

Abstract
Lung cancer is one of the most common malignancies. In spite of the progress made in past decades, further studies to improve current therapy for lung cancer are required. Dynamically controlled by methyltransferases and demethylases, methylation of lysine and arginine residues on histone proteins regulates chromatin organization and thereby gene transcription. Aberrant alterations of histone methylation have been demonstrated to be associated with the progress of multiple cancers including lung cancer. Inhibitors of methyltransferases and demethylases have exhibited anti-tumor activities in lung cancer, and multiple lead candidates are under clinical trials. Here, we summarize how histone methylation functions in lung cancer, highlighting most recent progresses in small molecular inhibitors for lung cancer treatment.
Keywords: Histone methylation, Histone demethylation, Lung cancer, Histone methyltransferase, Histone demethylase, Inhibitors
Abbreviations: ALK, anaplastic lymphoma kinase; DUSP3, dual-specificity phosphatase 3; Elk1, ETS-domain containing protein; EMT, epithelial-to-mesenchymal transition; HDAC, histone deacetylase; IHC, immunohistochemistry; KDMs, lysine demethylases; KLF2, Kruppel-like factor 2; KMTs, lysine methyltransferases; LSDs, lysine specific demethylases; MEP50, methylosome protein 50; NSCLC, non-small cell lung cancer; PAD4, peptidylarginine deiminase 4; PCNA, proliferating cell nuclear antigen; PDX, patient-derived xenografts; PRC2, polycomb repressive complex 2; PRMTs, protein arginine methyltrasferases; PTMs, posttranslational modifications; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl-L-methionine; SCLC, small cell lung cancer; TIMP3, tissue inhibitor of metalloproteinase 3
1. Introduction
Lung cancer is one of the most prevalent malignancies and the leading cause of cancer-related death in the US and in China [1, 2]. Among patients with lung cancer, non-small cell lung cancer (NSCLC) accounts for about 85% and small cell lung cancer (SCLC) accounts for the remaining 15% [3]. According to the pathological phenotypes, NSCLC includes adenocarcinoma, squamous cell carcinoma, and large cell lung carcinoma [4]. The past decades have witnessed the clinical introduction of several small molecular inhibitors, which significantly improved overall prognosis of lung cancer [[5], [6], [7]]. However, many patients still suffer from poor response to drug therapy due to individually differences in genetic, epigenetic, phenotypic or psychosocial features [8, 9]. Therefore, precision medicine aiming to provide personalized targeted treatment becomes an attractive strategy to improve the drug efficacy against lung cancer [[10], [11], [12]]. Epigenetic differences among patients have been considered as a critical factor in the development of precision medicine [13], and in various malignancies including lung cancer, epigenetic dysregulation has been identified to play a crucial role in the tumorigenicity and heterogeneity [[14], [15], [16], [17]].
Epigenetic dysregulation is usually resulted from aberrant changes in DNA and histone modifications. In eukaryotic cell, genomic DNA is wrapped around a protein octamer which contains four core histones (H2A, H2B, H3, H4), forming the structure of the nucleosome [18]. Each of the histone proteins possesses a tail, which is a classic location where various posttranslational modifications (PTMs) function [19, 20]. Through changing the charge density between DNA and histones, DNA methylation and histone PTMs (acetylation, methylation, and phosphorylation) can regulate the loosening of the nucleosome, affecting the access of transcription factors and RNA polymerase to their target genes [[21], [22], [23], [24]]. Currently, DNA methylation has been widely accepted as important biomarkers in the clinical management of lung cancer, since DNA methylation-based biomarkers provide useful information in distinct clinical questions about early diagnosis, staging, prognosis and therapy-response prediction [25]. However, questions about whether other epigenetic modifications can be explored as lung cancer therapeutic targets never stopped during the last decade. Histone acetylation has been demonstrated to play a vital role in lung cancer development by activating gene transcription [16]. Although some histone deacetylase (HDAC) inhibitors, such as Vorinostat and Panobinostat, have gained optimistic results in pre-clinical and clinical trials on NSCLC, further studies of HDAC inhibitors in lung cancer are necessary for evaluating their anti-tumor effect [16, 23]. Histone methylation, one of the most well studied patterns among histone modifications, can either promote or inhibit transcription at different gene loci, thus plays a rather complex role in lung cancer [21]. It is believed that the methylation of lysine (K) and arginine (R) residues on histone tails largely determines the chromatin configurations and, hence, biological outcomes [19]. Like other histone modifications, histone methylation is a dynamic process regulated by a series of ‘eraser’ and ‘writer’ enzymes. Methylation ‘erasers’ and ‘writers’ respectively remove and add specific methyl marks crucial for gene expression, genomic stability and cell fate [19]. Methyltransferase ‘writers’ and the corresponding demethylase ‘erasers’ for histone lysine residue are termed as histone lysine methyltransferases/demethylases (KMTs/KDMs for short respectively). For histone arginine residues, the ‘writers’ and ‘erasers’ are histone arginine methyltransferases and histone arginine demethylases respectively.
Some of these histone methylation modifiers have been identified in cancers with altered activities, suggesting their oncogenic or tumor-suppressor roles [19, 26]. Aberrations of histone methylation modifiers have been closely intertwined with lung cancers as well [14, 27]. Moreover, along with the deeper understanding of the patterns and functions of histone methylation in lung cancer, several inhibitors targeting histone methylation modifiers have entered clinical trials [22]. It may be a right time to review and rethink the potential of histone methylation for developing lung cancer therapy, however, there lacks a systematic review about this issue. Here, we discussed the functions and related structural foundations of histone methylation modifiers in lung cancer, and highlighted the most recent progresses in lung cancer therapy targeting histone methylation.
2. KMTs and their Roles in Lung Cancer
KMTs can remove methyl groups on lysine residues of histones or non-histone substrates [28, 29]. Based on the similarity of structural organization and catalytic domain, KMTs are divided into two categories, SET domain-containing KMTs and the only non-SET-domain-containing KMT DOT1L (Fig. 1A and B) [30, 31]. The first histone KMTs identified in human is the H3K9 methyltransferase SUV39H1, a mammalian homologue of Drosophila Su(var 3–9) [32]. Since then, more histone KMTs have been discovered, which target H3K4 [33], H3K9 [26, 34], H3K27 [35, 36], H3K36 [37], H3K79 [38, 39] or H4K20 [40]. In addition to their essential roles in physiologic activities, such methyltransferases are found to closely associate with variant cancers. Here, the structures and functions of representative histone methyltransferases and their therapeutic potentials for lung cancer are summarized (Table 1).
Fig. 1.

Schematic representations and structures of representative histone methylation ‘writers’. (A) Schematic representations and structure of EZH2. EZH2 belongs to SET-containing KMTs and catalyzes methylation at lysine residues via SET domain (PDB ID 4MI5). (B) Schematic representations and structure of DOT1L, the only non-SET-containing KMT enzyme (PDB ID 1NW3). (C) Schematic representations and structure of PRMT1 (PDB ID 1OR8). SANT (Swi3, Ada2, N-Cor, and TFIIIB), a domain that allows chromatin remodeling proteins to interact with histones. CXC, a cys-rich region preceding the SET domain. DNMTs, DNA methyltransferases.
Table 1.
Histone methyltransferases with reported functions in lung cancer.
| Name | Target | Links to lung cancer |
|---|---|---|
| MLL2 | H3K4 | Loss of expression and deleterious mutations in NSCLC [63] |
| G9a | H3K9 | Overexpressed in lung cancers [185] |
| EZH2 | H3K27 | Overexpressed in lung cancers [50, 51] |
| SMYD2 | H3K36 | Contributed to NSCLC cell growth [73] |
| SETD2 | H3K36 | Deleterious mutations in primary NSCLC [74] |
| WHSC1L1 | H3K36 | Over expressed in lung cancer [186] |
| DOT1L | H3K79 | Contributed to NSCLC cell growth [84] |
| SETD8/PRSET7 | H4K20 | Overexpressed in lung cancer [76] |
| SUV4-20H1/2 | H4K20 | H4K20me3 decreased during tumor progression [187] |
| PRMTs | Arginine on H3 and H4 | Contributed to NSCLC cell growth and overexpressed in TKI-resistant NSCLC [135] |
2.1. SET Domain-Containing KMTs
The SET domain comprises approximately 130 residues, and is regarded as the evolutionarily conserved catalytic motif of KMTs (Fig. 1A). It was originally identified from three Drosophila proteins, i.e. Suppressor of variegation 3–9 (Su(var) 3–9), Enhancer of zeste (E (z)) and Trithorax (Trx), which involve in epigenetic process [41]. The SET domains of most of histone KMTs bind to histones as well as methyl donors (S-adenosyl-L-methionine, also known as AdoMet or SAM) and reaction products (S-adenosyl-L-homocysteine, also known as AdoHcy or SAH) [42]. Most SET-containing histone KMTs function SAM-dependently or SAH-dependently. A knot-like structure within the SET domain contributes to form the methyltransferase active site where lysine methylation tends to occur [43]. Documented aberrant SET-domain containing KMTs in lung cancer are reviewed below.
2.1.1. EZH2
EZH2, the human homologue of Drosophila En (zeste), is the key catalytic component of the Polycomb repressive complex 2 (PRC2). With the help of the cofactors SUZ12 and EED in a SAM-dependent manner, EZH2 plays a pivotal role of transferring one, two and three methylation marks to H3K27 (H3K27me1, me2, me3) (Fig. 3A) [[44], [45], [46]].
Fig. 3.

Cartoons of representative histone methylation and demethylation reaction catalyzed by different enzymes. (A) SET-containing EZH2 as a PRC2 core component methylates H3K27 via SET domain. (B) Non-SET-domain-containing DOT1L methylates H3K79 SAM-dependently. (C) SAM-dependent arginine methylation on histone H3 and H4. (D) FAD-dependent H3K4me2/me1 demethylation by non-JmjC KDM LSD1. (E) JmjC-containing KDM4A demethylates H3K36me3/me2 and H3K9me3/me2 via JmjC-domain-mediated reaction involving αKG and Fe(II).
High levels of EZH2 and correlated H3K27me3 are closely related to the poor clinical outcome of cancers, including lower overall survival and disease-free survival [[47], [48], [49], [50], [51]]. Additionally, advanced NSCLC patients with positive EZH2 expression compared with those with negative EZH2, showed resistance to platinum-based chemotherapy [52].
Over-expressed EZH2 promotes lung cancer progression in multiple ways involving proliferation, apoptosis inhibition, migration and metastasis. Studies demonstrated a mutual regulation between EZH2 and the vascular endothelial growth factor-A (VEGF-A) signaling pathway and AKT phosphorylation, which closely linked to enhanced cell proliferation, migration and metastasis [53, 54]. Increased EZH2 expression was also found to be closely associated with either E2F amplification or loss of RB1, which induced disruption of the E2F/Rb pathway, in 96% SCLC samples [55]. Aberrant methylation of PRC2 target genes contribute to generate a ‘stem-cell like’ hypermethylation signature in SCLC, leading to highly aggressive tumor phenotype such as rapid cell growth [55]. Moreover, elevated EZH2 expression promoted SCLC progression, by suppressing apoptosis through epigenetically silencing TGF-β type II receptor (TβRII) [56]. Coordinately, silencing EZH2 inhibited lung cancer cell growth by cell cycle disruption and triggering cell death [[57], [58], [59]]. The oncogenic role of EZH2 in lung cancer was now clearly demonstrated by all these works, and several EZH2 inhibitors under clinical trials exhibited potential to be applied as novel targeted therapy or as an aid to current drug therapy for lung cancer (more detailed discussion in 5.1, 5.2) [60, 61]. The results of clinical trials and further mechanistic investigations would be essential for the potential application of EZH2 inhibitors in lung cancer treatment.
2.1.2. MLL2
The MLL (KMT2) methyltransferases family members, which specifically methylate H3K4, are implicated in various cancers either by dysregulation or loss of function [62]. A whole-exome sequencing identified MLL2 as one of the most frequent mutated genes in Chinese NSCLC patients [63]. Loss of MLL2 expression was commonly observed in NSCLC, and deleterious mutations of MLL2 were found in 11.4% NSCLC patients [63]. MLL2 mutations in human SCLC cell lines was associated with reduced MLL2 protein levels and reduced monomethylation of H3K4, and frequent inactivated mutations of MLL2 were also identified in some of primary SCLC clinical samples and SCLC cell lines [64]. However, a recent study found that MLL2 mutation was associated with reduced survival in NSCLC but not in SCLC, indicating that MLL2 may act differently in different lung cancer subtypes [65]. Since MLL2 mutations usually resulted in genome instability [66], mutant MLL2 might drive the initiation of lung cancer, yet this assumption needed to be verified with more evidences.
2.1.3. G9a
G9a is a KMT responsible for the mono- and di-methylation of H3K9 (H3K9me1, me2) [67]. Highly expressed G9a was observed in aggressive lung cancer cells and the progression of mouse lung cancer induced by urethane [68, 69]. RNAi-mediated G9a silencing reduced cell migration and invasion in vitro and in vivo [68]. Mechanistic investigations revealed that G9a knockdown suppressed the cell adhesion molecule Ep-CAM by reducing the levels of H3K9me2 and disrupting the recruitment of transcriptional cofactors at the Ep-CAM promoter [68]. Similar mechanism was found between G9a and EMT-related proteins, according to another study which demonstrated the metastasis-promoting role of G9a in lung cancer cells [70]. G9a also silenced caspase 1 (CASP1) by increasing the levels of H3K9me2 around CASP1 promoter, thereby promoted NSCLC cell growth and invasion [71]. Moreover, over-expression of G9A or low expression of CASP1 was strongly correlated with poor overall survival in lung cancer [71]. Apart from the oncogenic role of G9a in lung cancer discussed above, G9a inhibition was reported to potentiate the anti-tumor activity of DNA double-strand break (DSB) inducing agents [72]. Further studies may be necessary to investigate whether G9a plays a role in resistance to chemotherapy in lung cancer.
2.1.4. SMYD2 and SETD2
SET and MYND domain-containing 2 (SMYD2) is one of the H3K36-specific methyltransferases, which methylates lysine residues in anaplastic lymphoma kinase (ALK) and contributes to oncogenic ALK activation. Combination treatment of a SMYD2 inhibitor LLY-507 and an ALK inhibitor crizotinib exhibited significantly enhanced suppression on NSCLC cell growth compared with mono-treatment of either agent [73].
Interestingly, unlike SMYD2, another H3K36-specific methyltransferase SETD2 act as a tumor suppressor in lung cancer. Deleterious mutations SETD2 were detected in primary NSCLC tumors [74]. Loss of SETD2 and subsequent decrease of H3K36me3 led to significant tumor-promoting consequences, accelerating both early- and late-stage lung adenocarcinoma tumors in mice [75]. The results indicate that SETD2 may be further explored as a diagnostic or prognostic marker for NSCLC. Moreover, the examples of different roles of H3K36-specific KMTs in lung cancer implicate a complex and precisely orchestrated regulation network for different target genes mediated by the same H3K36 methylation.
2.1.5. SETD8
SETD8, known as KMT5A or SET8, specifically targets H4K20 for methylation and has been implicated in multiple cancer processes [76]. Previous studies revealed that SETD8 was related with proper cell cycle progression, DNA damage response, and transcriptional regulation. Elevated expression of SETD8 stimulated S-phase progression via methylating a non-histone protein proliferating cell nuclear antigen (PCNA), thus promoted proliferation of lung cancer cells [76]. Additionally, SETD8 was directly inhibited by a tumor suppressor miR-382 in NSCLC cells, which led to inhibition in NSCLC cell tumorigenesis and metastasis [77]; whereas restoration of SETD8 can enhance NSCLC cell proliferation, migration and invasion in vitro [77]. SETD8 was also reported to reprogram cancer cell metabolism via hypoxia-inducible factor-1α (HIF-1α) mediated process by stabilizing HIF-1α protein through post-transcriptional regulation [78]. Meanwhile, SETD8 was also capable of monomethylating the tumor suppressor p53 on lysine 382, which can attenuate the pro-apoptotic and growth arrest functions of p53 [79]. Taken together, SETD8 seems to function in lung tumorigenesis and metastasis beyond a mere histone methyltransferase role.
2.2. DOT1L, a Non-SET-Domain-Containing KMT
The catalytic activity of SET domain is not the only determining factor of KMTs function [22, 80]. DOT1L is the only known H3K79 methyltransferase, and shares structural similarities with type I protein arginine methyltrasferases (PRMTs) [81, 82]. Through a spatial arrangement at its N terminal, DOT1L catalyzes SAM-dependent methylation on nucleosomal substrates (Fig. 1B and 3B) [81]. DOT1L and H3K79me3 were identified as promising targets for the treatment of acute myelocytic leukemia (AML), but played a rather unclear role in lung cancer [83].
H3K79 methylation was up-regulated in lung cancer cell lines and clinical tumor tissues. DOT1L knockdown reduced H3K79 methylation and led to disturbed cell proliferation; additionally, chromosomal missegregation occurred in DOT1L-deficient lung cancer cells, resulting in cell cycle arrest at G1 phase and subsequent senescence [84]. Interestingly, during the process of TGF-β1-induced epithelial-to-mesenchymal transition (EMT) in lung cancer, H3K79me3 was decreased without association with DOT1L expression; and DOT1L inhibitors, EPZ5676 and SGC0946, were not effective on EMT-related genes [85]. These seemingly contradictory results indicate a complex mechanism for H3K79 methylation in lung cancer, more studies are required to establish a link between different roles of DOT1L and H3K79 methylation.
2.3. Other KMTs
Reports about the role of other KMTs in lung cancer are relatively rare. Studies in cancer cell lines might provide a glimpse of their roles, revealing directions to further investigations. For examples, H4K20-specific KMTs SUV4-20H1/2 played an important role in maintaining genomic stability through methylating H4K20 to H4K20me2 and H4K20me3 [86]; WHSC1L1, a KMT for H3K36, enhanced activation of ERK pathway by mono-methylating lysine 721 of the tyrosine kinase domain of epidermal growth factor receptor (EGFR) [87]. These discoveries suggested that there are remain multiple unknowns about KMTs in lung cancer. More KMTs, like above-mentioned, with unrevealed yet study-worthy functions in lung cancer will be identified in future research.
3. KDMs and their Roles in Lung Cancer
Methylation on protein lysine or arginine residues was not regarded as a reversible PTM until the discovery of the first histone demethylase in 2004 [88, 89]. Based on the oxidative mechanism of the demethylation reaction and the structure of the catalytic domains, KDMs can be categorized into lysine specific demethylases (LSDs, or KDM1 subfamily) and Jumonji (JmjC)-domain-containing demethylases (JmjC KDMs, or KDM2–7 subfamilies). So far, >20 KDMs have been discovered and characterized, and many of them have been reported to be dysregulated in multiple diseases [90, 91]. Here, we summarize representative aberrant KDMs in lung cancer (Table 2).
Table 2.
Histone demethylases with reported functions in lung cancer.
| Name | Target | Links to lung cancer |
|---|---|---|
| KDM1A,LSD1 | H3K4me2/me1, H3K9me2/me1 | Overexpressed in lung cancer [97, 98] |
| KDM2A | H3K36me2/me1 | Overexpressed in NSCLC [105] |
| KDM3A | H3K9me2/me1 | Overexpressed in NSCLC [108] |
| KDM4A | H3K9me3/me2,H3K36me3/me2 | Overexpressed in lung cancer [115] |
| KDM4C | H3K9me3/me2,H3K36me3/me2 | Overexpressed in lung sarcomatoid carcinoma [188] |
| KDM4D | H3K9me3/me2/me1,H3K36me3/me2 | Overexpressed in lung cancer [115] |
| KDM5A | H3K4me3/me2 | Overexpressed in lung cancer [117] |
| KDM6A | H3K27me3/me2/me1 | Loss led to lung tumorigenesis [123] |
| JMJD6 | Arginine on H3 and H4 | Overexpressed in lung cancer [137] |
| PAD4 | Arginine on H3 and H4 | Overexpression led to gefitinib resistance in NSCLC [143] |
3.1. Non-JmjC-Domain-Containing KDMs
The LSD family members demethylate lysine residues with a cofactor flavin adenine dinucleotide (FAD) (Fig. 2A) [92, 93]. Interestingly, because the forming of an imine intermediate requires protonated amine during the demethylation process, the LSD family members are only capable of demethylating mono- and dimethyl lysine residues (me1, me2), but not trimethylated (me3) lysine residues (Fig. 3D) [89].
Fig. 2.

Schematic representations and structures of representative histone methylation ‘erasers’. (A) Schematic representations and structure of LSD1, which functions without JmjC domain (PDB ID 2DW4). (B) Schematic representations and structure of KDM4A, a classic JmjC family demethylase. KDM4A can remove methyl groups from either H3K9 or H3K4 via reactions on different sites (JmjC domain and H3K9me3, PDB ID 2OX0. Tudor domain and H3K4me3, PDB ID 2GFA). (C) Secondary structures of histone arginine ‘demethylases’ PAD4 (PDB ID 3APN) and JMJD6 (PDB ID 3LD8). The schematic arrangement of PAD4 is relatively insufficient, and JMJD6 shares similar schematic structure with other JmjC family demethylases. SWIRM (Swi3, Rsc, and Moira) domain, a proposed anchor site for histone molecules. PHD, hydrophobic cage of residues that bind methylated peptides.
LSD1 (or KDM1A), the first reported and most studied KDM demethylase, belongs to the non-JmjC-domain-containing LSD family [88]. LSD1 usually functions on H3K4 as a key component of the CoREST complex, yet it may change target to H3K9 with the presence of the androgen receptor, thus acting either as a transcriptional corepresssor or a coactivator [94, 95].
As a H3K4 and H3K9 KDM enzyme, LSD1 demonstrated aberrant overexpression and acted as a classic oncogene in various cancers including lung cancer [96]. Overexpressed LSD1 was closely correlated with shorter overall survival of NSCLC patients [97]. Consistently, LSD1 silencing resulted in significant suppression of proliferation of lung cancer cell lines [96]; moreover, SCLC was sensitive to a LSD1 inhibitor GSK-2879552 [98]. LSD1 was recruited to Kruppel-like factor 2 (KLF2) or E-cadherin promoters via binding with several lncRNAs in NSCLC cells, resulting in promoted tumor proliferation and EMTs [99, 100]. Expression of tissue inhibitor of metalloproteinase 3 (TIMP3) was repressed by LSD1-mediated H3K4me2 demethylation at TIMP3 promoter, which consequently enhanced MMP2 expression as well as JNK phosphorylation, and eventually promoted the metastasis of NSCLC cells [101]. A LSD1/ integrin β3 axis was also reported to attribute to tumor progression and invasiveness in lung adenocarcinoma [102]. Due to its critical roles in promoting lung cancer as well as various developed specific inhibitors, LSD1 is regarded as a highly promising target for treating lung cancer.
3.2. JmjC KDMs
Unlike LSD family, JmjC KDMs have been proven to remove all three methylation states from lysine residues [103]. The JmjC KDMs catalyze demethylation utilizing Fe(II) as a cofactor, and 2-oxoglutarate (2-OG) and α-ketoglutarate (αKG) as co-substrates (Fig. 3E) [90, 92]. The JmjC domain, folding into eight β-sheets, provides an active pocket for αKG and Fe(II) binding [93]. Dysregulation of JmjC KDMs has been observed in different cancers. Representative JmjC KDMs associated with lung cancer are discussed below.
3.2.1. KDM2A
KDM2 catalyzes demethylation on H3K36, which is associated with gene activation [104]. KDM2A, but not its homologue KDM2B, was reported to be associated with lung cancer. KDM2A was found highly dysregulated in 54 NSCLC cell lines according to Affymetrix microarray gene expression data, and its mRNA and protein levels are significantly higher in primary NSCLC tumor samples than in adjacent normal lung tissues [105]. KDM2A-catalyzed H3K36me2 demethylation occurred gene-specifically at the promoter region of cancer-related genes including dual-specificity phosphatase 3 (DUSP3), which in turn antagonized DUSP3-mediated ERK1/2 dephosphorylation and consequently promoted lung tumorigenesis [105]. KDM2A also transcriptionally repressed the histone deacetylase 3 (HDAC3) by demethylating H3K36me2 at the HDAC3 promoter, thereby up-regulated HDAC3 target genes including the cell invasion-associated NANOS1 and the cell cycle-related CDK6 in KDM2A-overexpressing NSCLC cells [106]. However, the lack of ideal inhibitors to KDM2A limits the potential amplification of KDM2A-based therapy. Recent discovery of highly selective inhibitor of KDM2A might provide opportunities to develop KDM2A targeted therapy for lung cancer [107].
3.2.2. KDM3A
The expression level of KDM3A, a H3K9-specific KDM demethylase, was found upregulated in more than half of the NSCLC cases [108]. KDM3A activated Homeobox A1 (HOXA1) transcription by removing methyl groups from H3K9me2 at HOXA1 promoter, stimulated the activation of HOXA1 downstream target gene CCND1, an essential factor of the cell cycle progression, thus positively regulated G1/S transition in A549 cell line [108]. Additionally, KDM3A knockdown decreased the expression of tumor-promoting EZH2 and increased the anti-tumor miRNA let-7c expression, thus inhibited tumorigenesis in NSCLC cell lines and xenograft model [109].Recently, KDM3A was also found to facilitate the immune evasion of A549 cells by promoting Foxp3 transcription [110], which provides another evidence that KDM3A acts as an oncogene in lung cancer. Interestingly, KDM3A showed an anti-apoptotic function by erasing monomethylaion from p53K372, thus disturbing the stability of chromatin-bound p53 in cancer and promoted drug resistance [111], which brings new insight to understanding the role of KDM3A in lung cancer.
3.2.3. KDM4A and KDM4D
KDM4 subfamily can demethylate H3 at K4, K9 and K36 residues. KDM4B and KDM4C are structurally and catalytically similar to KDM4A, which is well studied, whereas KDM4D is unique for its lack of PHD and Tudor domains [112]. The JmjC domain in KDM4A is responsible for demethylating H3K9me3/me2 and H3K36me3/me2, and the substrate of Tudor domain is H3K4me3 (Fig. 2B) [113]. Aberration of KDM4 subfamily members were discovered in various cancers including breast cancer and prostate cancer, yet reports on the link between KDM4 subfamily members and lung cancer are relatively rare [22, 114]. Soini et al. found that both KDM4A and KDM4D appeared to significantly relate to the metastasis of lung cancer. They observed more nuclear and cytoplasmic KDM4A expression in the tumors with lymph node metastasis than in tumors with no metastasis, and the same tendency also occurred in nuclear expression of KDM4D. They also discovered an association between cytoplasmic KDM4A expression and poor prognosis both in survival and recurrence free interval [115]. To further understand these observations, additional studies are required to establish a mechanistic link between KDM4A and its oncogenic role in lung cancer.
3.2.4. KDM5A
KDM5A, a H3K4-spesific KDM demethylase, was found to be implicated in developing drug resistance in a study on the epigenetic basis of cancer drug resistance [116], and it was enriched in lung cancer tissues as well as drug-resistant cells [117]. KDM5A bound directly to the promoters of integrin-β1, which was reported to mediate cell-matrix interaction [118, 119], thereby promoted cell migration and invasion. Meanwhile, KDM5A bound to the promoters of cell-cycle-related genes cyclin D1 and p27, promoting cell proliferation directly by repressing p27 and activating cyclin D1 and indirectly up-regulating cyclin E1 expression. Additionally, a selective inhibitor of KDM5A, YUKA1, suppressed cell proliferation and prevented drug resistance in various cancer cell lines including A549 [120], implicating its application potential in lung cancer treatment.
3.2.5. KDM6A
The link between lung cancer and KDM6A, a H3K27-specific demethylase which usually acts antagonistically to EZH2, was previously studied in NSCLC cells with controversial outcomes: KDM6A epigenetically antagonized TGF-β-induced EMT process [121]; whereas its inhibitor GSKJ4 demonstrated anticancer-effect on a set of NSCLC cell lines [122]. However, a recent study unveiled KDM6A as an important tumor suppressor gene in lung cancer with evidence gained from human lung cancer specimens and transgenic NSCLC mouse models [123]. KDM6A knockout resulted in an increased EZH2 level and an up-regulated H3K27me3 level, and significantly promoted lung tumorigenesis in vivo [123].
Interestingly, the KDM6A-knockout lung tumors appeared to be more sensitive to EZH2 inhibitor, indicating that NSCLC patients with KDM6A loss may get more benefit from EZH2 inhibition therapy [123]. Such preferentially sensitivity was also identified in other malignant diseases. For examples, loss of KDM6A amplified PRC2-regulated transcriptional repression of IGFBP3 in urothelial bladder cancer and promoted tumor growth, and sensitized bladder cancer cells and tumors to EZH2 inhibition [124]; moreover, loss of KDM6A led to malignant phenotype in multiple myeloma via deactivating the expression of multiple genes including IRG4 and c-MYC, and EZH2 inhibitors performed better anti-tumor effects in KDM6A loss cases by rebalancing H3K27me3 levels at specific genes [125]. This balance between KDM6A and EZH2 may be important in guiding therapeutic strategy in multiple diseases, including lung cancer.
3.3. Other KDMs
Although other JmjC KDM subfamilies members are supposed to involve in tumoringenesis, for example KDM6B in T-cell acute lymphoblastic leukemia (T-ALL) [126], reports of their roles in lung cancer are very limited. Additionally, there is no report that relates KDM7 subfamily with lung cancer. Future analysis in clinical samples may help identify their roles in lung cancer.
4. Histone Arginine Methylation/Demethylation in Lung Cancer
Histone arginine methylation participates in epigenetic regulation largely by cross-talks with other epigenetic modifications [127]. With the capacity to prevent or enhance the binding of important transcriptional factors, histone arginine methylation is found in both repressed and active chromatin states [128].
4.1. Histone Arginine Methylation
Methylation on arginine residues is catalyzed by protein arginine methyltransferases (PRMTs) family, which transfers the methyl group from SAM to the guanidino group of arginine [129]. The catalytic core of PRMT consists of a β-barrel (unique to PRMT), a methyltransferase domain, and a dimerization arm (conserved in type I PRMT) [130]. PRMTs are categorized into two types according to their structural similarity. Type I PRMTs, such as PRMT1–4, 6 and 8, catalyze methylation SAM-dependently with a SAM-binding site like DOT1L (Fig. 1C) [131]. The only identified type II PRMT is PRMT5, which forms a protein complex with methylosome protein 50 (MEP50) to exert its catalytic function (Fig. 3C) [21, 128, 132].
PRMT1, PRMT4 and PRMT6 demonstrated higher expression in lung cancer tissues, while abrogation of each resulted in growth suppression [133, 134]. Moreover, PRM1 played a role in lung cancer metastasis. For instance, silencing PRMT1 decreased a mitogenic factor called Neuromedin B receptor while increased epithelial markers cytokeratins 7 and 8, which consequently resulted in reduced cell proliferation and enhanced tumor differentiation [134]. Additionally, up-regulated PRMT1 repressed E-cadherin activity and promoted EMT in erlotinib-resistant NSCLC cells (erlotinib is a tyrosine kinase inhibitor against NSCLC) [135].
4.2. Histone Arginine Demethylation
4.2.1. JMJD6
Arginine methylation is very stable, and whether it can be directly demethylated by enzymes remained unclear until the discovery of a putative histone arginine demethylase, JMJD6 [128, 136]. The JmjC-containing JMJD6 was previously known as a phosphatidylserine receptor (Fig. 2C) [128]. Significantly high expression of JMJD6 in lung adenocarcinoma was found positively correlated with tumor size and pleural invasion, and led to significantly poor clinical outcomes [137]. Moreover, an elevated level of JMJD6 was positively associated with pathological grade, pT status and pN status, indicating its potential to be a clinical diagnostic and prognostic marker for NSCLC [137]. Notably, targeting JMJD6 may provide additional ways to treat lung cancer, since suppressing JMJD6 via acetylating its upstream transcriptional factor HOXB9 resulted in a decrease in tumor growth and migration in xenograft models [138].
4.2.2. PAD4
Peptidylarginine deiminase 4 (PAD4) targets arginine sites on histone H3 and H4 [139, 140]. Methylation marks on histone arginine residues occasionally convert into citrullination marks via PAD4-mediated hydrolysis, representing another form of ‘demethylation’ (Fig. 2C) [141].
PAD4 exacerbated TNF-α-induced lung inflammation [142]. In addition, PAD4 expression decreased significantly in gefitinib-resistant NSCLC cells (gefitinib, a widely-used tyrosine kinase inhibitor against NSCLC) [143]. Overexpression of PAD4 inhibited EMT activity by suppressing ETS-domain containing protein (Elk1), which was reported to regulate EMT process [144], and thus restrained gefitinib resistance [143].
5. Representative Histone Methyltransferase/Demethylase Inhibitors
Inhibitors targeting either histone methyltransferases or demethylases have been widely reported to exert anti-tumor activities for multiple malignancies either in singe-agent therapy or combination therapy. In pre-clinical studies, for examples, a classic G9a inhibitor BIX-01294 induced autophagy-associated cell death and impaired tumor growth in breast cancer [145], oral squamous cell carcinoma [146] and hepatocellular carcinoma [[145], [146], [147]]; GSKJ4, the selective inhibitor of KDM6A and KDM6B, not only effectively suppressed tumor progression in AML [148], breast cancer [149], ovarian cancer [150] and castration-resistant prostate cancer [151], but also enhanced the radiosensitivity of multiple tumor cell lines [152]. Some of the inhibitors have entered clinical trials after their anti-cancer potentials identified in pre-clinical studies. For single-agent therapy, some LSD1 inhibitors such as TCP/ATRA are under phase I/II clinical trials for AML therapy (Trial number: NCT0273102, NCT2267). For combination therapy, the most potent DOT1L inhibitor EPZ5676 showed synergy with daunorubicin and cytarabine, two standard agents in current chemotherapy for AML [153, 154]; an EZH2 inhibitor EPZ6438 (tazemetostat), in combination with prednisolone, entered phase II clinical trials for diffuse large B-cell lymphoma (Trial number: NCT01897571). In general, more and more histone methylation modifier inhibitors have been identified as potential reagents aiding cancer therapy. For lung cancer, to the best of our knowledge, inhibitors targeting EZH2 and LSD1 demonstrated most prominent anti-tumor effects (Table 3).
Table 3.
Representative inhibitors of EZH2 or LSD1 in lung cancer.
| Compound | Structure | Mechanism and potency | Clinical Trial Number | Ref |
|---|---|---|---|---|
| DZNep | 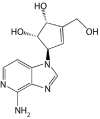 |
SAH hydrolase inhibitor (Ki = 50 pM) |
N/A | [155, 160] |
| GSK2816126 (GSK126) | 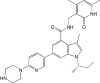 |
SAM-competitive EZH2 inhibitor (IC50 = 9.9 nM) |
NCT02082977 | [165, 172] |
| EPZ6438 (Tazemetostat) | 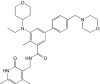 |
SAM-competitive EZH2 inhibitor (Ki = 2.5 nM) (IC50 = 11 nM) |
NCT01897571 NCT02601950 NCT02601937 |
[167, 172] |
| CPI1205 | 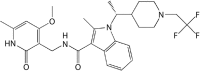 |
SAM-competitive EZH2 inhibitor |
NCT02395601 | [168, 172] |
| GSK-2879552 | 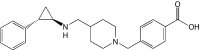 |
FAD-dependent irreversible LSD1 inhibitor (Kiapp = 1.7 μM) |
NCT02034123 | [98, 176] |
| RG6016 (ORY-1001) | 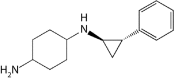 |
FAD-dependent irreversible LSD1 inhibitor (IC50 < 20 nM) |
NCT02913443 | [177] |
N/A: Not available.
5.1. EZH2 Inhibitors
The structure of the conserved SET domain in EZH2 predicts two critical binding pockets for inhibitors: the key methyl donor SAM and the H3K27 substrate [155]. So far, almost all the small molecular inhibitors targeting EZH2 are SAM-competitive [156]. Although such EZH2 inhibitors are usually obtained via diverse independent screens, most of them share a common structure namely pyridine group [22]. Recently, a series of 4-amino-2,2′,6,6′-tetramethylpiperidine analogues, which inhibited EZH2 in a SAM-competitive manner, were identified yet demonstrated weaker cellular potency comparing to the pyridone-based inhibitors [157, 158]. EZH2 inhibitors are widely applied in pre-clinical and clinical trials of various cancers due to the remarkable link of EZH2 dysregulation to oncogenesis in multiple tissue types [159]. For lung cancer, some EZH2 inhibitors stood out with promising therapeutic potentials.
3-dezaneplanocin-A (DZNep) was the first and best known EZH2 inhibitor identified through drug screening [160]. It is a SAH hydrolase inhibitor, which indirectly inhibits the methylation reaction via interfering with SAM and SAH metabolism [155]. Investigations on NSCLC cell lines revealed a significant dose-dependent growth inhibitory effect of DZNep [161]. Further investigation using a whole-body physiologically based pharmacokinetic (PBPK) models predicted that DZNep administration at proper dose could exert anti-tumor effect in vivo [162]. However, the lack of specificity, which may lead to unwanted SAM-dependent reactions, along with its short half-life, limited the clinical translation of DZNep [163].
Ever since then, more selective EZH2 inhibitors with better anti-lung cancer effects have been identified. For example, GSK2816126 was obtained through optimization of a previously identified compound GSK-A [164], which was SAM-competitive but noncompetitive versus histones [165]. GSK2816126 was highly selective to both WT and mutant EZH2 and showed slow inhibitor-enzyme dissociation [165, 166]. GSK2816126 not only inhibited migration of A549 dose-dependently and showed similar effect to gefitinib at the same doses, it also inhibited angiogenesis in vitro and in vivo [60].
Other representative optimized EZH2 specific pyridone-based inhibitors include EPZ6438 [167] and CPI1205 [168], which showed selective inhibition for EZH2 with improved oral bioavailability [169]. EPZ6438, CPI1205 and GSK2816126 have already entered clinical trials (Trial number: NCT01897571, NCT02601950, NCT02601937, NCT02395601, and NCT02082977) and achieved satisfying positive results in solid tumors, indicating their potential for further investigation in lung cancer [[170], [171], [172]]. Moreover, pharmacological EZH2 inhibition also sensitized lung tumors to other inhibitors. For examples, either DZNep or GSK126 administration promoted the anti-tumor effect of TopoII inhibitor doxorubicin against BRG1 and EGFR mutant lung cancer, suggesting an opportunity for combination of traditional chemotherapy medicines and EZH2 inhibitors in NSCLC [61].
5.2. LSD1 Inhibitors
The catalytic domain of LSD1 contains a FAD-binding site. It is a highly conserved functional region indispensible for LSD1-mediated demethylation process [94]. Despite varied chemical characters of presently known LSD1 inhibitors, all LSD1 inhibitors in clinical trials are FAD-dependent irreversible LSD1 inhibitors [173]. Two most potent LSD1 inhibitors, GSK-2879552 and RG6016 (also known as ORY-1001), share a privileged scaffold called tranylcypromine (TCP), indicating that TCP is critical for further designing of LSD1 inhibitors [173]. Although accumulating reports linked LSD1 aberrations with multiple malignancies, the most optimizing results of LSD1 inhibition therapy were obtained in AML and SCLC [174, 175]. Both GSK-2879552 and RG6016 represented a promising novel epigenetic approach for SCLC therapy.
GSK-2879552 was first discovered and characterized in 2015, which irreversibly inactivated the catalytic activity of LSD1 [98]. GSK-2879552 led to enhanced H3K4 methylation at loci of LSD1 target genes in a dose and time dependent manner, thus increased the activation of genes important for cell-development, leading to significant cytostatic effect in various SCLC cell lines and anti-tumor activity in three SCLC xenograft models [98]. In patient-derived xenografts (PDX) models, stronger methylation signatures were associated with increased sensitivity to GSK-2879552 [98, 176]. Following preclinical validation, GSK-2879552 has stepped into Phase I clinical trials for relapsed/refractory SCLC (Trial number: NCT02034123). Similarly, RG6016 inhibited the proliferation of multiple SCLC cell lines and xenograft growth, and has also entered Phase I clinical trials for SCLC therapy (Trial number: NCT02913443). Interestingly, RG6016 treatment was only effective on cell lines and xenografts with certain similar gene expression pattern which also observed in SCLC patient samples, indicating that RG6016 responsive gene signature may help identifying SCLC patients who may benefit from LSD1-based therapy [177].
6. Challenges and Future Directions
Alteration of histone methylation patterns is widely proved to play a vital role in multiple malignancies. Recent progress made in this field has drawn attention to the role of histone methylation in lung cancer. Dysregulation of histone methylation ‘writers’ and ‘erasers’ are closely linked to clinical outcomes in lung cancer patients through a variety of cellular pathways relating proliferation, invasion, EMT etc. Some histone methylation modifiers, such as SETD8, KDM2A and KDM6A, were identified as oncogenes or tumor suppressor genes in lung cancer [76, 105, 123]; some demonstrated potential link with drug resistance to lung cancer therapy, for example KDM5A and KDM3A [111, 117]. JMJD6 and KDM4A might serve as biomarkers for lung cancer although further mechanistic investigations were necessary [115, 137]. Lung tumors with KDM6A loss was more sensitive to EZH2 inhibition, indicating lung cancer patients with specific histone methylation features might benefit from specific epigenetic therapy [123]. Despite that some questions, such as how H3K36-specific KMTs function differently in lung cancer, remain unclear, inhibitors targeting histone methylation modifiers have gradually entered clinical trials for lung cancer therapy and show optimizing anti-tumor effect in either mono-treatment or combination treatment [60, 61, 178]. All these progress indicate the potential importance of histone methylation for lung cancer.
Although there is no applications in everyday clinical practice yet, in depth studies on the histone methylation dysregulation of lung cancer not only help better understand the mechanism of tumorigenesis and development of lung cancer, but also lead to discovery of potential molecular targets, biomarkers, and therapeutic candidates for lung cancer. Additionally, lung cancer with certain histone methylation features may provide new fine classifications in lung cancer that eventually leads to precise personal medicine. Moreover, the development of histone methylation modifier inhibitors, such as EZH2 or LSD1 inhibitors, not only provides promising therapeutic choices for lung cancer treatment, but also may benefit patients who are resistant to current targeted therapies like tyrosine kinase (e.g. EGFR) inhibition by combining with chemotherapy [61, 173].
Meanwhile, challenges still exist and have revealed directions for future research. First, targeting one histone methyl residue or one certain related enzyme may result in diverse unpredictable effects. This is because the interactions between histones, histone methyltransferases and histone demethylases appear to be influenced by the subtle environment surrounding the residue, and crosstalk between different methylated sites are also involved [179]. For example, H3K9me3 prevented SET7-dependent mono-methylation on H3K4 [180], and H3K4me3 prevented G9a- or other H3K9 KMTs-mediated methylation on H3K9 [179, 181]. Second, the lack of complete information and thorough understanding about histone methylation remains an obstacle to further developing epigenetic strategies for lung cancer. For instance, until now, H3K79-specific KDMs have not been reported yet [182]. Additionally, the regulatory mechanism behind the precise assignment of different methyltransferas/demethylasessjbe is not fully explained [183]. The third challenge is to develop more specific inhibitors with clinical application potential, since most histone methyltransferases or demethylases share similar structures and catalytic domains. Take SET domain for example, it participates in a number of reactions, whereas a majority of KMTs contain this domain [42]. A number of inhibitors targeting SET domain were found lacking specificity, which may lead to unwanted effects [184]. The first two challenges require further mechanistic investigations, which are important to complete the map of histone methylation in lung cancer. To deal with the third challenge, rational drug design is necessary to develop relative inhibitors with high selectivity and satisfactory pharmacokinetics. In addition, more pre-clinical and clinical studies are also required to evaluate the anti-tumor efficacy and side effects of new drugs.
In summary, targeting histone methylation is a promising therapeutic strategy for lung cancer treatment. Ever since the recognition of the significance of epigenetic dysregulation in lung cancer, extensive studies have revealed complex but precisely orchestrated regulations mediated by different histone methyltransferases/demethylases. Along with the inhibitors under study or in trials, these efforts have paved the way for an era of better lung cancer therapy.
Acknowledgments
Acknowledgements
We sincerely appreciate the investigators and authors who have contributed to this field, and apologize that we could not discuss and cite all of their works in this mini-review due to space limitations. This work was supported by the Natural Science Foundation of China (31471208, 31671195, 81573461, & 81703552), the Natural Science Foundation of Hubei Province (2017CFB651), the Academic Frontier Youth Team Project of HUST (2016001), Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST) and the Natural Science Foundation of the Self-dependent Innovation of HUST (2016YXMS144), the Fundamental Research Funds for the Central Universities, HUST (2018).
Conflict of Interest
The authors declare no conflict of interest regarding this work.
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Chen W. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Wakelee H., Kelly K., Edelman M.J. 50 Years of progress in the systemic therapy of non-small cell lung cancer. Am Soc Clin Oncol Educ Book. 2014:177–189. doi: 10.14694/EdBook_AM.2014.34.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gridelli C. Non-small-cell lung cancer. Nat Rev Dis Primers. 2015;1:15009. doi: 10.1038/nrdp.2015.9. [DOI] [PubMed] [Google Scholar]
- 5.Reck M. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382(9893):709–719. doi: 10.1016/S0140-6736(13)61502-0. [DOI] [PubMed] [Google Scholar]
- 6.Piperdi B., Merla A., Perez-Soler R. Targeting angiogenesis in squamous non-small cell lung cancer. Drugs. 2014;74(4):403–413. doi: 10.1007/s40265-014-0182-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lovly C.M. Combating acquired resistance to tyrosine kinase inhibitors in lung cancer. Am Soc Clin Oncol Educ Book. 2015:e165–e173. doi: 10.14694/EdBook_AM.2015.35.e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hensing T. A personalized treatment for lung cancer: molecular pathways, targeted therapies, and genomic characterization. Adv Exp Med Biol. 2014;799:85–117. doi: 10.1007/978-1-4614-8778-4_5. [DOI] [PubMed] [Google Scholar]
- 9.Sun Z. New development of inhibitors targeting the PI3K/AKT/mTOR pathway in personalized treatment of non-small-cell lung cancer. Anticancer Drugs. 2015;26(1):1–14. doi: 10.1097/CAD.0000000000000172. [DOI] [PubMed] [Google Scholar]
- 10.Larry Jameson, J. And D.L. Longo, Precision medicine—personalized, problematic, and promising. Obstet Gynecol Surv, 2015. 70(10): p. 612–614. [DOI] [PubMed]
- 11.Sampsonas F. Molecular testing and personalized treatment of lung cancer. Curr Mol Pharmacol. 2014;7(1):22–32. doi: 10.2174/187446720701150105171219. [DOI] [PubMed] [Google Scholar]
- 12.Gadgeel S.M. Personalized therapy of non-small cell lung Cancer (NSCLC) Adv Exp Med Biol. 2016;890:203–222. doi: 10.1007/978-3-319-24932-2_11. [DOI] [PubMed] [Google Scholar]
- 13.Chen C. Five critical elements to ensure the precision medicine. Cancer and Metastasis Reviews. 2015;34(2):313–318. doi: 10.1007/s10555-015-9555-3. [DOI] [PubMed] [Google Scholar]
- 14.Schiffmann I. Epigenetic therapy approaches in non-small cell lung cancer: update and perspectives. Epigenetics. 2016;11(12):858–870. doi: 10.1080/15592294.2016.1237345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J Physiol. 2014;592(3):505–521. doi: 10.1113/jphysiol.2013.266411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petta V. Histones and lung cancer: are the histone deacetylases a promising therapeutic target? Cancer Chemother Pharmacol. 2013;72(5):935–952. doi: 10.1007/s00280-013-2223-9. [DOI] [PubMed] [Google Scholar]
- 17.Huang J. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics. 2015;10(1):62–72. doi: 10.4161/15592294.2014.990780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang W. Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy. 2017;13(5):941–954. doi: 10.1080/15548627.2017.1293768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Audia J.E., Campbell R.M. Histone modifications and cancer. Cold Spring Harb Perspect Biol. 2016;8(4):a019521. doi: 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Ding Y., Zheng L. Transgenerational Epigenetics. 2014. Chapter 9 – histone-mediated transgenerational epigenetics; pp. 87–103. [Google Scholar]
- 21.Cheng X. Structural and functional coordination of DNA and histone methylation. Cold Spring Harb Perspect Biol. 2014:6(8). doi: 10.1101/cshperspect.a018747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mcgrath J., Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther. 2015;150:1–22. doi: 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Damaskos C. Histone deacetylase inhibitors as a novel targeted therapy against non-small cell lung Cancer: where are we now and what should we expect? Anticancer Res. 2018;38(1):37–43. doi: 10.21873/anticanres.12189. [DOI] [PubMed] [Google Scholar]
- 24.Ding Y. DNA hypomethylation of inflammation-associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. Int J Obes (Lond) 2014;38(2):198–204. doi: 10.1038/ijo.2013.98. [DOI] [PubMed] [Google Scholar]
- 25.Fleischhacker M. The role of DNA methylation as biomarkers in the clinical management of lung cancer. Expert Rev Respir Med. 2013;7(4):363–383. doi: 10.1586/17476348.2013.814397. [DOI] [PubMed] [Google Scholar]
- 26.Kimura H. Histone modifications for human epigenome analysis. J Hum Genet. 2013;58(7):439–445. doi: 10.1038/jhg.2013.66. [DOI] [PubMed] [Google Scholar]
- 27.Song J.S. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Int. 2012;62(3):182–190. doi: 10.1111/j.1440-1827.2011.02776.x. [DOI] [PubMed] [Google Scholar]
- 28.Moore K.E., Gozani O. An unexpected journey: lysine methylation across the proteome. Biochim Biophys Acta. 2014;1839(12):1395–1403. doi: 10.1016/j.bbagrm.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson S.M. Proteome-wide enrichment of proteins modified by lysine methylation. Nat Protoc. 2014;9(1):37–50. doi: 10.1038/nprot.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen A.T., Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25(13):1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen A.T. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rea S. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 33.Deb M. Chromatin dynamics: H3K4 methylation and H3 variant replacement during development and in cancer. Cell Mol Life Sci. 2014;71(18):3439–3463. doi: 10.1007/s00018-014-1605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mozzetta C. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol. 2015;16(8):499–513. doi: 10.1038/nrm4029. [DOI] [PubMed] [Google Scholar]
- 35.Nichol J.N. H3K27 methylation: a focal point of epigenetic deregulation in Cancer. Adv Cancer Res. 2016;131:59–95. doi: 10.1016/bs.acr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conway E., Healy E., Bracken A.P. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42–48. doi: 10.1016/j.ceb.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Rogawski D.S., Grembecka J., Cierpicki T. H3K36 methyltransferases as cancer drug targets: rationale and perspectives for inhibitor development. Future Med Chem. 2016;8(13):1589–1607. doi: 10.4155/fmc-2016-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vlaming H., van Leeuwen F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma. 2016;125(4):593–605. doi: 10.1007/s00412-015-0570-5. [DOI] [PubMed] [Google Scholar]
- 39.Farooq Z. The many faces of histone H3K79 methylation. Mutat Res Rev Mutat Res. 2016;768:46–52. doi: 10.1016/j.mrrev.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng X., Collins R.E., Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jenuwein T. SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell Mol Life Sci. 1998;54(1):80–93. doi: 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian C., Zhou M.M. SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell Mol Life Sci. 2006;63(23):2755–2763. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs S.A. The active site of the SET domain is constructed on a knot. Nat Struct Biol. 2002;9(11):833–838. doi: 10.1038/nsb861. [DOI] [PubMed] [Google Scholar]
- 44.Cao R., Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15(1):57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Garcia E., Licht J.D. Deregulation of H3K27 methylation in cancer. Nat Genet. 2010;42(2):100–101. doi: 10.1038/ng0210-100. [DOI] [PubMed] [Google Scholar]
- 46.Wan D. MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating Wnt signaling. J Mol Cell Biol. 2017;9(4):325–337. doi: 10.1093/jmcb/mjx027. [DOI] [PubMed] [Google Scholar]
- 47.Varambally S. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 48.Kleer C.G. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100(20):11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weikert S. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int J Mol Med. 2005;16(2):349–353. [PubMed] [Google Scholar]
- 50.Wan L. Quantitative analysis of EZH2 expression and its correlations with lung cancer patients' clinical pathological characteristics. Clin Transl Oncol. 2013;15(2):132–138. doi: 10.1007/s12094-012-0897-9. [DOI] [PubMed] [Google Scholar]
- 51.Sato T. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep. 2013;3:1911. doi: 10.1038/srep01911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu C. Expression of the enhancer of zeste homolog 2 in biopsy specimen predicts chemoresistance and survival in advanced non-small cell lung cancer receiving first-line platinum-based chemotherapy. Lung Cancer. 2014;86(2):268–273. doi: 10.1016/j.lungcan.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 53.Geng J. EZH2 promotes tumor progression via regulating VEGF-A/AKT signaling in non-small cell lung cancer. Cancer Lett. 2015;359(2):275–287. doi: 10.1016/j.canlet.2015.01.031. [DOI] [PubMed] [Google Scholar]
- 54.Riquelme E. VEGF/VEGFR-2 upregulates EZH2 expression in lung adenocarcinoma cells and EZH2 depletion enhances the response to platinum-based and VEGFR-2-targeted therapy. Clin Cancer Res. 2014;20(14):3849–3861. doi: 10.1158/1078-0432.CCR-13-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coe B.P. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One. 2013;8(8) doi: 10.1371/journal.pone.0071670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murai F. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-beta-Smad-ASCL1 pathway. Cell Discov. 2015;1:15026. doi: 10.1038/celldisc.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia H. EZH2 silencing with RNA interference induces G2/M arrest in human lung cancer cells in vitro. Biomed Res Int. 2014;2014:348728. doi: 10.1155/2014/348728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hubaux R. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol. 2013;8(8):1102–1106. doi: 10.1097/JTO.0b013e318298762f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H. MiR-138 inhibits tumor growth through repression of EZH2 in non-small cell lung cancer. Cell Physiol Biochem. 2013;31(1):56–65. doi: 10.1159/000343349. [DOI] [PubMed] [Google Scholar]
- 60.Chen Y.T. The novel EZH2 inhibitor, GSK126, suppresses cell migration and angiogenesis via down-regulating VEGF-A. Cancer Chemother Pharmacol. 2016;77(4):757–765. doi: 10.1007/s00280-016-2990-1. [DOI] [PubMed] [Google Scholar]
- 61.Fillmore C.M. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520(7546):239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Denissov S. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141(3):526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 63.Yin S. Exome sequencing identifies frequent mutation of MLL2 in non-small cell lung carcinoma from Chinese patients. Sci Rep. 2014;4:6036. doi: 10.1038/srep06036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Augert A. Small cell lung Cancer exhibits frequent inactivating mutations in the histone methyltransferase KMT2D/MLL2: CALGB 151111 (alliance) J Thorac Oncol. 2017;12(4):704–713. doi: 10.1016/j.jtho.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ardeshir-Larijani F. KMT2D mutation is associated with poor prognosis in non-small-cell lung Cancer. Clin Lung Cancer. 2018;19(4):e489–e501. doi: 10.1016/j.cllc.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Kantidakis T. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016;30(4):408–420. doi: 10.1101/gad.275453.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xue W. Histone methyltransferase G9a modulates hepatic insulin signaling via regulating HMGA1. Biochim Biophys Acta. 2018;1864(2):338–346. doi: 10.1016/j.bbadis.2017.10.037. [DOI] [PubMed] [Google Scholar]
- 68.Chen M.W. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule ep-CAM. Cancer Res. 2010;70(20):7830–7840. doi: 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- 69.Pandey M. Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: potential targets for cancer control. Toxicol Appl Pharmacol. 2014;280(2):296–304. doi: 10.1016/j.taap.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 70.Ma Y.N. SATB2 suppresses non-small cell lung cancer invasiveness by G9a. Clin Exp Med. 2018;18(1):37–44. doi: 10.1007/s10238-017-0464-3. [DOI] [PubMed] [Google Scholar]
- 71.Huang T. G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death Dis. 2017;8(4) doi: 10.1038/cddis.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Agarwal P., Jackson S.P. G9a inhibition potentiates the anti-tumour activity of DNA double-strand break inducing agents by impairing DNA repair independent of p53 status. Cancer Lett. 2016;380(2):467–475. doi: 10.1016/j.canlet.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang R. Effects of SMYD2-mediated EML4-ALK methylation on the signaling pathway and growth in non-small-cell lung cancer cells. Cancer Sci. 2017;108(6):1203–1209. doi: 10.1111/cas.13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hao C. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett. 2015;357(1):179–185. doi: 10.1016/j.canlet.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walter D.M. Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res. 2017;77(7):1719–1729. doi: 10.1158/0008-5472.CAN-16-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takawa M. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012;72(13):3217–3227. doi: 10.1158/0008-5472.CAN-11-3701. [DOI] [PubMed] [Google Scholar]
- 77.Chen T. miR-382 inhibits tumor progression by targeting SETD8 in non-small cell lung cancer. Biomed Pharmacother. 2017;86:248–253. doi: 10.1016/j.biopha.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Huang R. Monomethyltransferase SETD8 regulates breast cancer metabolism via stabilizing hypoxia-inducible factor 1alpha. Cancer Lett. 2017;390:1–10. doi: 10.1016/j.canlet.2016.12.038. [DOI] [PubMed] [Google Scholar]
- 79.Veschi V., Thiele C.J. High-SETD8 inactivates p53 in neuroblastoma. Oncoscience. 2017;4(3–4):21–22. doi: 10.18632/oncoscience.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Volkel P., Angrand P.O. The control of histone lysine methylation in epigenetic regulation. Biochimie. 2007;89(1):1–20. doi: 10.1016/j.biochi.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 81.Min J. Structure of the catalytic domain of human DOT1L, a non-SET domain Nucleosomal histone methyltransferase. Cell. 2003;112(5):711–723. doi: 10.1016/s0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 82.Dlakic M. Chromatin silencing protein and pachytene checkpoint regulator Dot1p has a methyltransferase fold. Trends Biochem Sci. 2001;26(7):405–407. doi: 10.1016/s0968-0004(01)01856-4. [DOI] [PubMed] [Google Scholar]
- 83.Rau R.E. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood. 2016;128(7):971–981. doi: 10.1182/blood-2015-11-684225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim W. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem. 2012;287(8):5588–5599. doi: 10.1074/jbc.M111.328138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Evanno E. Tri-methylation of H3K79 is decreased in TGF-beta1-induced epithelial-to-mesenchymal transition in lung cancer. Clin Epigenetics. 2017;9:80. doi: 10.1186/s13148-017-0380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jorgensen S., Schotta G., Sorensen C.S. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41(5):2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saloura V. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci Rep. 2017;7:40664. doi: 10.1038/srep40664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 89.Shi Y.G., Tsukada Y., The discovery of histone demethylases Cold Spring Harb Perspect Biol. 2013;5(9) doi: 10.1101/cshperspect.a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Walport L.J., Hopkinson R.J., Schofield C.J. Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol. 2012;16(5–6):525–534. doi: 10.1016/j.cbpa.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 91.Kooistra S.M., Helin K. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol. 2012;13(5):297–311. doi: 10.1038/nrm3327. [DOI] [PubMed] [Google Scholar]
- 92.Thinnes C.C. Targeting histone lysine demethylases - progress, challenges, and the future. Biochim Biophys Acta. 2014;1839(12):1416–1432. doi: 10.1016/j.bbagrm.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klose R.J., Kallin E.M., Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 94.Forneris F. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282(28):20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 95.Metzger E. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- 96.Hayami S. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011;128(3):574–586. doi: 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- 97.Lv T. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mohammad H.P. A DNA Hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell. 2015;28(1):57–69. doi: 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 99.Zang C. Long non-coding RNA LINC01133 represses KLF2, P21 and E-cadherin transcription through binding with EZH2, LSD1 in non small cell lung cancer. Oncotarget. 2016;7(10):11696–11707. doi: 10.18632/oncotarget.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.He R., Zhang F.H., Shen N. LncRNA FEZF1-AS1 enhances epithelial-mesenchymal transition (EMT) through suppressing E-cadherin and regulating WNT pathway in non-small cell lung cancer (NSCLC) Biomed Pharmacother. 2017;95:331–338. doi: 10.1016/j.biopha.2017.08.057. [DOI] [PubMed] [Google Scholar]
- 101.Kong L. KDM1A promotes tumor cell invasion by silencing TIMP3 in non-small cell lung cancer cells. Oncotarget. 2016;7(19):27959–27974. doi: 10.18632/oncotarget.8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lim S.Y. LSD1 modulates the non-canonical integrin beta3 signaling pathway in non-small cell lung carcinoma cells. Sci Rep. 2017;7(1):10292. doi: 10.1038/s41598-017-09554-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ozer A., Bruick R.K. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3(3):144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- 104.Mosammaparast N., Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 105.Wagner K.W. KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J Clin Invest. 2013;123(12):5231–5246. doi: 10.1172/JCI68642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dhar S.S. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J Biol Chem. 2014;289(11):7483–7496. doi: 10.1074/jbc.M113.521625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gerken P.A. Discovery of a highly selective cell-active inhibitor of the histone lysine demethylases KDM2/7. Angew Chem Int Ed Engl. 2017;56(49):15555–15559. doi: 10.1002/anie.201706788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho H.S. The JmjC domain-containing histone demethylase KDM3A is a positive regulator of the G1/S transition in cancer cells via transcriptional regulation of the HOXA1 gene. Int J Cancer. 2012;131(3):E179–E189. doi: 10.1002/ijc.26501. [DOI] [PubMed] [Google Scholar]
- 109.Zhan M. JMJD1A promotes tumorigenesis and forms a feedback loop with EZH2/let-7c in NSCLC cells. Tumour Biol. 2016;37(8):11237–11247. doi: 10.1007/s13277-016-4999-9. [DOI] [PubMed] [Google Scholar]
- 110.Li Y. KDM3A promotes inhibitory cytokines secretion by participating in TLR4 regulation of Foxp3 transcription in lung adenocarcinoma cells. Oncol Lett. 2017;13(5):3529–3537. doi: 10.3892/ol.2017.5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramadoss S., Guo G., Wang C.Y. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene. 2017;36(1):47–59. doi: 10.1038/onc.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berry W.L., Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73(10):2936–2942. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang Y. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312(5774):748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 114.Guerra-Calderas L. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015;208(5):215–224. doi: 10.1016/j.cancergen.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 115.Soini Y., Kosma V.M., Pirinen R. KDM4A, KDM4B and KDM4C in non-small cell lung cancer. Int J Clin Exp Pathol. 2015;8(10):12922–12928. [PMC free article] [PubMed] [Google Scholar]
- 116.Sharma S.V. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Teng Y.C. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013;73(15):4711–4721. doi: 10.1158/0008-5472.CAN-12-3165. [DOI] [PubMed] [Google Scholar]
- 118.Buck C.A., Horwitz A.F. Integrin, a transmembrane glycoprotein complex mediating cell-substratum adhesion. J Cell Sci Suppl. 1987;8:231–250. doi: 10.1242/jcs.1987.supplement_8.13. [DOI] [PubMed] [Google Scholar]
- 119.Buck C.A., Horwitz A.F. Cell surface receptors for extracellular matrix molecules. Annu Rev Cell Biol. 1987;3:179–205. doi: 10.1146/annurev.cb.03.110187.001143. [DOI] [PubMed] [Google Scholar]
- 120.Gale M. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget. 2016;7(26):39931–39944. doi: 10.18632/oncotarget.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Terashima M. Epigenetic regulation of epithelial-mesenchymal transition by KDM6A histone demethylase in lung cancer cells. Biochem Biophys Res Commun. 2017;490(4):1407–1413. doi: 10.1016/j.bbrc.2017.07.048. [DOI] [PubMed] [Google Scholar]
- 122.Watarai H. Impact of H3K27 demethylase inhibitor GSKJ4 on NSCLC cells alone and in combination with metformin. Anticancer Res. 2016;36(11):6083–6092. doi: 10.21873/anticanres.11198. [DOI] [PubMed] [Google Scholar]
- 123.Wu Q. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc Natl Acad Sci U S A. 2018;115(17):E3978–E3986. doi: 10.1073/pnas.1716589115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ler L.D., Ghosh S. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci Transl Med. 2017;9(378) doi: 10.1126/scitranslmed.aai8312. [DOI] [PubMed] [Google Scholar]
- 125.Ezponda T. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017;21(3):628–640. doi: 10.1016/j.celrep.2017.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ntziachristos P. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514(7523):513–517. doi: 10.1038/nature13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Molina-Serrano D., Schiza V., Kirmizis A. Cross-talk among epigenetic modifications: lessons from histone arginine methylation. Biochem Soc Trans. 2013;41(3):751–759. doi: 10.1042/BST20130003. [DOI] [PubMed] [Google Scholar]
- 128.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585(13):2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lee H.W., Kim S., Paik W.K. S-adenosylmethionine: protein-arginine methyltransferase. Purification and mechanism of the enzyme. Biochemistry. 1977;16(1):78–85. doi: 10.1021/bi00620a013. [DOI] [PubMed] [Google Scholar]
- 130.Zhang X. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 2003;12(1):177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baldwin R.M., Morettin A., Cote J. Role of PRMTs in cancer: could minor isoforms be leaving a mark? World J Biol Chem. 2014;5(2):115–129. doi: 10.4331/wjbc.v5.i2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Friesen W.J. A novel WD repeat protein component of the methylosome binds Sm proteins. J Biol Chem. 2002;277(10):8243–8247. doi: 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- 133.Yoshimatsu M. Dysregulation of PRMT1 and PRMT6, type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011;128(3):562–573. doi: 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- 134.Elakoum R. CARM1 and PRMT1 are dysregulated in lung cancer without hierarchical features. Biochimie. 2014;97:210–218. doi: 10.1016/j.biochi.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 135.Iderzorig T. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem Biophys Res Commun. 2018;496(2):770–777. doi: 10.1016/j.bbrc.2018.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hong X. Interaction of JMJD6 with single-stranded RNA. Proc Natl Acad Sci U S A. 2010;107(33):14568–14572. doi: 10.1073/pnas.1008832107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang J. High expression of JMJD6 predicts unfavorable survival in lung adenocarcinoma. Tumour Biol. 2013;34(4):2397–2401. doi: 10.1007/s13277-013-0789-9. [DOI] [PubMed] [Google Scholar]
- 138.Wan J. PCAF-mediated acetylation of transcriptional factor HOXB9 suppresses lung adenocarcinoma progression by targeting oncogenic protein JMJD6. Nucleic Acids Res. 2016;44(22):10662–10675. doi: 10.1093/nar/gkw808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cuthbert G.L. Histone deimination antagonizes arginine methylation. Cell. 2004;118(5):545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 140.Wang Y. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004;306(5694):279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 141.Thompson P.R., Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1(7):433–441. doi: 10.1021/cb6002306. [DOI] [PubMed] [Google Scholar]
- 142.Bawadekar M. Tumor necrosis factor alpha, citrullination, and peptidylarginine deiminase 4 in lung and joint inflammation. Arthritis Res Ther. 2016;18(1):173. doi: 10.1186/s13075-016-1068-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Duan Q. Overexpression of PAD4 suppresses drug resistance of NSCLC cell lines to gefitinib through inhibiting Elk1-mediated epithelial-mesenchymal transition. Oncol Rep. 2016;36(1):551–558. doi: 10.3892/or.2016.4780. [DOI] [PubMed] [Google Scholar]
- 144.Obacz J. The role of AGR2 and AGR3 in cancer: similar but not identical. Eur J Cell Biol. 2015;94(3–4):139–147. doi: 10.1016/j.ejcb.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 145.Kim Y. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy. 2013;9(12):2126–2139. doi: 10.4161/auto.26308. [DOI] [PubMed] [Google Scholar]
- 146.Ren A. Inhibition of H3K9 methyltransferase G9a induces autophagy and apoptosis in oral squamous cell carcinoma. Biochem Biophys Res Commun. 2015;459(1):10–17. doi: 10.1016/j.bbrc.2015.01.068. [DOI] [PubMed] [Google Scholar]
- 147.Yokoyama M. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget. 2017;8(13):21315–21326. doi: 10.18632/oncotarget.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J Cancer Res Clin Oncol. 2018;144(6):1065–1077. doi: 10.1007/s00432-018-2631-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yan N. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp Cell Res. 2017;359(2):405–414. doi: 10.1016/j.yexcr.2017.08.024. [DOI] [PubMed] [Google Scholar]
- 150.Sakaki H. GSKJ4, a selective Jumonji H3K27 demethylase inhibitor, effectively targets ovarian Cancer stem cells. Anticancer Res. 2015;35(12):6607–6614. [PubMed] [Google Scholar]
- 151.Morozov V.M. Inhibitor of H3K27 demethylase JMJD3/UTX GSK-J4 is a potential therapeutic option for castration resistant prostate cancer. Oncotarget. 2017;8(37):62131–62142. doi: 10.18632/oncotarget.19100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rath B.H. Inhibition of the histone H3K27 demethylase UTX enhances tumor cell Radiosensitivity. Mol Cancer Ther. 2018;17(5):1070–1078. doi: 10.1158/1535-7163.MCT-17-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shafer D., Grant S. Update on rational targeted therapy in AML. Blood Rev. 2016;30(4):275–283. doi: 10.1016/j.blre.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Klaus C.R. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. 2014;350(3):646–656. doi: 10.1124/jpet.114.214577. [DOI] [PubMed] [Google Scholar]
- 155.Tan J.Z. EZH2: biology, disease, and structure-based drug discovery. Acta Pharmacol Sin. 2014;35(2):161–174. doi: 10.1038/aps.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Frankel A.E., Liu X., Minna J.D. Developing EZH2-targeted therapy for lung Cancer. Cancer Discov. 2016;6(9):949–952. doi: 10.1158/2159-8290.CD-16-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nasveschuk C.G. Discovery and optimization of Tetramethylpiperidinyl Benzamides as inhibitors of EZH2. ACS Med Chem Lett. 2014;5(4):378–383. doi: 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Garapaty-Rao S. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20(11):1329–1339. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 159.Han Li C., Chen Y. Targeting EZH2 for cancer therapy: progress and perspective. Curr Protein Pept Sci. 2015;16(6):559–570. doi: 10.2174/1389203716666150409100233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tan J. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kikuchi J. Epigenetic therapy with 3-deazaneplanocin A, an inhibitor of the histone methyltransferase EZH2, inhibits growth of non-small cell lung cancer cells. Lung Cancer. 2012;78(2):138–143. doi: 10.1016/j.lungcan.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun F. Preclinical pharmacokinetic studies of 3-deazaneplanocin A, a potent epigenetic anticancer agent, and its human pharmacokinetic prediction using GastroPlus. Eur J Pharm Sci. 2015;77:290–302. doi: 10.1016/j.ejps.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 163.Miranda T.B. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8(6):1579–1588. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Diaz E. Development and validation of reagents and assays for EZH2 peptide and nucleosome high-throughput screens. J Biomol Screen. 2012;17(10):1279–1292. doi: 10.1177/1087057112453765. [DOI] [PubMed] [Google Scholar]
- 165.McCabe M.T. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 166.Van Aller G.S. Long residence time inhibition of EZH2 in activated polycomb repressive complex 2. ACS Chem Biol. 2014;9(3):622–629. doi: 10.1021/cb4008748. [DOI] [PubMed] [Google Scholar]
- 167.Knutson S.K. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Vaswani R.G. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J Med Chem. 2016;59(21):9928–9941. doi: 10.1021/acs.jmedchem.6b01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Soumyanarayanan U., Dymock B.W. Recently discovered EZH2 and EHMT2 (G9a) inhibitors. Future Med Chem. 2016;8(13):1635–1654. doi: 10.4155/fmc-2016-0096. [DOI] [PubMed] [Google Scholar]
- 170.Wee S. Targeting epigenetic regulators for cancer therapy. Ann N Y Acad Sci. 2014;1309:30–36. doi: 10.1111/nyas.12356. [DOI] [PubMed] [Google Scholar]
- 171.Yan W., Herman J.G., Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8(1):119–133. doi: 10.2217/epi.15.84. [DOI] [PubMed] [Google Scholar]
- 172.Gulati N., Beguelin W., Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk Lymphoma. 2018:1–12. doi: 10.1080/10428194.2018.1430795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fu X., Zhang P., Yu B. Advances toward LSD1 inhibitors for cancer therapy. Future Med Chem. 2017;9(11):1227–1242. doi: 10.4155/fmc-2017-0068. [DOI] [PubMed] [Google Scholar]
- 174.Przespolewski A., Wang E.S. Inhibitors of LSD1 as a potential therapy for acute myeloid leukemia. Expert Opin Investig Drugs. 2016;25(7):771–780. doi: 10.1080/13543784.2016.1175432. [DOI] [PubMed] [Google Scholar]
- 175.Zheng Y.C. Irreversible LSD1 inhibitors: application of tranylcypromine and its derivatives in Cancer treatment. Curr Top Med Chem. 2016;16(19):2179–2188. doi: 10.2174/1568026616666160216154042. [DOI] [PubMed] [Google Scholar]
- 176.Stewart C.A., Byers L.A. Altering the course of small cell lung Cancer: targeting Cancer stem cells via LSD1 inhibition. Cancer Cell. 2015;28(1):4–6. doi: 10.1016/j.ccell.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Milletti F. Abstract 4708: neuroendocrine gene transcript expression is associated with efficacy to lysine-specific demethylase-1 inhibitor RG6016 in small cell lung cancer-derived cell lines. Cancer Res. 2016;76(14 Supplement):4708. [Google Scholar]
- 178.Mohammad H.P., Kruger R.G. Antitumor activity of LSD1 inhibitors in lung cancer. Mol Cell Oncol. 2016;3(2) doi: 10.1080/23723556.2015.1117700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Binda O. On your histone mark, SET, methylate! Epigenetics. 2014;8(5):457–463. doi: 10.4161/epi.24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang H. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8(6):1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 181.Binda O. Trimethylation of histone H3 lysine 4 impairs methylation of histone H3 lysine 9: regulation of lysine methyltransferases by physical interaction with their substrates. Epigenetics. 2010;5(8):767–775. doi: 10.4161/epi.5.8.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wood K., Tellier M., Murphy S. DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules. 2018:8(1). doi: 10.3390/biom8010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Park S.Y., Park J.W., Chun Y.S. Jumonji histone demethylases as emerging therapeutic targets. Pharmacol Res. 2016;105:146–151. doi: 10.1016/j.phrs.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 184.Schapira M. Structural chemistry of human SET domain protein methyltransferases. Curr Chem Genomics. 2011;5(Suppl. 1):85–94. doi: 10.2174/1875397301005010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Watanabe H. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008;8:15. doi: 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kang D. The histone methyltransferase wolf-Hirschhorn syndrome candidate 1-like 1 (WHSC1L1) is involved in human carcinogenesis. Genes Chromosomes Cancer. 2013;52(2):126–139. doi: 10.1002/gcc.22012. [DOI] [PubMed] [Google Scholar]
- 187.Van Den Broeck A. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin Cancer Res. 2008;14(22):7237–7245. doi: 10.1158/1078-0432.CCR-08-0869. [DOI] [PubMed] [Google Scholar]
- 188.Italiano A. Molecular cytogenetic characterization of a metastatic lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genet Cytogenet. 2006;167(2):122–130. doi: 10.1016/j.cancergencyto.2006.01.004. [DOI] [PubMed] [Google Scholar]