

Abstract
Aims:
Despite vast clinical experience linking diabetes and atherosclerosis, the molecular mechanisms leading to accelerated vascular damage are still unclear. Here, we investigated the effects of nuclear factor of activated T-cells inhibition on plaque burden in a novel mouse model of type 2 diabetes that better replicates human disease.
Methods & Results:
IGF-II/LDLR–/–ApoB100/100 mice were generated by crossbreeding low-density lipoprotein receptor–deficient mice that synthesize only apolipoprotein B100 (LDLR–/–ApoB100/100) with transgenic mice overexpressing insulin-like growth factor-II in pancreatic β cells. Mice have mild hyperglycaemia and hyperinsulinaemia and develop complex atherosclerotic lesions. In vivo treatment with the nuclear factor of activated T-cells blocker A-285222 for 4 weeks reduced atherosclerotic plaque area and degree of stenosis in the brachiocephalic artery of IGF-II/LDLR–/–ApoB100/100 mice, as assessed non-invasively using ultrasound biomicroscopy prior and after treatment, and histologically after termination. Treatment had no impact on plaque composition (i.e. muscle, collagen, macrophages). The reduced plaque area could not be explained by effects of A-285222 on plasma glucose, insulin or lipids. Inhibition of nuclear factor of activated T-cells was associated with increased expression of atheroprotective NOX4 and of the anti-oxidant enzyme catalase in aortic vascular smooth muscle cells.
Conclusion:
Targeting the nuclear factor of activated T-cells signalling pathway may be an attractive approach for the treatment of diabetic macrovascular complications.
Keywords: Atherosclerosis, oxidative stress, type 2 diabetes, nuclear factor of activated T-cells, hyperglycaemia, ApoB100
Introduction
Diabetes-induced macro- and microvascular complications are the major cause of morbidity and mortality in diabetic patients. Based on current trends, the rising incidence of diabetes (expected to reach 700 million people worldwide by 2025) will undoubtedly equate to increased cardiovascular mortality.1 Diabetic patients have a much more widespread and aggressive form of atherosclerosis and, therefore, higher risk for myocardial infarction, peripheral vascular disease and stroke, but the molecular mechanisms leading to accelerated damage are still unclear.2,3
Previous studies from our laboratory identified the calcium/calcineurin-dependent transcription factor nuclear factor of activated T-cells (NFAT) as a potential mediator of vascular disease in the context of hyperglycaemia. NFAT is expressed in the vascular wall and is readily activated by modest elevations of extracellular glucose in conduit4 and medium-sized resistance arteries5 as well as in the microvasculature.6 Once activated, NFAT induces the expression of the pro-atherogenic cytokine osteopontin (OPN) in the arterial wall, as well as the expression of inflammatory mediators, such as cyclooxygenase-2 (COX-2), interleukin 6 (IL-6), vascular cell adhesion molecule 1 (VCAM-1), tissue factor (TF) and allograft inflammatory factor-1 (AIF-1).4,7–11 Clinical and experimental studies have implicated all these NFAT-targets in the development of vascular disease.4,7–11 More recently, we showed that in vivo treatment of streptozotocin (STZ)-induced diabetic female ApoE–/– mice with the NFAT blocker A-285222 for 4 weeks abrogated- hyperglycaemia induced atherosclerosis in the aorta.10
A large number of studies over the past years have focused on the generation of oxidative stress as the pathogenic mechanism linking diabetes to atherosclerosis.12 A shift in the balance between oxidant [i.e. reactive oxygen and nitrogen species (ROS and RNS)] and anti-oxidant or detoxifying agents [i.e. catalase and glyoxalase I (GLO1)] results in chronically elevated oxidative stress levels in diabetes.13 Key players in the generation of ROS in the arterial wall are the vascular isoforms of the NADPH oxidase (NOX) family of proteins.14 In particular, NOX1 and NOX2 have been shown to be induced and promote atherosclerosis in the context of diabetes, while NOX4 has recently emerged as an atheroprotective isoform in both humans and in mouse.15,16 Interestingly, in mouse kidney fibroblasts, the calcineurin/NFAT pathway has been shown to regulate NOX expression and activity in response to high glucose.17
Here, we investigated whether inhibition of NFAT had any impact on atherosclerosis in the context of type 2 diabetes, using a novel mouse model with a metabolic profile and plaque characteristics that better mimic human disease than other mouse models currently available.18 Moreover, we explore the potential link between NFAT signalling and oxidative stress in this setting.
Methods
Animals
IGF-II/LDLR–/–ApoB100/100 mice were originated by crossbreeding low-density lipoprotein (LDL) receptor–deficient mice expressing only apolipoprotein B100 (LDLR–/–ApoB100/100; C57BL/6x129/SvJae background) with C57BL6/SJL mice overexpressing IGF-II in pancreatic β-cells.19 Mice were either bred at the National Laboratory Animal Centre at the University of Eastern Finland or at Taconic Europe (Ejby, Denmark) after re-derivation from the University of Eastern Finland. Mice are characterized by hyperglycaemia, mild hyperinsulinaemia, a human-like hypercholesterolaemic lipid profile, and advanced and complex atherosclerotic lesions, hence representing a model that better replicates human disease and a more appropriate model for the study of macrovascular complications in the context of type 2 diabetes.19 Animals were housed in groups and maintained in a temperature- and humidity-controlled environment with a 12-h light/dark cycle. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were approved by the local ethics review board at Lund University and Malmö/Lund Animal Care and Use Committee (M29-12; M9-15), and by the National Animal Experiment Board in Finland (ESAVI-2012-001260).
Study protocol
The study protocol was carried out twice and referred to as Study I and Study II throughout the article (Figure 1(a)). Study I: Young (15–16 weeks old, N = 30) IGF-II/LDLR–/–ApoB100/100 mice bred at Taconic Europe were used at Lund University. Study II: Young (10 weeks old, N = 10) and old (46–74 weeks old, N = 17) IGF-II/LDLR–/–ApoB100/100 mice were bred and used at the University of Eastern Finland. Mice at Lund University, but not at the University of Eastern Finland were housed in individually ventilated cages. All animals were fed ad libitum and switched from a normal chow to high fat Western diet (TD 88137, Harlan Tekland: 42% of calories from fat and 0.15% from cholesterol) at the start of the experiments. After 4 weeks on high-fat diet, mice were randomized based on blood glucose, body weight and plasma cholesterol to receive daily intraperitoneal (i.p.) injections of the NFAT blocker A-285222 (0.29 mg/kg body weight) or vehicle (saline) for an additional 4 weeks. In Study II, brachiocephalic artery plaque size measured by ultrasound biomicroscopy (see below) before the start of A-285222 treatment was also included as parameter in the randomization. A-285222 inhibits all NFAT family members and was kindly provided by Abbott Laboratories (Abbott Park, IL). This drug belongs to a series of 3,5-bis (trifluoromethyl) pyrazole (BTP) derivatives demonstrated to maintain NFAT in a phosphorylated state, blocking its nuclear import and subsequent transcription, without affecting NF-kB or AP-1 activation, or calcineurin phosphatase activity.20 Body weight, fasting (16 h) and non-fasting venous blood glucose (saphenous or tail vein) were measured using Contour Glucometer (Bayer) before, during and at termination of the experiments. All fluid and tissue processing and analyses for both studies I and II were done at Lund University to avoid inter-lab variability.
Figure 1.

NFAT inhibition reduces plaque area and degree of stenosis in IGF-II/LDLR–/–ApoB100/100 mice. (a) Study protocol used for all conducted in vivo experiments (both for Study I and II): IGF-II/LDLR–/–ApoB100/100 mice were fed high-fat diet (HFD, 42% of calories from fat and 0.15% from cholesterol) for 8 weeks and received daily i.p. injections of A-285222 (0.29 mg/kg body weight) or vehicle (saline; control) for the last 4 weeks of the HFD period. Arrows indicate the time of the ultrasound biomicroscopy measurements (US) performed in Study II. (b–c) Histologically determined plaque size and degree of stenosis in the brachiocephalic arteries of young (10–16 weeks old) mice. Data represent merged results from Studies I and II, normalized to each control (N = 10 mice/condition, except N = 9 in control females). (d) Representative Elastin van Gieson stained sections from the brachiocephalic artery of young male mice in Study II after treatment with A-285222 or vehicle. Scale bar = 200 µm. (e) Plaque size determined non-invasively by ultrasound biomicroscopy in young mice before and after treatment with A-285222. (f) Change in plaque size from week 4 to week 8 for control and A-285222 treated mice. N = 4–6 mice/group; *p < 0.05.
Ultrasound measurements
Brachiocephalic artery plaque size and lesion progression were assessed non-invasively using ultrasound biomicroscopy as previously described and validated21,22 (Vevo2100, Visualsonics, Toronto, Canada), with a transducer frequency of 40MHz (providing a theoretical resolution of 40 µm at a frame rate of 32Hz). Prior to the scan, animals were anaesthetized with isoflurane (4.5% isoflurane 450 mL air for induction, 2% isoflurane 200 mL air for maintenance; Baxter International Inc., IL, USA). The brachiocephalic artery was visualized in a cross-sectional short axis view and atherosclerotic lesion size was measured within the proximal 200 µm from its bifurcation from the aortic arch. All measurements were performed blinded by one operator (intra-individual coefficient of variance = 2.7%). Loops of at least 100 frames were stored and the frame with the largest plaque was chosen for off-line, blinded measurements. At the end of the experimental protocol, mice were anaesthetized with ketamine hydrochloride and xylazine (i.p.; 7.5 mg and 2.5 mg/100 g body weight, respectively) and euthanized by exsanguination through cardiac puncture for blood collection and perfused with phosphate-buffered saline before tissue harvesting. Depth of anaesthesia was assessed by toe-pinch procedure and absence of muscular tone. All efforts were made to minimize suffering.
Plasma cholesterol, triglycerides, insulin and OPN measurements
Total plasma cholesterol and triglycerides levels were measured in overnight fasting blood samples before and after treatment by colorimetric assays (InfinityTM-Cholesterol and InfinityTM-Triglyceride; Thermo Scientific, Middletown, VA, USA). Plasma insulin and OPN were determined after overnight fasting at the end of the treatment using Mercodia mouse insulin enzyme-linked immunosorbent assay (ELISA) kit (Mercodia AB, Uppsala, Sweden) and Quantikine mouse OPN ELISA kit (R&D Systems, Abingdon UK), respectively. All assays were performed according to the manufacturer’s instructions and the lower limits of detection were 0.2 µg/L for insulin and 5.7 pg/mL for OPN.
Histological evaluations of plaque size and plaque composition
Brachiocephalic arteries and hearts were immersion-fixed with 4% buffered paraformaldehyde (PFA, pH 7.4) and thereafter embedded in paraffin. Plaque and media contents of smooth muscle, collagen and elastin were evaluated in cross-sections (5 µm) stained with haematoxylin and eosin (H&E), as well as with Elastin van Gieson (EvG). Plaque macrophage contents were evaluated both in cross-sections (5 µm) of the brachiocephalic artery and of the subvalvular region of the aortic root, using rat anti-MOMA-2 (monocytes/macrophage 2) primary antibody (1 µg/mL, BMA Biomedicals, Augst, Switzerland), biotinylated rabbit anti-rat secondary IgG antibody (1:70, BA-40011, Vector Laboratories, Burlingame, CA) and immPACT-DAB (Vector Laboratories, Burlingame, CA) for visualization. No staining was detected when primary or secondary antibodies were omitted from the protocols. Sections were counterstained with Mayer’s haematoxylin. Plaque and media areas (mm2), degree of stenosis and media thickness (µm) were measured under blinded conditions using computer-aided morphometry (BioPix iQ2.3.1 software, Biopix AB, Gothenburg, Sweden). Stenosis is expressed as the percentage of area within the internal elastic lamina occupied by atherosclerotic plaque. For media thickness, tiff-files were imported into NIS Elements BR Analysis 3.2 64-bit software and a wheel (Rose binary) grid with 16 spokes was centred in the lumen. An average of the media thickness measured at each of the 16 positions where the spokes met the media was calculated.
Cell culture
Vascular smooth muscle cells (VSMCs) were obtained by outgrowth from aorta explants from BalB/c NFATc3–/– and wild-type (WT; NFATc3+/+) mice23 as previously described.8 Briefly, mice were anaesthetized with ketamine hydrochloride and xylazine (i.p.; 7.5 mg and 2.5 mg/100 g body weight, respectively) and euthanized by cervical dislocation. After reaching confluence, VSMCs (passages 10–15) were seeded in 24-well plates (7.5 × 104 cells/well) and cultured for 24 h in Dulbecco’s Modified Eagle Medium (DMEM) containing 5 mmol/L d-glucose and supplemented with 10% FBS before stimulations. Hydrogen peroxide (H2O2, 500 µmol/L; Sigma-Aldrich, Darmstadt, Germany) or 3-Morpholinosydnonimine hydrochloride (SIN-1, 100 µmol/L, Sigma-Aldrich) were used to induce oxidative stress. After stimulation, cells were harvested for gene expression analyses. Oxidative stress levels were measured in culture media using OxiSelect™ In Vitro ROS/RNS Assay Kit (Cells Biolab, Inc., San Diego, CA, USA), according to the manufacturer’s instructions. H2O2 levels were determined in culture medium using Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit [Molecular probes, Eugene, OR, USA] in VSMCs cultured as above or in human microvascular endothelial cells [HMEC-1, Centers for Disease Control and Prevention (CDC)/Emory University] cultured in M200 medium supplemented with low serum growth supplement (LSGS).
RT-qPCR
Total RNA was extracted from intact, snap-frozen aortas from IGF-II/LDLR–/–ApoB100/100 mice using TRI Reagent BD (Sigma-Aldrich, St Louis, MO, USA) or from cultured VSMCs using RNeasy® Plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. cDNA synthesis (RevertAid M-MuLV Reverse Transcriptase; Fermentas GMBH, St Leon-Rot, Germany) and real-time PCR were performed as previously described24 using TaqMan Gene Expression assays for OPN (Mm00436763), IL-6 (Mm00446190_m1), ICAM (Mm00516023), CD68 (Mm03047340_m1), TF (Mm00436948_m1), GLO1 (Mm00844954_s1), NOX1 (Mm00549170_m1), NOX2 (Mm01287743_m1), NOX4 (Mm00479246_m1) and catalase (Mm00437992_m1) with HPRT (Mm0046968_m1) and β-actin (Mm00607939_s1) as endogenous controls.
Statistics
Results are expressed as mean ± SEM unless otherwise specified. Analyses of distributions were performed before statistical tests were performed using GraphPad software (Prism 7.0). For parametric data, significance was determined using Student’s t-test, one- or two-way analysis of variance (ANOVA), followed by Bonferroni post-tests. Non-parametric data were analysed using Mann–Whitney or Kruskal–Wallis test followed by Dunn’s post-test.
Results
In vivo inhibition of NFAT effectively leads to plaque regression in the brachiocephalic artery of IGF-II/LDLR–/–ApoB100/100 mice
Histological evaluation of the brachiocephalic arteries of young mice revealed reduced plaque area and degree of stenosis after 4 weeks of treatment with the NFAT blocker A-285222 (Figure 1(b) to (d)). The effects of the blocker were significant in the males only (Figure 1(b) to (d)). The same pattern was observed in Study I and II (Supplemental Figure 1(A) and (B)). No significant differences in media area or thickness were observed after NFAT inhibition (Supplemental Figure 1(C) to (H)). Male IGF-II/LDLR–/–ApoB100/100 mice exhibited larger atherosclerotic plaques than female mice. Interestingly, despite being younger, male mice in Study II had considerably larger plaques than males in Study I, maybe due to different breeding or housing environments.
Non-invasive longitudinal ultrasound measurements in Study II revealed that the effects of NFAT inhibition on atherosclerosis may not only be due to limited plaque progression but also to plaque regression, as evidenced by smaller plaque size after treatment with A-285222 when compared to plaque size in the same mice before treatment (Figure 1(e) and (f)). Ultrasound experiments also showed significantly smaller brachiocephalic artery plaque size in mice treated with A-285222 when compared to control mice (Figure 1(e)), in line with the histological results.
The reduction in plaque size was unlikely due to lowering effects of A-285222 on plasma glucose, cholesterol or triglyceride levels, since these were not significantly reduced by the treatment (Table 1). In male mice, we even observed a small elevation in fasting blood glucose in the A-285222 treated group (Table 1), which would instead counteract any beneficial effect of the NFAT blocker. We have seen a similar effect of the NFAT blocker on blood glucose in STZ-induced diabetic BalB/c mice: an effect that was no longer evident after lowering the dose of A-285222 from 0.29 to 0.15 mg/kg.4 We have not observed this effect in other diabetic mouse models or strains.10 This could potentially be attributed to direct effects of A-285222 on pancreatic β-cells, where the involvement of NFAT in the regulation of insulin transcription is well established.25 This effect was only evident under fasting conditions and no effects on plasma insulin or body weight were detected after treatment with A-285222 (Table 1). Male mice had higher body weight than female mice and higher plasma triglycerides: this last, potentially contributing to their larger atherosclerotic plaques (Table 1).
Table 1.
Metabolic parameters of young IGF-II/LDLR–/–ApoB100/100 mice on HFD.
| Control | A-285222 | ||
|---|---|---|---|
| Glucose (mmol/L) | |||
| Fasted | Males | 7.16 ± 0.99 | 8.92 ± 2.09* |
| Females | 6.77 ± 1.19 | 6.69 ± 0.93 | |
| Fed | Males | 9.47 ± 3.14 | 9.17 ± 1.80 |
| Females | 8.46 ± 1.79 | 7.96 ± 1.69 | |
| Insulin (µg/L) | |||
| Males | 0.549 ± 0.37 | 0.492 ± 0.37 | |
| Females | 0.383 ± 0.12 | 0.323 ± 0.18 | |
| Triglycerides (mmol/L) | |||
| Males | 2.39 ± 0.99 | 2.28 ± 1.35 | |
| Females | 1.55 ± 0.50 | 1.28 ± 0.62 | |
| Cholesterol (mmol/L) | |||
| Males | 33.9 ± 6.81 | 33.7 ± 9.00 | |
| Females | 34.8 ± 9.44 | 33.9 ± 7.43 | |
| OPN (ng/mL) | |||
| Males | 151 ± 37 | 135 ± 25 | |
| Females | 136 ± 23 | 122 ± 19 | |
| Body weight (g) | |||
| Males | 34.4 ± 2.7 | 33.1 ± 2.5 | |
| Females | 24.5 ± 3.3 | 24.7 ± 2.6 | |
HFD: high-fat diet; OPN: osteopontin.
Values are expressed as mean ± SD (N = 8–11 mice/group from Study I and II) and represent values after overnight fasting measured at the end of the treatment (week 8 of study protocol). Blood glucose values under non-fasting conditions are also shown (measured during week 7 of study protocol).
p < 0.05 versus control. Two-way ANOVA for the effect of gender and treatment revealed significant effects of gender on plasma triglycerides (p = 0.0035) and body weight (p < 0.0001).
We also evaluated if inhibition of NFAT had any impact on plaque composition, but we did not detect any differences in plaque muscle, collagen or macrophage contents after treatment with A-285222 in Study I (Figure 2(a) to (d)). No significant differences were observed regarding media muscle, collagen or elastin contents (Figure 2(a) and (e) to (g)). Similarly, no significant differences were observed when corresponding measurements were performed in vessels from Study II (Supplemental Figure 2).
Figure 2.

Composition of atherosclerotic plaques. (a) Representative images showing plaque and media regions of brachiocephalic arterial sections from young mice in Study I, stained with Elastin van Gieson (left and right panels in each row) and MOMA-2 (middle panels). Sections from control mice are shown in the upper row, while sections from A-285222 treated mice are shown in the lower row. Graphs summarize morphometric data showing plaque (b) to (d) and media (e) and (f) composition. Muscle (b) and (e), collagen (c) and (f), macrophage (d) and elastin (g) contents in µm2 are expressed as percentage of plaque area. N = 7–8 mice/group; scale bars = 50 µm.
In vivo inhibition of NFAT signalling induces changes in the expression of oxidative stress and inflammatory targets in the arterial wall of IGF-II/LDLR–/–ApoB100/100 mice
In agreement with previous work, both NOX2 and NOX4 were expressed in mouse aorta.16 Treatment with the NFAT blocker A-285222 for 4 weeks significantly increased the expression of the atheroprotective isoform NOX4 in the aorta of male mice (Figure 3(a)), while changes in NOX2 were not significant (Figure 3(b)). We also examined the expression of the detoxifier protein glyoxalase I (GLO1), which metabolizes methylglyoxal (MG), a major source of intracellular and plasmatic advanced glycation end product (AGE) formation, into the unreactive d-lactate. But neither changes in GLO1 expression (Figure 3(c)) nor in the expression of another detoxifier enzyme, catalase (Figure 3(d)), were significant. Consistent with results obtained in our previous studies in which STZ-induced diabetic ApoE–/– female mice were treated with A-285222,10 expression of the macrophage marker CD68 was significantly reduced by NFAT inhibition in the aortas of IGF-II/LDLR–/–ApoB100/100 female mice (Figure 3(e)), along with tendencies to reduced IL-6 and OPN (Figure 3(f) to (g)). In line with previous studies,10 plasma OPN was not affected by treatment with A-285222 (Table 1), suggesting the effects of the blocker are targeted to the vessel wall and not due to lower systemic inflammation.
Figure 3.
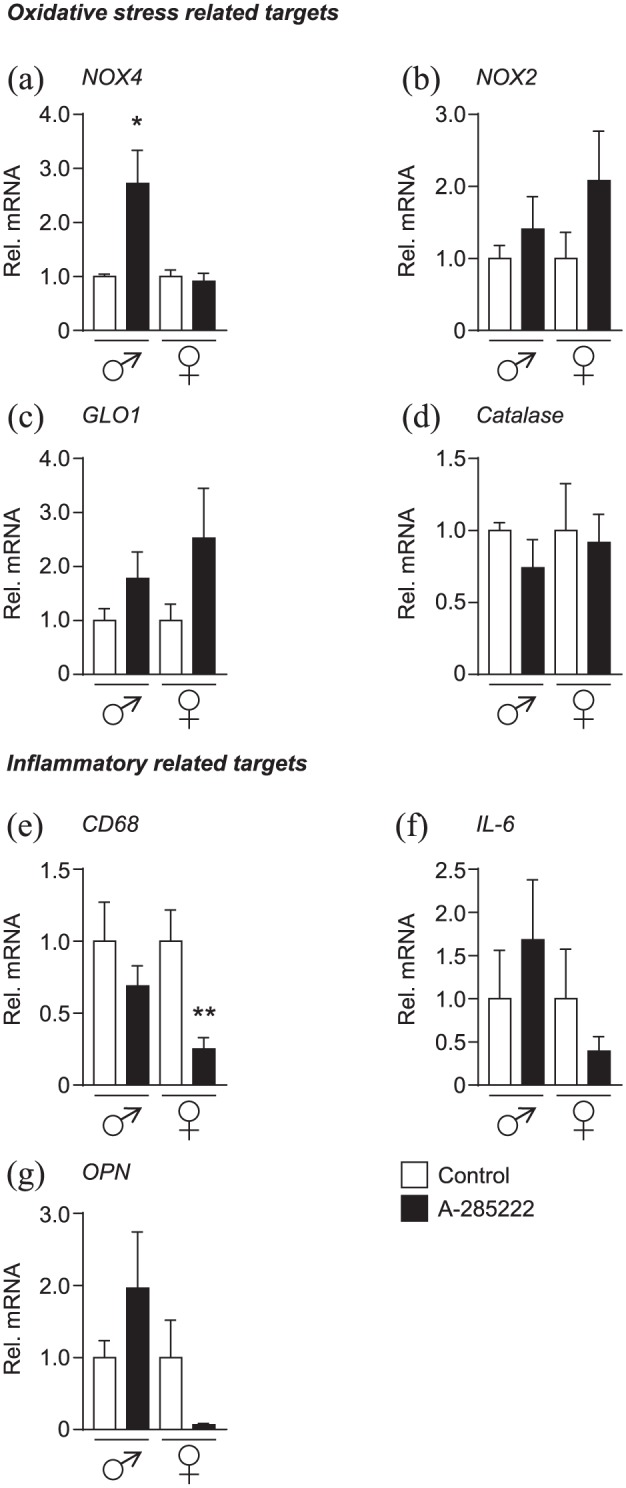
In vivo inhibition of NFAT modifies the expression of oxidative stress and inflammatory markers in the aorta of young IGF-II/LDLR–/–ApoB100/100 mice. Gene expression of oxidative stress targets (a) NOX4, (b) NOX2, (c) GLO1 and (d) catalase, as well as inflammatory targets (e) CD68, (f) IL-6 and (g) OPN were quantified by qPCR in the aortas of young IGF-II/LDLR–/–ApoB100/100 mice from Study I and II. HPRT and β-actin were used as endogenous controls and data were expressed relative to each control group. N = 5 and 7 for control and treated males, respectively; N = 8 for female groups.
*p < 0.05 and **p < 0.01.
The analysis of the expression data also revealed sex-dependent differences in the IGF-II/LDLR–/–ApoB100/100 model, as assessed in the aortas of vehicle treated mice. Levels of NOX2 were relatively higher in males than in females, while arteries from female mice expressed relatively higher levels of atheroprotective NOX4, catalase and GLO1 (Supplemental Figure 3): maybe underlying the differences in overall plaque burden observed between sexes (Supplemental Figure 1(A) and (B)).
NFAT modulates the expression of NOX4 and catalase in a powerful oxidative stress environment
Next, we explored whether inhibition of NFAT could alter the expression of oxidative stress related genes in VSMCs cultured under controlled oxidative stress conditions. VSMCs were incubated either with hydrogen peroxide (H2O2; 500 µmol/L) or with peroxynitrite donor 3-morpholinosydnonimine (SIN-1; 100 µmol/L) for 48 h to induce oxidative stress, in the presence or absence of A-285222 (1 µmol/L). Figure 4 shows that inhibition of NFAT effectively increased expression of NOX4 in VSMCs exposed to H2O2 and of catalase in VSMCs exposed to SIN-1, suggesting involvement of NFAT in the modulation of these genes under strong oxidative stress conditions. Inhibition of NFAT with A-285222 had no impact on H2O2 levels measured during culture of VSMCs or endothelial cells (Figure 4(c)). Interestingly, ROS/RNS levels are significantly lower in the culture medium of VSMCs from NFATc3 knockout (NFATc3–/–) mice cultured in high glucose (25 mmol/L) for 48 h, when compared to levels measured in the medium of NFATc3 competent (NFATc3+/+) cells (Figure 4(d)). H2O2 levels were below detection in the culture media of VSMCs from NFATc3–/– mice.
Figure 4.

In vitro inhibition of NFAT increased the expression of the anti-oxidant enzymes NOX4 and catalase in VSMCs. Mouse VMSCs were incubated with (a) H2O2 (500 µmol/L) or (b) SIN-1 (100 µmol/L) for 48 h in the presence or absence of A-285222 (1 µmol/L). The expression of oxidative stress targets NOX4, NOX1, NOX2, catalase and GLO1 was measured using qPCR and are expressed relative to untreated control cells. N = 4–7 experiments/condition *p < 0.05. (c) H2O2 levels were measured in the medium of VSMCs and HMEC-1 cells after 24 h in high glucose (25 mmol/L; HG) in the presence or absence of A-285222 (1 µmol/L). H2O2 levels were normalized to protein and expressed relative to control without A-285222 (N = 6–16 experiments/condition). (d) ROS/RNS levels, expressed as relative fluorescence units (RFU) were measured in the cell culture medium of VSMCs from NFATc3 competent (NFATc3+/+) and knockout (NFATc3–/–) mice in the presence of high glucose concentration (25 mmol/L) during 48 h. The dashed line shows ROS/RNS levels measured under low glucose conditions (5 mmol/L) as a reference (N = 9 experiments/condition).
In vivo inhibition of NFAT signalling in old IGF-II/LDLR–/–ApoB100/100 mice have modest effects on plaque size
Treatment of older mice for 4 weeks with A-285222 (0.29 mg/kg) resulted in reduced histologically assessed plaque area only in female mice (Figure 5(a)). This smaller plaque size was not translated in a significantly larger arterial lumen (Figure 5(b)). We were not able to detect differences in plaque size by ultrasound biomicroscopy (Figure 5(c) and (d)), maybe due to the lower sensitivity of the method when compared to histology. Old IGF-II/LDLR–/–ApoB100/100 mice had significantly lower macrophage infiltration in subvalvular aortic plaques than young mice, as expected to be the case during later phases of the atherogenic process (Figure 5(e) and (f)), but higher circulating levels of OPN (222 ± 70 ng/mL and 185 ± 42 ng/mL for males and females, respectively; Supplemental Table 1) reflecting increased plasma inflammatory burden when ageing.
Figure 5.

In vivo treatment with the NFAT blocker A-285222 reduces plaque size in the brachiocephalic artery of old female IGF-II/LDLR–/–ApoB100/100 mice. (a) Histologically determined plaque size and (b) stenosis in the brachiocephalic artery of old mice treated with A-285222 (0.29 mg/kg) or vehicle (saline) for 4 weeks according to the same protocol detailed in Figure 1(a). N = 3 males, N = 4–7 females; *p < 0.05. (c) and (d) Plaque size determined non-invasively by ultrasound biomicroscopy before (baseline, week 4 of the experiment) and after treatment (week 8 of the experiment; N = 7–10 mice/group). (d) Change in plaque size from week 4 to week 8 for each group. (e) Young IGF-II/LDLR–/–ApoB100/100 mice had higher proportion of macrophage infiltration in subvalvular aortic plaques when compared to old mice (N = 4–6 young mice, N = 7–10 old mice). (f) Representative MOMA-2 stained sections of the subvalvular aorta from untreated young and old mice. Scale bar = 400 µm. (g) and (h) Gene expression of oxidative stress targets NOX4, NOX2, catalase and GLO1 (g) and inflammatory targets OPN, IL6, ICAM-1 and TF (h) in the aorta of old mice (N = 2–7 mice/group and sex). HPRT and β-actin were used as endogenous controls. *p < 0.05; **p < 0.01; and ***p < 0.001.
Despite reduced expression of CD68 in the aortas of younger mice (Figure 3(e)), treatment with A-285222 did not translate into reduced macrophage infiltration in subvalvular plaques regardless the age of the mice (Figure 5(e)). This lack of effect at the level of the aortic root is in line with previous data in diabetic ApoE–/– mice.10 Treatment with the NFAT blocker had no impact on elastin or collagen contents in the subvalvular plaques (data not shown) or on elastin and collagen contents or vessel morphometry in the brachiocephalic arteries (Supplemental Figure 4). No significant differences in gene expression were found in the aortas of older mice treated with A-285222 when compared to vehicle-treated mice (Figure 5(g) and (h)). In line with findings in the young mice, no significant effects on body weight or on metabolic parameters that could explain changes in plaque size were found after treatment with A-285222 (Supplemental Table 1).
Discussion
This study demonstrates that inhibition of NFAT signalling in IGF-II/LDLR–/–ApoB100/100 mice reduces atherosclerosis in the brachiocephalic artery, a particularly disease-prone vascular segment in mice, independently of changes in plasma glucose, insulin or lipid levels. This may not only be due to limited plaque progression but also due to plaque regression, as evidenced in the ultrasound biomicroscopy experiments by smaller plaque size after treatment with A-285222 when compared to plaque size in the same mice before treatment. These findings are in line with previous work from our laboratory showing abrogation of diabetes-induced aggravation of atherosclerosis in the aortic arch of STZ-induced diabetic ApoE–/– mice. This study goes beyond a toxin-induced type 1 diabetes model and supports a role for NFAT in the development of atherosclerosis in the context of type 2 diabetes and in an animal model that better replicates human disease than the STZ model.
Experimental models of atherosclerosis and diabetes are very susceptible to the housing environment, often translating into differences in the severity of the disease phenotype (i.e. atherosclerotic plaque size, degree of hyperglycaemia) when the same strain is studied in different animal facilities.18 This issue adds to the complexity of using animal models in medical research, potentially having an impact on study results and their interpretation, and to some extent, contributing to the poor translation of preclinical in vivo results into successful clinical trials and higher approval rates of new drug candidates. Among other explanations for this limited translational value [e.g. shortcomings in the design of clinical trials, overoptimistic conclusions from methodologically flawed animal studies, animal models that do not reflect human disease (reviewed in Van der Worp et al.26)] is the fact that validation studies are not traditionally conducted in the animal experimental field (when it is an absolute requirement for all other steps leading to the establishment of new therapies). A strength of this study is that the inhibitory effect of the NFAT blocker on atherosclerosis was replicated in two independent mice cohorts undergoing the same experimental protocol at different animal facilities.
Plaque size is the preferred primary readout in animal models and is typically determined histologically at the end of the experiments. Advances in ultrasound techniques have made possible the measurement of plaque progression/regression non-invasively in rodents, allowing for longitudinal studies which help reduce the number of animals required for statistical power. At termination, complementary evaluations of plaque area have shown good correlation between the histological and the non-invasive measurements in the brachiocephalic artery, the aorta and carotid arteries.22,27 Here, we also found a good correlation between the two techniques in the brachiocephalic arteries of young mice but were not able to dissect the histological difference in plaque size observed in the old female mice (Figure 5), probably because the effect size of the NFAT blocker was smaller in older mice than in younger mice and due to lower number of mice in the older cohort. Nevertheless, ultrasound biomicroscopy should be considered as an attractive non-invasive method to assess the effects of interventions in rodents.
In the ApoE-deficient mouse, it is well established that atherosclerotic lesions progress with age through all phases of atherogenesis, from monocyte adhesion, then lipid deposition and formation of fatty streaks to more mature and fibrous plaques;28,29 and that the pace at which this happens can vary depending on the vascular segment (e.g. more advanced lesions in the aortic root vs the aortic arch;10 slowest progressing lesions in the coronary arteries29). Our previous work highlighted differential susceptibility to STZ-induced atherosclerosis depending on the age of the lesions, with more pronounced effects of hyperglycaemia on plaque size in early stage lesions, such as those in the aortic arch, when compared to the effects on more advanced plaques such as those in the aortic root.10 In line with NFAT being activated by hyperglycaemia as we previously established,4,5 the effects of A-285222 had a more distinct impact on the aortic arch than on the aortic root of STZ-diabetic ApoE–/– mice, stressing the need for a diabetes-driven process for NFAT-inhibition to play a role.10 Here, we found in the IGF-II/LDLR–/–ApoB100/100 mice, a model in which the extent and severity of the lesions progress with age,19 a clear effect of A-285222 in the younger cohorts but only modest effects of NFAT-inhibition in the older mice (Figure 1 vs 5). Young mice exhibited more dynamic lesions characterized by an active inflammatory process with higher degree of macrophage infiltration than old mice (Figure 5(e) and (f)). It is possible that the reduction in plaque size observed after inhibition of NFAT signalling could be, at least in part, driven by reduced inflammation. Even though the proportion of the plaque occupied by macrophages was not affected by A-285222 treatment (Figure 2(d)), the reduction in plaque size observed in the males inevitably translated into reduced total macrophage content (from 7474 ± 2497 µm2 to 2239 ± 939 µm2 in males, p = 0.08).
IGF-II/LDLR–/–ApoB100/100 mice included in this study have elevated fasting blood glucose levels in line with previously reported values;19,30,31 however, they exhibited only moderate non-fasting hyperglycaemia compared to the values originally reported when the model was generated (~16 mmol/L)19. This discrepancy could be attributed to genetic drift, which should not be neglected even in inbred strains. The combination of impaired glucose tolerance and impaired fasting glucose31 makes the IGF-II/LDLR–/–ApoB100/100 mice a relevant model of pre-diabetes, a condition associated with higher risk of developing type 2 diabetes and cardiovascular disease. In previous work, we showed a clear dose-dependency of glucose-induced NFAT transcriptional activity, with modest elevations of a few mmol/L of extracellular glucose being sufficient for significant NFATc3 activation in the arterial wall.5 In light of this dose-dependency and the need of a hyperglycaemia-driven process to occur for NFAT-inhibition to have effects, it was reasonable to find that the same treatment with A-285222 had a more profound inhibitory effect on the expression of inflammatory markers in the aorta of STZ-diabetic ApoE–/– mice (a model characterized by profound hyperglycaemia) than of IGF-II/LDLR–/–ApoB100/100 mice with their milder hyperglycaemic phenotype.
NOX4 has recently been shown to protect against endothelial dysfunction and atherosclerosis in LDLR deficient mice.32 It was suggested that hydrogen peroxide derived from NOX4 acted via pAKT1 phosphorylation on eNOS activation. Here, we showed that inhibition of NFAT signalling in VSMCs exposed to a strong oxidative stress environment, significantly increased NOX4 expression (Figure 4(a)). The cell experiments also suggest that the observed upregulation of NOX4 expression in the aorta of male IGF-II/LDLR–/–ApoB100/100 mice (Figure 3(a)) after treatment with A-285222 could at least in part be due to direct effects of the NFAT blocker on the vascular cells. A similar NFAT-dependent regulation of NOX4 has been demonstrated in immortalized human podocytes stimulated with insulin using three complementary approaches to inhibit NFAT-signalling (tacrolimus, cyclosporine A and VIVIT peptide).33 Using another inducer of oxidative stress (SIN-1), we also found that inhibition of NFAT signalling resulted in enhanced catalase expression in VSMCs (Figure 4(b)).
This study shows that inhibition of NFAT signalling in IGF-II/LDLR–/–ApoB100/100 mice not only limits the progression of atherosclerosis but, more importantly, it leads to atherosclerosis plaque regression. The effects are not due to changes in plasma glucose, insulin or lipid levels but may be attributed to reduced inflammatory burden and improved anti-oxidant defences such as NOX4 and catalase. Vascular NFAT may be considered as a novel therapeutic target for the treatment of diabetic macrovascular complications.
Supplemental Material
Supplemental material, dvdres-sep-2017-00131-File003 for In vivo inhibition of nuclear factor of activated T-cells leads to atherosclerotic plaque regression in IGF-II/LDLR–/–ApoB100/100 mice by Fabiana Blanco, Suvi E Heinonen, Erika Gurzeler, Lisa M Berglund, Anna-Maria Dutius Andersson, Olga Kotova, Ann-Cathrine Jönsson-Rylander, Seppo Ylä-Herttuala and Maria F Gomez in Diabetes & Vascular Disease Research
Supplemental Material
Supplemental material, dvdres-sep-2017-00131-File004 for In vivo inhibition of nuclear factor of activated T-cells leads to atherosclerotic plaque regression in IGF-II/LDLR–/–ApoB100/100 mice by Fabiana Blanco, Suvi E Heinonen, Erika Gurzeler, Lisa M Berglund, Anna-Maria Dutius Andersson, Olga Kotova, Ann-Cathrine Jönsson-Rylander, Seppo Ylä-Herttuala and Maria F Gomez in Diabetes & Vascular Disease Research
Acknowledgments
The authors would like to thank Anna Zetterqvist for valuable input on the project and manuscript and Anne-Christine Andréasson for histological assistance.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: This work was supported by the Swedish Heart and Lung Foundation (grant numbers 20130700, 20160872), the Swedish Research Council (grant numbers 2011-3900, 2014-03352), the Swedish Foundation for Strategic Research (grant number IRC15-0067), the Swedish Society for Medical Research, the Crafoord, Albert Påhlsson and Knut & Alice Wallenberg foundations: also by the Innovative Medicines Initiative Joint Undertaking (grant number 115006), comprising funds from the European Union’s Seventh Framework Programme (grant number FP7/2007-2013) and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies’ in kind contribution. F.B. received support from CSIC (Comisión Sectorial de Investigación Científica) and PEDECIBA (Programa de Desarrollo de Ciencias Básicas).
ORCID iDs: Lisa M Berglund  https://orcid.org/0000-0001-6440-116X
https://orcid.org/0000-0001-6440-116X
Maria F Gomez
https://orcid.org/0000-0001-6210-3142
References
- 1. NCD-RisC. Worldwide trends in diabetes since 1980: a pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016; 387: 1513–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Madonna R, De Caterina R. Cellular and molecular mechanisms of vascular injury in diabetes – part I: pathways of vascular disease in diabetes. Vascul Pharmacol 2011; 54: 68–74. [DOI] [PubMed] [Google Scholar]
- 3. Paneni F, Costantino S, Cosentino F. Insulin resistance, diabetes, and cardiovascular risk. Curr Atheroscler Rep 2014; 16: 419. [DOI] [PubMed] [Google Scholar]
- 4. Nilsson-Berglund LM, Zetterqvist AV, Nilsson-Ohman J, et al. Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia. Arterioscler Thromb Vasc Biol 2010; 30: 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nilsson J, Nilsson LM, Chen YW, et al. High glucose activates nuclear factor of activated T cells in native vascular smooth muscle. Arterioscler Thromb Vasc Biol 2006; 26: 794–800. [DOI] [PubMed] [Google Scholar]
- 6. Zetterqvist AV, Blanco F, Öhman J, et al. Nuclear factor of activated T cells is activated in the endothelium of retinal microvessels in diabetic mice. J Diabetes Res 2015; 2015: 428473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee MY, Garvey SM, Baras AS, et al. Integrative genomics identifies DSCR1 (RCAN1) as a novel NFAT-dependent mediator of phenotypic modulation in vascular smooth muscle cells. Hum Mol Genet 2010; 19: 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nilsson LM, Sun ZW, Nilsson J, et al. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol 2007; 292: C1167–C1178. [DOI] [PubMed] [Google Scholar]
- 9. Orr AW, Lee MY, Lemmon JA, et al. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 2008; 29: 225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zetterqvist AV, Berglund LM, Blanco F, et al. Inhibition of nuclear factor of activated T-cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice. PLoS ONE 2013; 8: e65020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berglund LM, Kotova O, Osmark P, et al. NFAT regulates the expression of AIF-1 and IRT-1: yin and yang splice variants of neointima formation and atherosclerosis. Cardiovasc Res 2012; 93: 414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamagishi SI, Nakamura K, Matsui T, et al. Role of postprandial hyperglycaemia in cardiovascular disease in diabetes. Int J Clin Pract 2007; 61: 83–87. [DOI] [PubMed] [Google Scholar]
- 13. Lubrano V, Balzan S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J Exp Med 2015; 5: 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 2010; 30: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Konior A, Schramm A, Czesnikiewicz-Guzik M, et al. NADPH oxidases in vascular pathology. Antioxid Redox Signal 2014; 20: 2794–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gray SP, Di Marco E, Kennedy K, et al. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol 2016; 36: 295–307. [DOI] [PubMed] [Google Scholar]
- 17. Williams CR, Gooch JL. Calcineurin Aβ regulates NADPH oxidase (Nox) expression and activity via nuclear factor of activated T cells (NFAT) in response to high glucose. J Biol Chem 2014; 289: 4896–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heinonen SE, Genove G, Bengtsson E, et al. Animal models of diabetic macrovascular complications: key players in the development of new therapeutic approaches. J Diabetes Res 2015; 2015: 404085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heinonen SE, Leppanen P, Kholova I, et al. Increased atherosclerotic lesion calcification in a novel mouse model combining insulin resistance, hyperglycemia, and hypercholesterolemia. Circ Res 2007; 101: 1058–1067. [DOI] [PubMed] [Google Scholar]
- 20. Trevillyan JM, Chiou XG, Chen YW, et al. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J Biol Chem 2001; 276: 48118–48126. [DOI] [PubMed] [Google Scholar]
- 21. Gan LM, Gronros J, Hagg U, et al. Non-invasive real-time imaging of atherosclerosis in mice using ultrasound biomicroscopy. Atherosclerosis 2007; 190: 313–320. [DOI] [PubMed] [Google Scholar]
- 22. Gronros J, Wikstrom J, Brandt-Eliasson U, et al. Effects of rosuvastatin on cardiovascular morphology and function in an ApoE-knockout mouse model of atherosclerosis. Am J Physiol Heart Circ Physiol 2008; 295: H2046–H2053. [DOI] [PubMed] [Google Scholar]
- 23. Oukka M, Ho IC, de la, Brousse FC, et al. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity 1998; 9: 295–304. [DOI] [PubMed] [Google Scholar]
- 24. Awla D, Zetterqvist AV, Abdulla A, et al. NFATc3 regulates trypsinogen activation, neutrophil recruitment, and tissue damage in acute pancreatitis in mice. Gastroenterology 2012; 143: 1352–1360. [DOI] [PubMed] [Google Scholar]
- 25. Heit JJ. Calcineurin/NFAT signaling in the beta-cell: from diabetes to new therapeutics. Bioessays 2007; 29: 1011–1021. [DOI] [PubMed] [Google Scholar]
- 26. Van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med 2010; 7: e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gronros J, Wikstrom J, Hagg U, et al. Proximal to middle left coronary artery flow velocity ratio, as assessed using color Doppler echocardiography, predicts coronary artery atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2006; 26: 1126–1131. [DOI] [PubMed] [Google Scholar]
- 28. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med 1999; 340: 115–126. [DOI] [PubMed] [Google Scholar]
- 29. Nakashima Y, Plump AS, Raines EW, et al. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994; 14: 133–140. [DOI] [PubMed] [Google Scholar]
- 30. Heinonen SE, Merentie M, Hedman M, et al. Left ventricular dysfunction with reduced functional cardiac reserve in diabetic and non-diabetic LDL-receptor deficient apolipoprotein B100-only mice. Cardiovasc Diabetol 2011; 10: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinnunen K, Heinonen SE, Kalesnykas G, et al. LDLR-/-ApoB100/100 mice with insulin-like growth factor II overexpression reveal a novel form of retinopathy with photoreceptor atrophy and altered morphology of the retina. Mol Vis 2013; 19: 1723–1733. [PMC free article] [PubMed] [Google Scholar]
- 32. Langbein H, Brunssen C, Hofmann A, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J 2016; 37: 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia S, Liu Y, Li X, et al. Insulin increases expression of TRPC6 channels in podocytes by a calcineurin-dependent pathway. Cell Physiol Biochem 2016; 38: 659–669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, dvdres-sep-2017-00131-File003 for In vivo inhibition of nuclear factor of activated T-cells leads to atherosclerotic plaque regression in IGF-II/LDLR–/–ApoB100/100 mice by Fabiana Blanco, Suvi E Heinonen, Erika Gurzeler, Lisa M Berglund, Anna-Maria Dutius Andersson, Olga Kotova, Ann-Cathrine Jönsson-Rylander, Seppo Ylä-Herttuala and Maria F Gomez in Diabetes & Vascular Disease Research
Supplemental material, dvdres-sep-2017-00131-File004 for In vivo inhibition of nuclear factor of activated T-cells leads to atherosclerotic plaque regression in IGF-II/LDLR–/–ApoB100/100 mice by Fabiana Blanco, Suvi E Heinonen, Erika Gurzeler, Lisa M Berglund, Anna-Maria Dutius Andersson, Olga Kotova, Ann-Cathrine Jönsson-Rylander, Seppo Ylä-Herttuala and Maria F Gomez in Diabetes & Vascular Disease Research