Baldauf M, Barbany G, Fioretos T, Giles F, Gjertsen BT, Hochhaus A, et al. Leukemic stem cell quantification in newly diagnosed patients with chronic myeloid leukemia predicts response to nilotinib therapy. Clin Cancer res. 2016;22(16):4030-4038. [DOI] [PubMed] [Google Scholar]
- 6.Hjorth-Hansen H, Stenke L, Söderlund S, et al. ; Nordic CML Study Group. Dasatinib induces fast and deep responses in newly diagnosed chronic myeloid leukaemia patients in chronic phase: clinical results from a randomised phase-2 study (NordCML006). Eur J Haematol. 2015;94(3):243-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mustjoki S, Richter J, Barbany G, et al. ; Nordic CML Study Group (NCMLSG). Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia. 2013;27(7):1520-1526. [DOI] [PubMed] [Google Scholar]
- 8.Brümmendorf TH, Holyoake TL, Rufer N, et al. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood. 2000;95(6):1883-1890. [PubMed] [Google Scholar]
- 9.Boultwood J, Fidler C, Shepherd P, et al. Telomere length shortening is associated with disease evolution in chronic myelogenous leukemia. Am J Hematol. 1999;61(1):5-9. [DOI] [PubMed] [Google Scholar]
- 10.Brümmendorf TH, Ersöz I, Hartmann U, et al. Telomere length in peripheral blood granulocytes reflects response to treatment with imatinib in patients with chronic myeloid leukemia. Blood. 2003;101(1):375-376. [DOI] [PubMed] [Google Scholar]
- 11.Drummond M, Lennard A, Brûmmendorf T, Holyoake T. Telomere shortening correlates with prognostic score at diagnosis and proceeds rapidly during progression of chronic myeloid leukemia. Leuk Lymphoma. 2004;45(9):1775-1781. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann U, Balabanov S, Ziegler P, et al. Telomere length and telomerase activity in the BCR-ABL-transformed murine Pro-B cell line BaF3 is unaffected by treatment with imatinib. Exp Hematol. 2005;33(5):542-549. [DOI] [PubMed] [Google Scholar]
- 13.Wenn K, Tomala L, Wilop S, et al. Telomere length at diagnosis of chronic phase chronic myeloid leukemia (CML-CP) identifies a subgroup with favourable prognostic parameters and molecular response according to the ELN criteria after 12 months of treatment with nilotinib. Leukemia. 2015;29(12):2402-2404. [DOI] [PubMed] [Google Scholar]
- 14.Brümmendorf TH, Balabanov S. Telomere length dynamics in normal hematopoiesis and in disease states characterized by increased stem cell turnover. Leukemia. 2006;20(10):1706-1716. [DOI] [PubMed] [Google Scholar]
- 15.Rufer N, Brümmendorf TH, Kolvraa S, et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med. 1999;190(2):157-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ventura Ferreira MS, Crysandt M, Ziegler P, et al. Evidence for a pre-existing telomere deficit in non-clonal hematopoietic stem cells in patients with acute myeloid leukemia. Ann Hematol. 2017;96(9):1457-1461. [DOI] [PubMed] [Google Scholar]
- 17.Hummel S, Ventura Ferreira MS, Heudobler D, et al. Telomere shortening in enterocytes of patients with uncontrolled acute intestinal graft-versus-host disease. Blood. 2015;126(22):2518-2521. [DOI] [PubMed] [Google Scholar]
- 18.Werner B, Beier F, Hummel S, et al. Reconstructing the in vivo dynamics of hematopoietic stem cells from telomere length distributions. eLife. 2015;4:e08687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beier F, Masouleh BK, Buesche G, et al. Telomere dynamics in patients with del (5q) MDS before and under treatment with lenalidomide [published online ahead of print 2015 September 2015]. Leuk Res. doi:10.1016/j.leukres.2015.09.003. S0145-2126(15)30380-5. [DOI] [PubMed] [Google Scholar]
- 20.Schneider RK, Schenone M, Ferreira MV, et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016;22(3):288-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saussele S, Richter J, Guilhot J, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19(6):747-757. [DOI] [PubMed] [Google Scholar]
- 22.Sloma I, Jiang X, Eaves AC, Eaves CJ. Insights into the stem cells of chronic myeloid leukemia. Leukemia. 2010;24(11):1823-1833. [DOI] [PubMed] [Google Scholar]
- 23.Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci USA. 1997;94(10):5320-5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eisterer W, Jiang X, Christ O, et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia. 2005;19(3):435-441. [DOI] [PubMed] [Google Scholar]
- 25.Giustacchini A, Thongjuea S, Barkas N, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med. 2017;23(6):692-702. [DOI] [PubMed] [Google Scholar]
- 26.Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789-799. [PubMed] [Google Scholar]
- 27.Hasford J, Pfirrmann M, Hehlmann R, et al. ; Writing Committee for the Collaborative CML Prognostic Factors Project Group. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. J Natl Cancer Inst. 1998;90(11):850-858. [DOI] [PubMed] [Google Scholar]
- 28.Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118(3):686-692. [DOI] [PubMed] [Google Scholar]
- 29.Pfirrmann M, Baccarani M, Saussele S, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. 2016;30(1):48-56. [DOI] [PubMed] [Google Scholar]
- 30.Fabarius A, Leitner A, Hochhaus A, et al. ; Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) and the German CML Study Group. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760-6768. [DOI] [PubMed] [Google Scholar]
- 31.Brummendorf TH, Ersoz I, Hartmann U, et al. Normalization of previously shortened telomere length under treatment with imatinib argues against a preexisting telomere length deficit in normal hematopoietic stem cells from patients with chronic myeloid leukemia. Ann N Y Acad Sci. 2003;996:26-38. [DOI] [PubMed] [Google Scholar]
- 32.Keller G, Brassat U, Braig M, Heim D, Wege H, Brümmendorf TH. Telomeres and telomerase in chronic myeloid leukaemia: impact for pathogenesis, disease progression and targeted therapy. Hematol Oncol. 2009;27(3):123-129. [DOI] [PubMed] [Google Scholar]
- 33.Ohyashiki K, Iwama H, Tauchi T, et al. Telomere dynamics and genetic instability in disease progression of chronic myeloid leukemia. Leuk Lymphoma. 2000;40(1-2):49-56. [DOI] [PubMed] [Google Scholar]
- 34.Vajen B, Thomay K, Schlegelberger B. Induction of chromosomal instability via telomere dysfunction and epigenetic alterations in myeloid neoplasia. Cancers (Basel). 2013;5(3):857-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braig M, Pällmann N, Preukschas M, et al. A ‘telomere-associated secretory phenotype’ cooperates with BCR-ABL to drive malignant proliferation of leukemic cells. Leukemia. 2014;28(10):2028-2039. [DOI] [PubMed] [Google Scholar]
- 36.Brassat U, Balabanov S, Bali D, Dierlamm J, Braig M, Hartmann U, Sirma H, Gunes C, et al. Functional p53 is required for effective execution of telomerase inhibition in BCR-ABL-positive CML cells. Exp Hematol. 2011;39(1):66-76.e1-2. [DOI] [PubMed] [Google Scholar]
- 37.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nat Protoc. 2006;1(5):2365-2376. [DOI] [PubMed] [Google Scholar]
- 38.Aubert G, Baerlocher GM, Vulto I, Poon SS, Lansdorp PM. Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PLoS Genet. 2012;8(5):e1002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samassekou O, Ntwari A, Hébert J, Yan J. Individual telomere lengths in chronic myeloid leukemia. Neoplasia. 2009;11(11):1146-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welner RS, Amabile G, Bararia D, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27(5):671-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baerlocher GM, Lansdorp PM. Telomere length measurements in leukocyte subsets by automated multicolor flow-FISH. Cytometry A. 2003;55(1):1-6. [DOI] [PubMed] [Google Scholar]
- 42.Beier F, Balabanov S, Amberger CC, et al. Telomere length analysis in monocytes and lymphocytes from patients with systemic lupus erythematosus using multi-color flow-FISH. Lupus. 2007;16(12):955-962. [DOI] [PubMed] [Google Scholar]
- 43.Nieborowska-Skorska M, Kopinski PK, Ray R, et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood. 2012;119(18):4253-4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samassekou O, Malina A, Hébert J, Yan J. Presence of alternative lengthening of telomeres associated circular extrachromosome telomere repeats in primary leukemia cells of chronic myeloid leukemia. J Hematol Oncol. 2013;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lansdorp PM. Maintenance of telomere length in AML. Blood Adv. 2017;1(25):2467-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vasko T, Kaifie A, Stope MB, Kraus T, Ziegler P. Telomeres and telomerase in hematopoietic dysfunction: prognostic implications and pharmacological interventions. Int J Mol Sci. 2017;18(11):2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.