

Abstract
Rationale:
Behçet disease (BD) is an inflammatory disorder characterized by recurrent oral aphthous ulcers, genital ulcers, ocular lesions, and skin lesions. Complication of amyloidosis in patients with BD is rare. Here, we report a case of BD with immunoglobulin light chain (AL)-amyloidosis manifested as hematochezia.
Patient concerns:
A 61-year-old man developed sudden hematochezia due to bleeding from multiple small colonic ulcers; AL-amyloid deposition was found on immunohistochemical examination of biopsy specimen of colonic ulcer. Systemic investigation revealed cardiac disfunction with cardiomegaly and progressive renal dysfunction, which indicated the presence of systemic AL-amyloidosis.
Diagnoses:
Based on the findings of colonic ulcers with cardiac and renal involvement, a diagnosis of systemic AL-amyloidosis complicated by incomplete BD was established.
Interventions:
He was treated with increased dose of oral prednisolone (20 mg/day), colchicine and mesalazine, because he was reluctant to receive aggressive chemotherapy (melphalan and dexamethasone) or autologous stem cell transplantation.
Outcomes:
Colonic ulcers completely diminished after treatment, however, he died because of severe urinary tract infection and progressive renal failure after one year of gastrointestinal (GI) manifestations.
Lessons:
Our case shows that patients with BD may have GI manifestations due not only to entero-BD but also due to GI amyloidosis.
Keywords: AL-amyloidosis, Behçet disease, gastrointestinal manifestation
1. Introduction
Behçet disease (BD) is an inflammatory disorder characterized by recurrent oral aphthous ulcers, genital ulcers, ocular lesions (uveitis), and skin lesions.[1,2] Gastrointestinal (GI) manifestations of BD are common; and they are of importance for association with significant morbidity and mortality.[3] Complication of amyloidosis in patients with BD is rare. The most common manifestation of amyloidosis with BD is renal amyloidosis, and these cases usually show amyloid A (AA)-type deposition in the kidney,[4–6] whereas GI manifestation of amyloidosis in patients with BD is extremely rare.[5] So far, no patients with BD with immunoglobulin light-chain (AL)-amyloidosis accompanied with GI manifestations have been reported. Here, we report a rare case of BD with AL-amyloidosis manifested as hematochezia due to colonic ulcers.
2. Case report
A 61-year-old man was referred to our hospital in December 2014 for dyspnea, progressive anemia, and renal dysfunction. At the age of 30 years, according to the criteria for BD,[1] he was diagnosed with an incomplete BD because of oral aphtha, genital ulcers, and superficial thrombophlebitis. He received 5 mg of oral prednisolone (PSL) for 30 years. Owing to poor control of BD, he repeatedly developed skin ulcers on lower legs despite PSL treatment. He had a history of bilateral femoral head necrosis and had underwent surgery for prosthetic replacement of both femoral heads. Additionally, he underwent lumbar-peritoneal shunt procedure for idiopathic normotensive hydrocephalus at the age of 44 years. At admission, his body temperature was 36.9°C and blood pressure was 140/89 mm Hg. A physical examination revealed coarse crackles in the left lung and oral aphtha and skin ulcers (approximately 1 cm diameter) on both the lower legs. There were no ophthalmologic symptoms. A blood examination demonstrated elevated C-reactive protein level (13.08 mg/dL, normal range: below 0.14 mg/dL) and erythrocyte sedimentation rate (74 mm at 1 hour, normal: 3–15 mm at 1 hour); he had anemia (hemoglobin, 9.8 g/dL, normal: 11.6–14.8 g/dL) and his creatinine levels were 2.1 mg/dL (normal: 0.46–0.79 mg/dL) with a positive urine test for protein (2+) and blood (1+). Serum IgG, IgA, and IgM levels were 414 mg/dL (normal: 861–1747 mg/dL), 153 mg/dL (normal: 93–393 mg/dL), and 78 mg/dL (normal: 50–269 mg/dL), respectively. Serum M-protein was not detected, and free light chain kappa/lambda ratio was within normal limits (1.433, normal range: 0.2–1.65). Antinuclear antibody and urine Bence Jones protein were negative. A chest X-ray revealed consolidation of both the lower lung lobes, and he was diagnosed with bacterial pneumonia and received an antibiotic treatment. Toward the end of December 2014, he suddenly developed a massive hematochezia. Occurrence of entero-BD was suspected. Emergency colonoscopy showed multiple, small, round-shaped ulcers extending from the ascending to the transverse colon (Fig. 1A, B). A biopsy from the margin of transverse colon ulcer revealed amorphous eosinophilic extracellular deposits in the vascular wall (Fig. 2A). The deposits stained positive on Congo red staining and showed apple-green birefringence under polarized light (Fig. 2B, C). Immunohistochemistry revealed that the deposits were positive for lambda light chain, but showed faint staining for kappa light chain, which seemed to be nonspecific reaction (Fig. 2D, E). Systemic amyloidosis was suspected, and echocardiography was performed to assess cardiac function. It showed diffuse cardiac hypertrophy with thickened interventricular septum (12.4 mm). Ventricular wall showed high echogenicity with decreased ejection fraction (39.1%), which indicated amyloid deposition in the heart. Brain-type natriuretic peptide levels were elevated to 939.2 pg/mL (normal: <18.4 pg/mL). Bone marrow examination showed hypocellular bone marrow with no evidence of proliferation of neoplastic plasma cells or lymphoid cells. Skin biopsy from his leg ulcer showed no evidence of amyloid deposition. Based on these findings, a diagnosis of systemic AL-amyloidosis complicated by incomplete BD was established.[7] However, he was reluctant to receive aggressive chemotherapy (melphalan and dexamethasone) or autologous stem-cell transplantation. Therefore, he received an increased dose of oral PSL (20 mg/day), oral colchicine (1 mg/day), and mesalazine (3000 mg/day). GI symptoms were ameliorated by treatment. Second colonoscopy performed after 8 months revealed complete resolution of colonic ulcers. However, renal dysfunction progressed, and he repeatedly suffered from infections. He died because of severe urinary tract infection and progressive renal failure after one year of GI manifestations.
Figure 1.
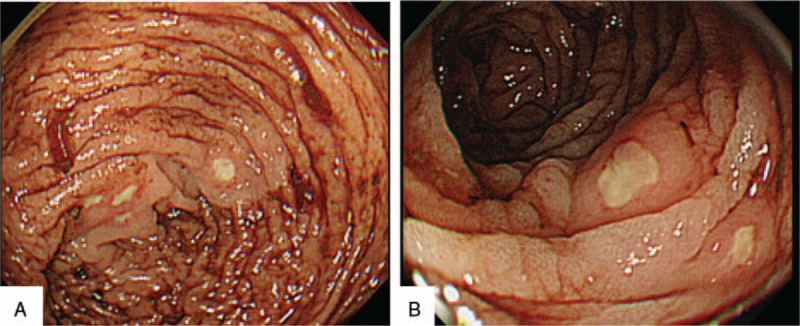
(A, B) Emergent colonoscopy was performed. Endoscopic images showing multiple, small round-shaped ulcers extending from ascending colon to transverse colon.
Figure 2.
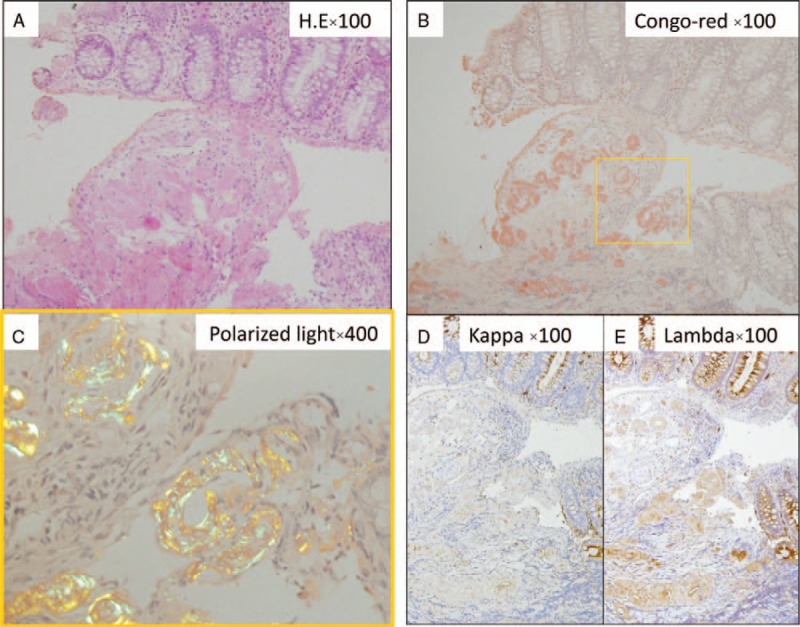
(A) Biopsy from a transverse colon ulcer revealed amorphous eosinophilic extracellular deposits in the vascular wall. (B) The deposits were positive for Congo red staining and (C) showed apple green birefringence under polarized light. On immunohistochemistry, the deposits were positive for lambda light chain (D), but stained faintly for kappa light chain (E), which seemed to be nonspecific reaction.
3. Discussion
This patient developed sudden hematochezia caused by multiple small colonic ulcers during the clinical course of BD, and AL-amyloid deposition was observed in the colon ulcers. We considered a possibility of local AL-amyloid deposition (local AL-amyloidosis) of the colon; however, local plasma cell proliferation was not observed in colon biopsies. Furthermore, concomitant cardiac and renal involvement was quite different from the features of local AL-amyloidosis. On the contrary, there was no histologic evidence of myeloma, clonal plasma cell proliferation, or hematologic malignancies in bone marrow aspiration. Although serum M-protein and skewed kappa/lambda ratios were not detected (long-term oral steroid therapy can be affected to serologic tests), a diagnosis of incomplete BD with systemic AL-amyloidosis was finally established according to the criteria for diagnosing AL-amyloidosis.[7] To the best of our knowledge, this is the first case of BD with AL-amyloidosis which developed hematochezia as an initial manifestation of GI involvement. Amyloidosis is a rare complication of BD. The reported prevalence of amyloidosis in patients with BD ranges from 0.04% to 3%.[4,5] The most common manifestation of amyloidosis is renal amyloidosis, which often manifests as nephrotic syndrome.[6] GI manifestation of patients with BD is mainly derived from the disease itself (entero-BD) and not from GI amyloidosis.[3] Till date, only 4 patients (age 33–54 years) with BD with prominent GI symptoms due to amyloidosis have been reported.[8–11] Four out of 5 patients were males (including our case, Table 1). The interval from BD onset to GI manifestations ranged from 7 to 31 years. The main symptom of GI amyloidosis with BD was diarrhea (4/5 cases, 80%), whereas hematochezia was our case only. Most cases (80%) showed deposition of AA-amyloid in the GI tract, which was thought to reflect secondary amyloidosis (inflammation-associated amyloidosis) due to chronic inflammation associated with BD.[9] However, Chiba et al reported that about a half of the patients with BD complicated with amyloidosis (10/19 cases) showed amyloid deposition in the GI tract,[9] which suggests that patients with BD with amyloidosis often develop deposition of amyloid (AA-type) in the GI tract without major GI symptoms. AL-amyloid (called primary amyloid) refers to the immunoglobulin light-chain associated amyloid, a plasma-cell dyscrasia related to multiple myeloma; the clonal plasma cells in the bone marrow produce amyloidogenic immunoglobulins.[12] AA-type amyloid refers to the inflammation-associated amyloid (secondary amyloid). The most common form of systemic amyloidosis is AL-amyloidosis and patients with AL-amyloidosis often develop heart and kidney involvement; on the contrary, GI involvement is relatively rare (8% of patients with AL-amyloidosis).[13] In our case, entero-BD was initially suspected because of GI manifestations; however, endoscopic findings differed from those typically observed in entero-BD.[3] For instance, Skef et al reported that multisegmental and diffuse colonic involvement were rare in entero-BD (6%) and 85% of these patients had <6 ulcers (67% had single ulcer; 18% had 2–5 ulcers); furthermore, ulcer size is usually large (>1 cm).[3] On the contrary, endoscopic features of AL-amyloidosis are nonspecific and include erythema, erosions/ulcerations, granular or plaque-like mucosa of the stomach or small intestine, and polypoid protrusions.[13]
Table 1.
Reported patients with Behçet disease with prominent gastrointestinal symptoms due to amyloidosis.

4. Conclusion
The patients with BD may have GI manifestations due to both entero-BD and GI amyloidosis. Further investigation of such cases is needed to clarify the pathogenesis of AL-amyloid deposition in patients with BD.
Author contributions
Conceptualization: Shuzo Sato.
Data curation: Shuzo Sato, Naoki Matsuoka, Satoshi Kawana, Tomoyuki Asano.
Formal analysis: Satoshi Kawana.
Investigation: Shuzo Sato, Makiko Yashiro, Satoshi Kawana, Tomoyuki Asano, Kazuhiro Tasaki, Yuko Hashimoto, Kiyoshi Migita.
Methodology: Satoshi Kawana, Kazuhiro Tasaki.
Project administration: Yuko Hashimoto.
Supervision: Kiyoshi Migita.
Validation: Hiroko Kobayashi, Hiroshi Watanabe, Yuko Hashimoto, Kiyoshi Migita.
Writing – original draft: Shuzo Sato, Satoshi Kawana.
Writing – review & editing: Makiko Yashiro, Naoki Matsuoka, Tomoyuki Asano, Kazuhiro Tasaki, Hiroko Kobayashi, Hiroshi Watanabe, Yuko Hashimoto, Kiyoshi Migita.
Footnotes
Abbreviations: AA-type = amyloid-A type, AL-amyloidosis = immunoglobulin light chain amyloidosis, BD = Behçet disease, GI = gastrointestinal, PSL = prednisolone.
S. Sato had full access to the data in this study and mainly drafted the manuscript. All authors contributed to drafting the manuscript and the critical revision. All authors read and approved the final version of the manuscript to be published.
Informed consent was obtained from the patient. Because of a case report of single patient, ethical approval was waived for institutional review board in Fukushima Medical University.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Weichsler B, Davatchi F, Mizushima Y, et al. Criteria for diagnosis of Behçet's disease. International Study Group for Behçet's disease. Lancet 1990;335:1078–80. [PubMed] [Google Scholar]
- [2].Sakane T, Takeno M, Suzuki N, et al. Behçet's disease. N Engl J Med 1999;341:1284–91. [DOI] [PubMed] [Google Scholar]
- [3].Skef W, Hamilton MJ, Arayssi T. Gastrointestinal Behçet's disease: a review. World J Gastroenterol 2015;21:3801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dilşen N, Koniçe M, Aral O, et al. Behçet's disease associated with amyloidosis in Turkey and in the world. Ann Rheum Dis 1988;47:157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Melikoğlu M, Altiparmak MR, Fresko I, et al. A reappraisal of amyloidosis in Behçet's syndrome. Rheumatology (Oxford) 2001;40:212–5. [DOI] [PubMed] [Google Scholar]
- [6].Akpolat T, Akpolat I, Kandemir B. Behcet's disease and AA-type amyloidosis. Am J Nephrol 2000;20:68–70. [DOI] [PubMed] [Google Scholar]
- [7].Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol 2005;79:319–28. [DOI] [PubMed] [Google Scholar]
- [8].Wechsler B, Le Thi HD, Hamza M, et al. Renal and digestive amyloidosis and Behçet's disease. Apropos of a case [in French]. Rev Med Interne 1986;7:361–4. [DOI] [PubMed] [Google Scholar]
- [9].Chiba M, Inoue Y, Arakawa H, et al. Behçet's disease associated with amyloidosis. Gastroenterol Jpn 1987;22:487–95. [DOI] [PubMed] [Google Scholar]
- [10].Hamza M, Wechsler B, Godeau P, et al. Intestinal amyloidosis: an unusual complication of Behçet's disease. Am J Gastroenterol 1988;83:793–4. [PubMed] [Google Scholar]
- [11].Erten S, Perçınel S, Olmez U, et al. Behçet's disease associated with diarrhea and secondary amyloidosis. Turk J Gastroenterol 2011;22:106–7. [DOI] [PubMed] [Google Scholar]
- [12].Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003;349:583–96. [DOI] [PubMed] [Google Scholar]
- [13].Petre S, Shah IA, Gilani N. Review article: gastrointestinal amyloidosis - clinical features, diagnosis and therapy. Aliment Pharmacol Ther 2008;27:1006–16. [DOI] [PubMed] [Google Scholar]