

Unlike typical respiratory oxidases, alternative oxidase (Aox) does not directly contribute to energy conservation, and its activity would presumably reduce the efficiency of respiration and associated ATP production. Aox has been identified in certain bacteria, a majority of which are marine associated. The presence of Aox in these bacteria poses the interesting question of how Aox function benefits bacterial growth and survival in the ocean. Using the genetically tractable marine bacterium Vibrio fischeri, I have identified a role for Aox in reduction of stress under conditions where electron flux through the aerobic respiratory pathway is inhibited. These results suggest that Aox activity could positively impact longer-term bacterial fitness and survival under stressful environmental conditions.
KEYWORDS: Aliivibrio fischeri, RNA-seq, Fenton chemistry, Vibrio fischeri, nitric oxide, oxidase, respiration
ABSTRACT
Alternative oxidase (Aox) is a non-energy-conserving respiratory oxidase found in certain eukaryotes and bacteria, whose role in physiology is not entirely clear. Using the genetically tractable bacterium Vibrio fischeri as a model organism, I have identified a role for Aox to reduce levels of stress in cells exposed to oxygen and nitric oxide (NO). In V. fischeri lacking the NO-detoxifying enzyme flavohemoglobin (Hmp), deletion of aox in cells grown in the presence of oxygen and NO results in alterations to the transcriptome that include increases in transcripts mapping to stress-related genes. Using fluorescence-based reporters, I identified corresponding increases in intracellular reactive oxygen species and decreases in membrane integrity in cells lacking aox. Under these growth conditions, activity of Aox is linked to a decrease in NADH levels, indicating coupling of Aox activity with NADH dehydrogenase activity. Taken together, these results suggest that Aox functions to indirectly limit production of ferrous iron and damaging hydroxyl radicals, effectively reducing cellular stress during NO exposure.
IMPORTANCE Unlike typical respiratory oxidases, alternative oxidase (Aox) does not directly contribute to energy conservation, and its activity would presumably reduce the efficiency of respiration and associated ATP production. Aox has been identified in certain bacteria, a majority of which are marine associated. The presence of Aox in these bacteria poses the interesting question of how Aox function benefits bacterial growth and survival in the ocean. Using the genetically tractable marine bacterium Vibrio fischeri, I have identified a role for Aox in reduction of stress under conditions where electron flux through the aerobic respiratory pathway is inhibited. These results suggest that Aox activity could positively impact longer-term bacterial fitness and survival under stressful environmental conditions.
INTRODUCTION
Alternative oxidase (AOX [eukaryotes] or Aox [bacteria]) is a terminal respiratory oxidase present in all plants as well as certain other eukaryotes (1). The activity of this protein does not directly contribute to the generation of a proton motive force (2), a characteristic that is atypical of respiratory oxidases. AOX has been well studied in plants, where its non-energy-conserving activity is known to reduce the energy efficiency of plant respiration. Although seemingly wasteful, AOX has been linked to useful activities such as heat production in thermogenic plants (3). The role of this protein in nonthermogenic plants is not as clear, but it is believed to play a role in modulating metabolic state and/or signaling during stress (4), as well as affecting energy and carbon metabolism in plants under nonstress conditions (5). Understanding the specific roles of AOX in nonthermogenic plants and other eukaryotic organisms is currently an active area of research.
The discovery of Aox in bacteria is relatively more recent, coinciding with the availability of increased amounts of genomic and environmental metagenomic sequencing data (6–8). The number of bacteria predicted to encode Aox continues to grow as more genomes and environmental samples are sequenced, and based on database searches, a majority of aox-like genes are found in marine environment-associated bacterial isolates and marine metagenome samples. Examples include an abundance of aox-like sequences in the metagenomic data sets from the Global Ocean Sampling expedition (9) and the Sargasso Sea (7) and the presence of aox in a cultivated member of the SAR11 group of numerically dominant marine bacteria (Pelagibacter spp.) (10). In addition, based on genome sequence data, it appears that aox-like genes are fairly widespread, although not ubiquitous, in Vibrio spp. (reference 11 and data not shown). The presence of aox-like genes in these data sets suggests an importance of Aox function for the growth and survival of certain bacteria in the ocean. However, the role of Aox in bacterial physiology is not yet clear, as detailed studies have been limited by availability of isolates and/or genetically manipulable isolates. We previously discovered an Aox-encoding gene in the marine bacterium Vibrio fischeri (Aliivibrio fischeri) strain ES114 (12) and are using this genetically tractable bacterium to better understand the physiological benefit of Aox function in bacteria.
V. fischeri ES114 was isolated from the Hawaiian bobtail squid, Euprymna scolopes (13), and is used as a model strain in studying this mutualistic light organ symbiosis (14). Aox is expressed in V. fischeri ES114 in response to nitric oxide (NO) and, unlike the other terminal oxidases in this bacterium, is resistant to inhibition by NO (12). It is known that V. fischeri encounters host-produced NO during symbiotic colonization (15), and therefore, the response to NO is physiologically relevant in the natural environments this bacterium inhabits.
Based on these previous results, I hypothesized that the physiological benefit of Aox function is relevant to conditions where both oxygen and NO are present and there is flux through the aerobic respiratory pathways. Under these conditions, NO would be expected to inhibit the NO-sensitive respiratory oxidases while inducing expression of the NO-resistant Aox. Aox could then function to allow electron flow, which could positively impact the cell in various ways. Here, I demonstrate that in a strain lacking the main aerobic NO-detoxifying protein Hmp (16), Aox contributes to reducing NADH levels while also decreasing the levels of reactive oxygen species and membrane damage in cells exposed to NO. These results indicate that a physiological benefit of Aox function in V. fischeri is to reduce cellular stress during NO exposure, which could positively impact longer-term fitness and survival.
RESULTS
A lack of aox in a V. fischeri hmp mutant background affects growth in the presence of nitric oxide.
Although a strain lacking aox did not display phenotypic differences from the wild type under previously tested growth conditions (12), deletion of this gene in a V. fischeri strain lacking the aerobic NO-detoxifying enzyme flavohemoglobin (Hmp) (16) resulted in an alteration in the growth phenotype of cells grown in glucose mineral salts medium containing the NO-generating compound dipropylenetriamine (DPTA)-NONOate (Fig. 1). This growth phenotype could be complemented by providing aox in trans under the control of an inducible promoter. A similar growth phenotype was observed in mineral salts medium containing NO and either N-acetylglucosamine or glycerol as the carbon source (data not shown).
FIG 1.
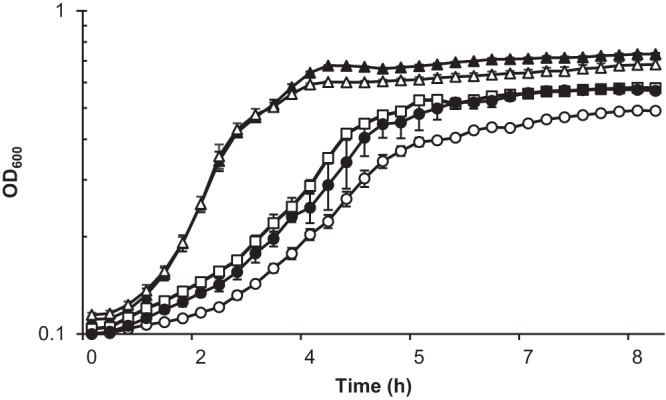
A V. fischeri strain lacking hmp and aox displays an altered growth phenotype in mineral salts medium with nitric oxide. V. fischeri strains ES114 (wild type; white triangles), AKD915 (Δhmp; white squares), AKD916 (Δhmp Δaox; white circles), and AKD916 containing the aox-complementing plasmid pAKD601Baox (black circles) were grown in mineral salts medium and treated with DPTA-NONOate. Data are also shown for the wild-type strain grown in the absence of DPTA-NONOate (black triangles). There was no difference in growth between the wild type, AKD915, and AKD916 when grown in the absence of DPTA-NONOate (data not shown). Experiments were repeated three times, and the results of one representative experiment are shown.
V. fischeri cells lacking aox and hmp have higher plating efficiency on solid medium after NO treatment.
To determine whether the growth phenotype observed during NO treatment was due to differences in the number of cells present in the cultures, AKD915 (Δhmp) and AKD916 (Δhmp Δaox) were grown in mineral salts medium and samples removed 6 h after NO treatment for direct counting and plating to determine recoverable CFU (Fig. 2B). No significant difference between the numbers of cells of the two strains were observed by direct counting (P > 0.05). However, a significantly higher number of CFU were observed from the cultures lacking aox, suggesting that these cells were better able to cope with the transition from mineral salts liquid medium to rich solid medium. No significant differences in direct counts or plating efficiency were observed between the wild-type strain (ES114) and AKD915 and AKD916 in the absence of NO treatment (Fig. 2A), indicating that this plating efficiency phenotype was linked to NO treatment. Previous work in Escherichia coli identified that when cells were transferred from liquid to solid medium, global stress responses were induced (17). If V. fischeri cells lacking aox experienced higher levels of stress when treated with NO in liquid medium, then it is plausible that these stress response pathways would be upregulated and help the cells transition from liquid to solid medium.
FIG 2.
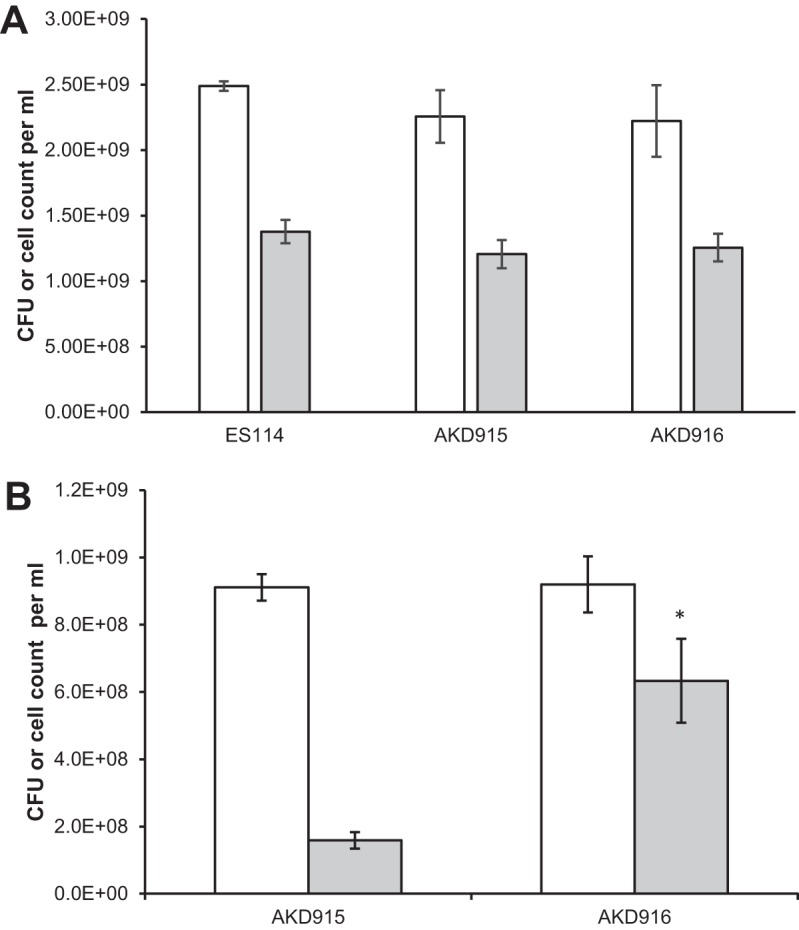
Cell recovery via direct count versus plating of the wild-type strain (ES114), AKD915 (Δhmp), and AKD916 (Δhmp Δaox) after growth in mineral salts medium either without (A) or with (B) nitric oxide treatment. Cells were grown in mineral salts medium and, where indicated, treated with DPTA-NONOate. After 6 h, samples were removed for direct counts using a Petroff-Hausser counter (white bars) and plating onto rich medium (gray bars) to determine recoverable CFU. (A) In the absence of nitric oxide, there was no significant difference (P > 0.05) between strains in direct cell counts or CFU recovered after plating. (B) After treatment with nitric oxide, more CFU of a V. fischeri strain lacking hmp and aox (AKD916) than of the parental strain lacking hmp (AKD915) were recovered. Significance for comparisons between direct counts of AKD915 and AKD916 or between plating recovery for AKD915 and AKD916 was measured using Student's t test. *, P < 0.04. No significant difference was detected between direct counts or plating recovery for wild-type cells grown in the presence or absence of NO (P > 0.05) (data not shown). Data shown are averages (±standard error) from one representative experiment containing three independent cultures. Experiments were repeated three times.
Transcriptome analysis identifies alterations in gene expression in AKD916 (Δhmp Δaox), including genes likely involved in stress responses.
To determine whether there were significant alterations in the transcriptomes of AKD915 (Δhmp) and AKD916 (Δhmp Δaox) grown in the presence of nitric oxide, cells were grown in glucose mineral salts medium with DTPA-NONOate for 6 h prior to harvesting total RNA for transcriptome sequencing (RNA-seq) analysis. Analysis of these data using Rockhopper (18) identified 602 differentially regulated transcripts (410 upregulated and 192 downregulated) (see Data Set S1 in the supplemental material). Of the nine sense transcripts identified as having a 4.5-fold or greater induction in AKD916 compared to AKD915, two mapped to open reading frames (ORFs) annotated as encoding hypothetical proteins (VF_A0006 and VF_A0314), and five mapped to ORFs annotated as encoding stress-related proteins (VF_1685, VF_A0005, VF_A0311, VF_A0312, and VF_A0313) (Table 1). RNA-seq results were verified for a subset of Rockhopper-predicted upregulated and downregulated genes using quantitative reverse transcriptase PCR (qRT-PCR) (Fig. 3).
TABLE 1.
Sense transcripts with 4.5-fold or greater induction in V. fischeri AKD916 (Δhmp Δaox) compared to AKD915 (Δhmp)
| ORF | Annotation | Fold induction |
|---|---|---|
| VF_1685 | recR; recombination protein RecR | 5.2 |
| VF_1735 | acpP; acyl carrier protein | 4.9 |
| VF_A0005 | msrAB; peptide-methionine (S)-S-oxide reductase | 5.7 |
| VF_A0006 | Hypothetical protein | 5.8 |
| VF_A0311 | pspA; regulatory protein for phage shock protein operon | 4.5 |
| VF_A0312 | pspB; phage shock protein B | 5.6 |
| VF_A0313 | pspC; phage shock protein C | 5.5 |
| VF_A0314 | ycjX; hypothetical protein | 5.1 |
| VF_A0364 | Transporter | 5.1 |
FIG 3.
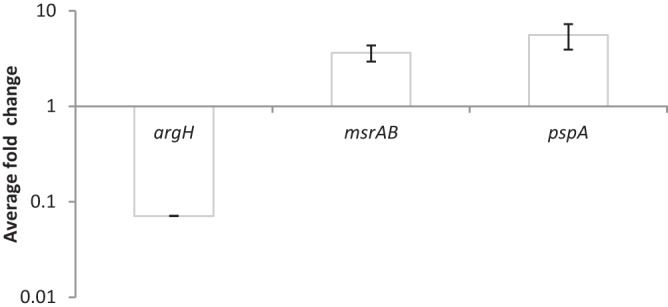
Quantitative real-time PCR results for selected genes identified as differentially expressed in the Rockhopper analysis of RNA-seq data. The results demonstrate relative transcript levels of argH (VF_2303), msrAB (VF_A0005), and pspA (VF_A0311) in AKD916 (Δhmp Δaox) compared to AKD915 (Δhmp) after 6 h of growth in mineral salts medium containing the nitric oxide generator DPTA-NONOate. Relative gene expression was measured using the relative standard curve method and ftsK (VF_0905) as an endogenous control. The trend in gene expression levels agrees with the Rockhopper analysis (Table 1; see Data Set S1 in the supplemental material). Data are the averages from three separate PCR runs, one for each of three independent replicates.
Aox function under NO treatment reduces intracellular reactive species levels and improves membrane integrity.
Transcriptome analysis suggested that V. fischeri cells lacking a functional Aox were experiencing more cellular stress when treated with NO. To attempt to measure increased levels of stress, cells grown in mineral salts medium were harvested 6 h after NO treatment and stained with fluorescent indicators for reactive oxygen species (aminophenyl fluorescein [APF]) or membrane integrity (SYTO 9 and propidium iodide [LIVE/DEAD BacLight assay]). Compared to AKD915 (Δhmp) cells, AKD916 (Δhmp Δaox) cells had significantly higher levels of fluorescence when treated with APF and higher levels of red fluorescence when treated with SYTO 9 and propidium iodide, indicating higher levels of hypochlorite, peroxynitrite, and/or the hydroxyl radical and increased membrane damage, respectively (Table 2). These results support the hypothesis that cells lacking Aox experience higher levels of cellular stress when treated with NO.
TABLE 2.
Fluorescence-based assays for reactive oxygen species production and membrane integrity in V. fischeri strains treated with nitric oxide
| Straina | Reactive oxygen species (APF) |
Membrane integrity (LIVE/DEAD) |
||
|---|---|---|---|---|
| Avg fluorescence (SE) | P valueb | Avg green/red fluorescence (SE) | P value | |
| AKD915 (Δhmp) | 635 (32) | 0.018 | 129 (11) | 0.026 |
| AKD916 (Δhmp Δaox) | 909 (33) | 88 (8) | ||
| AKD916 with pAKD601Baox | ||||
| Without IPTG | 782 (13) | 0.0007 | 91 (3) | 0.011 |
| With 0.1 mM IPTG | 646 (11) | 128 (7) | ||
Representative data shown for the comparison of AKD915 and AKD916 were collected in an experiment separate from that for the comparison of AKD916(pAKD601Baox) with and without IPTG.
P value calculated using Student's t test comparing AKD915 with AKD916 or comparing AKD916(pAKD601Baox) with and without IPTG.
V. fischeri AKD916 (Δhmp Δaox) has higher levels of NADH when grown in the presence of NO.
Previously, we showed that V. fischeri Aox is induced in response to nitric oxide and functions as a NO-resistant oxidase terminating the electron transport chain (12). If activity of Aox was coupled to the activity of a NADH dehydrogenase in the electron transport chain, a lack of Aox under these conditions should result in higher cellular NADH levels. To test this, samples of AKD915 (Δhmp) and AKD916 (Δhmp Δaox) were harvested after 6 h of growth in mineral salts medium containing DPTA-NONOate, and the levels of NAD+ and NADH were assayed. After 6 h of growth, AKD916 had significantly higher levels of NADH than AKD915, indicating that under these conditions, Aox activity is coupled to the activity of an NADH dehydrogenase (Fig. 4). It has been previously shown that increased levels of NADH can promote oxidative DNA damage in E. coli (19), suggesting a possible link between Aox function, NADH levels, and cellular stress in V. fischeri.
FIG 4.
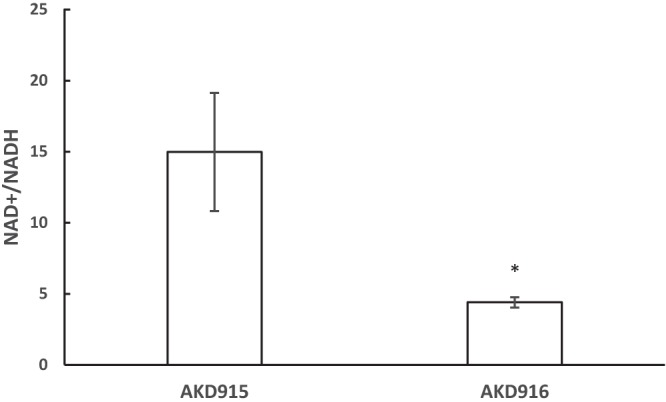
A V. fischeri strain lacking hmp and aox has elevated NADH levels 6 h after treatment with nitric oxide. V. fischeri strains AKD915 (Δhmp) and AKD916 (Δhmp Δaox) were grown in mineral salts medium and treated with DPTA-NONOate. Samples were taken 6 h after treatment and assayed for NAD+ and NADH levels. AKD915 samples had an average of 1.25 (±0.098) μM NAD+ plus NADH and AKD916 samples had an average of 1.68 (±0.16) μM NAD+ plus NADH. Data shown are the averages from three experiments, each containing three independent replicates. Significance was measured using Student's t test. *, P < 0.02.
Addition of the iron chelator 2,2′-bipyridyl or the antioxidant glutathione to cultures can partially reverse the NO-induced aox mutant growth phenotype.
Oxidative DNA damage in E. coli in response to increased levels of NADH is mediated through Fenton chemistry (19), where NADH indirectly contributes to the reduction of iron to the ferrous state. The ferrous iron then participates in the Fenton reaction to generate the DNA-damaging hydroxyl radical. If a similar situation was occurring in V. fischeri cells lacking Aox, then addition of the ferrous iron chelator 2,2′-bipyridyl or the antioxidant glutathione to NO-treated cultures should result in less stress and a less pronounced growth phenotype. Addition of either of these compounds did reduce the magnitude of the growth phenotype of cells lacking Aox (Fig. 5), indicating that under these conditions, Aox functions to reduce NADH levels and therefore presumably indirectly reduces production of ferrous iron and damaging hydroxyl radicals.
FIG 5.
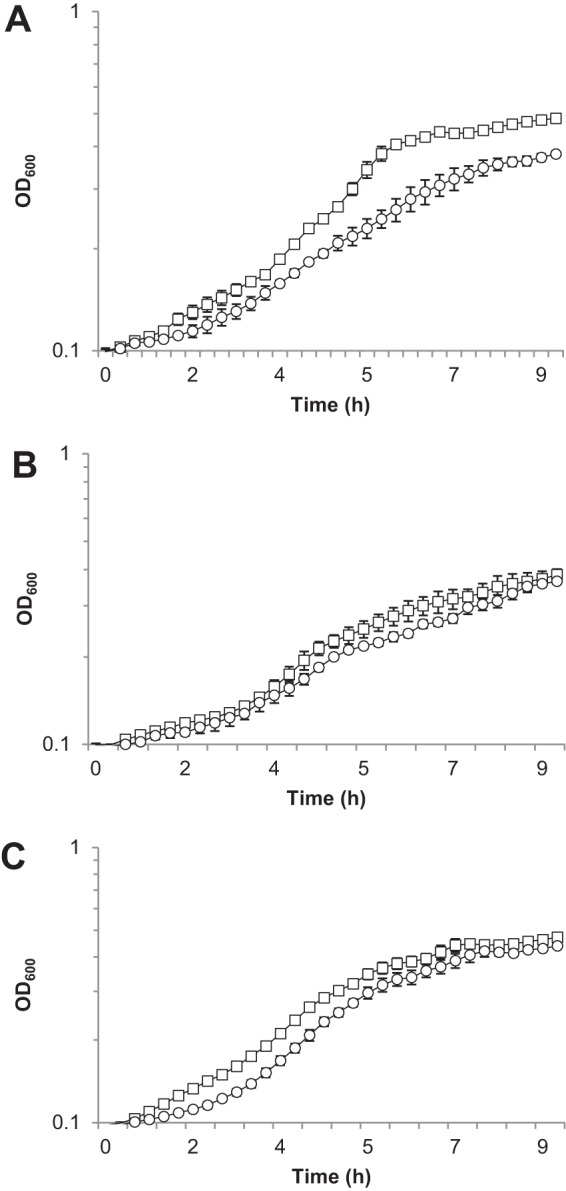
The nitric oxide-induced growth phenotype of the V. fischeri strain lacking hmp and aox can be partially complemented by providing the iron chelator 2,2′-bipyridyl or the antioxidant glutathione. AKD915 (Δhmp; squares) and AKD916 (Δhmp Δaox; circles) were grown in mineral salts medium containing DPTA-NONOate alone (A) or with the addition of 50 μM 2,2′-bipyridyl (B) or 1 mM glutathione (C). Experiments were repeated three times, and the results of one representative experiment are shown.
DISCUSSION
The results of this study identify a function for bacterial alternative oxidase in reduction of cellular stress under conditions where electron flux through the aerobic respiratory pathway is inhibited. We previously demonstrated that in V. fischeri ES114, addition of NO to cultures reversibly inhibits activity of the typical respiratory oxidases while at the same time inducing expression of Aox, an NO-resistant oxidase (12). This induction of Aox activity would allow electron flow through the respiratory pathway in the presence of NO. This would likely be at a lower energy efficiency because Aox activity is not directly linked to the generation of the proton motive force, but it presumably would have other cellular benefits under these growth conditions. To test this hypothesis, I used a V. fischeri strain lacking a main aerobic NO-detoxifying enzyme, Hmp, to create a situation where NO was not removed as quickly in cells to allow better observation of the effect of Aox activity on cellular processes. Using this Δhmp strain background, I observed that Aox activity did have significant positive impacts on the cells, resulting in lower levels of NADH, lower levels of reactive oxygen species, and less cellular membrane damage.
V. fischeri ES114 has flexible respiratory pathways (20) and could couple oxidase activity with several different dehydrogenases during respiration. However, under these conditions, my results suggest that Aox activity is coupled to the activity of an NADH dehydrogenase, as lack of Aox results in higher levels of cellular NADH (Fig. 3). Excess NADH could contribute to increased cellular stress similarly to the case in E. coli, where higher NADH levels promoted oxidative DNA damage (19). In E. coli, it was demonstrated that NADH indirectly contributes to the reduction of iron to the ferrous state, which then can participate in a Fenton chemistry reaction to produce DNA-damaging hydroxyl radicals (19). Several lines of experimental evidence support this scenario in V. fischeri. Transcriptome analysis identified increased levels of transcripts mapping to genes whose products could be involved in stress responses, including DNA damage repair (recR) (Table 1; see Data Set S1 in the supplemental material) and membrane stress (pspA, pspB, and pspC) (Table 1 and Data Set S1) (21). In addition, cells lacking Aox had a significantly higher level of fluorescence when treated with the reactive oxygen species indicator APF, which detects hypochlorite, peroxynitrite, and the hydroxyl radical (Table 2). Finally, addition of the ferrous iron chelator 2,2′-bipyridyl or the antioxidant glutathione reduced the severity of the growth phenotype in the strain lacking Aox (Fig. 5). Taken together, these data support the function of Aox to allow electron flow under conditions where the aerobic electron transport chain activity is inhibited, effectively reducing intracellular stress. Further studies will explore how Aox activity contributes to longer-term fitness and survival.
It remains to be determined whether Aox regulation and function are similar in other marine bacteria. Our unpublished preliminary data using quantitative RT-PCR suggest that NO does induce aox transcript levels in other marine bacteria, including alphaproteobacterial and gammaproteobacterial isolates. In the case of V. fischeri, it is known that NO is encountered during host colonization (15), which might not be relevant to the lifestyle(s) of other marine bacteria. However, it is known that denitrifying bacteria can produce NO that affects surrounding microbes (22), which could be relevant in certain marine environments inhabited by bacteria encoding Aox. Even if NO does not universally induce Aox expression in all Aox-containing bacteria, it is possible that other conditions that impact normal electron flow through the respiratory pathway induce Aox expression, resulting in a similar protective mechanism against cellular stress. The results reported here lay the foundation for further exploration of the conservation of Aox function in diverse bacteria and how activity of this interesting protein impacts bacterial growth and survival in marine environments.
More broadly, understanding the role of Aox in bacteria will allow functional comparisons to eukaryotic AOX and investigations into the evolutionary history of this protein. In certain plants, AOX activity is induced in response to stress and may contribute to reduction in the production of reactive oxygen species (23), suggesting that there may be similarities in Aox function in bacteria and eukaryotes. Studying AOX in plants has been complicated by the presence of multiple genes encoding different versions of the protein which can be used under different situations with different outcomes (24), making studies involving the physiological role of AOX difficult. Bacteria typically contain a single copy of an aox-like gene, with certain isolates being more amenable to genetic manipulation, which can simplify functional studies. In addition, it has been postulated that AOX was acquired from bacteria by eukaryotes during endosymbiosis events that created mitochondria (7). Therefore, studying Aox function and distribution in bacteria also has implications for understanding the evolutionary history of this protein.
MATERIALS AND METHODS
Strains, plasmids, and growth conditions.
The strains and plasmids used in this study are listed in Table 3. Escherichia coli was grown in LB medium (25) with 40 μg/ml kanamycin where noted or in HiVeg Special Infusion broth (Himedia, Mumbai, India) containing 150 μg/ml erythromycin. Vibrio fischeri was grown either in LB medium with added salt (LBS) (26) or in mineral salts medium with 20 mM glucose as the carbon source, containing the following (per liter): 378 ml 1 M NaPO4 (pH 7.5), 50 ml 1 M Tris (pH 7.5), 3 mg FeSO4 · 7H2O, 0.59 g NH4Cl, 13.6 g MgSO4 · 7H2O, 0.83 g KCl, 1.62 g CaCl2, 1 g Casamino Acids (Difco, vitamin assay), and, where noted, 100 μg/ml kanamycin. Where indicated, dipropylenetriamine (DPTA)-NONOate (Cayman Chemical, Ann Arbor, MI) was added to cultures to a final concentration of 200 μM. DPTA-NONOate is a nitric oxide (NO) donor with a half-life of 5 h at 22 to 25°C and pH 7.4 to liberate 2 mol of NO per mol of parent compound. For complementation assays, 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Research Products International), 50 μM 2,2′-bipyridyl (Sigma, St. Louis, MO), or 1 mM glutathione (Sigma) was added to cultures where indicated.
TABLE 3.
Strains and plasmids
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| Vibrio fischeri | ||
| ES114 | Wild type | 13 |
| AKD915 | Δhmp (allele exchanged from pAKD915 into ES114) | This study |
| AKD916 | Δhmp Δaox (allele exchanged from pAKD780 into AKD915) | This study |
| Escherichia coli | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR supE44 hsdR17 recA1 endA1 gyrA96 thi-1 relA | 31 |
| DH5αλpir | λpir derivative of DH5α | 32 |
| CC118λpir | Δ(ara-leu) araD ΔlacX74 galE galK phoA20 thi-1 rpsE rpoB argE(Am) recA; lysogenized with λpir | 27 |
| Plasmids | ||
| pAKD915 | Δhmp allele, R6Kγ and ColE1 replication origins, RP4 oriT, Ermr Kanr | This study |
| pAKD780 | Δaox allele, R6Kγ and ColE1 replication origins, RP4 oriT, Ermr Kanr | 12 |
| pAKD601Baox | Plasmid containing lacIq and an IPTG-inducible promoter for controlled expression of Aox in V. fischeri, Kanr | 12 |
| pEVS104 | Conjugative helper plasmid, R6Kγ replication origin, Kanr | 27 |
Ermr, erythromycin resistance; Kanr, kanamycin resistance (aph).
General cloning procedures were used with E. coli, and constructs containing the RP4 origin of transfer were introduced into V. fischeri using triparental mating (27). E. coli DH5α λpir was used for plasmid construction, and E. coli DH5α, DH5α λpir, or CC118 λpir was used for conjugative transfer of plasmids into V. fischeri. Plasmids were constructed using standard molecular biology techniques and restriction enzymes from New England BioLabs (Ipswich, MA). PCR products were amplified using Phusion high-fidelity DNA polymerase (New England BioLabs) with primers from Integrated DNA Technologies (Coralville, IA). Primer sequences are listed in Table 4.
TABLE 4.
Primers used in this study
| Primer name | Primer sequence (5′→3′) | Use |
|---|---|---|
| hmpupF | GAGTTACCACGCTGAATCGCT | Forward primer for upstream fragment for hmp deletion construct |
| hmpupR | TTATAAAAATTAGTCGACGTTGAGCATATTGTTTACCTTTAAAGGTGTA | Reverse primer for upstream fragment for hmp deletion construct |
| hmpdownF | CAATATGCTCAACGTCGACTAATTTTTATAAAGTAGAAATGCAAAAACGCCCA | Forward primer for downstream fragment for hmp deletion construct |
| hmpdownR | CACGGTACTTACCCTTACGTAACATC | Reverse primer for downstream fragment for hmp deletion construct |
| hmpRepF | TACTACCTAGGACGCTTGCTTTGCTTTCGCT | Used with hmpTestR to screen for hmp deletion mutants |
| hmpTestR | TCTGTTACTGCTGATGGACGTAAACAT | |
| msrAB F | CGCAAGAAGAGGGAACAGAA | Forward primer for qRT-PCR of VF_A0005 |
| msrAB R | CCAACCAGTCCCGGATTTATAC | Reverse primer for qRT-PCR of VF_A0005 |
| pspA F | CCAACAAACTGCACAGCATAG | Forward primer for qRT-PCR of VF_A0311 |
| pspA R | CTCATCAATTCGGCGTTCATATT | Reverse primer for qRT-PCR of VF_A0311 |
| argH F | ATTGGCTGATACCGTGACTTC | Forward primer for qRT-PCR of VF_2303 |
| argH R | CCATATACACGTCCACACTTACC | Reverse primer for qRT-PCR of VF_2303 |
| ftsZ F | GTGCGTGTCGTTGAAGTTATTC | Forward primer for qRT-PCR of VF_0905 |
| ftsZ R | GGCTACCTACAACATCAGAGAAG | Reverse primer for qRT-PCR of VF_0905 |
V. fischeri strains lacking hmp (VF_2316) or aox (VF_0578) were constructed using allelic exchange (28). Briefly, approximately 1.6 kb of DNA upstream of each target was PCR amplified and fused to an approximately 1.6-kb DNA fragment downstream of each gene using an engineered restriction site. The PCR products included the respective start and stop codons and additional flanking codons as needed for primer design. For the hmp deletion construct, two codons after the start were included, and for the aox deletion construct, two codons prior to the stop were included. These constructs resulted in replacement of the genes with several codons and a 6-bp restriction enzyme recognition site. Primers are listed in Table 4.
Microplate-based growth curves.
V. fischeri strains were grown overnight in LBS medium, and 12 μl was subcultured into 18-mm culture tubes containing 3 ml of glucose mineral salts medium. Tubes were incubated with vigorous shaking at 28°C. After 1.5 h, 200-μl samples were removed and placed into the wells of a lidded 96-well microtiter dish (Falcon 351172 flat-bottom sterile polystyrene plate). For samples exposed to NO, DPTA-NONOate was added to a final concentration of 200 μM. For complementation assays, IPTG, 2,2′-bipyridyl, or glutathione was added. The plate was incubated at 28°C with double orbital shaking (slow speed, 282 cpm, 3-mm radius) in a BioTek Synergy H1 plate reader (Winooski, VT). Absorbance at 600 nm was measured every ∼16 min for ∼9 h.
Flask-based growth.
For experiments involving direct cell counts and CFU recovery, transcriptome analysis, fluorescence-based assays for reactive oxygen species and membrane integrity, and NAD+/NADH assays, flask-based cultures were used. Strains were grown overnight in LBS medium, and 60 μl was subcultured into 125-ml flasks containing 15 ml of glucose mineral salts medium. Flasks were incubated with vigorous shaking at 28°C. After 1.5 h, where indicated, DPTA-NONOate was added to each flask to a final concentration of 200 μM. Samples were collected 6 h after addition of DPTA-NONOate. Absorbance at 600 nm was measured using a Bio-Rad SmartSpec Plus spectrophotometer (Hercules, CA).
Direct cell counts and CFU recovery.
Cultures were grown as described above for flask-based growth. Three independent replicates of the wild-type strain ES114, AKD915, and AKD916 were grown in mineral salts medium. Samples were collected at 7.5 h for experiments without DPTA-NONOate (Fig. 2A) (ES114 average optical density at 600 nm [OD600], 2.17 ± 0.02; AKD915 average OD600, 2.19 ± 0.01; AKD916 average OD600, 2.09 ± 0.05) or 6 h after addition of DPTA-NONOate (Fig. 2B) (ES114 average OD600, 1.97 ± 0.07; AKD915 average OD600, 0.82 ± 0.01; AKD916 average OD600, 0.73 ± 0.005). Cells were directly counted using a Petroff-Hausser counting chamber (Hausser Scientific Co., Horsham, PA). For each sample, the bacteria within three 50-μm squares were counted and averaged. This number was multiplied by 20,000,000 according to the manufacturer's directions to determine the number of bacteria per milliliter. For CFU recovery, samples were spot dilution plated (29) onto LBS medium and incubated overnight at 28°C prior to counting. Data presented are the average for three independent cultures from one representative experiment, with error bars indicating standard error. Experiments were repeated three times with similar results. Significance was measured using Student's t test.
Transcriptome analysis using RNA-seq.
Cultures were grown as described above for flask-based growth. Three independent replicates each of AKD915 and AKD916 were grown in mineral salts medium. Samples were collected at 6 h after addition of DPTA-NONOate (AKD915 average OD600, 0.84 ± 0.01; AKD916 average OD600, 0.69 ± 0.003). Two-milliliter culture samples were treated with RNAprotect Bacteria Reagent (Qiagen) following the manufacturer's protocol, and the resultant pellets were resuspended in 2 ml of TRIzol reagent (Life Technologies). RNA was isolated using the Zymo Direct-zol RNA miniprep kit, following the manufacturer's protocol (Zymo Research, Irvine, CA). To remove contaminating DNA, RNA samples were treated with the Turbo DNA-free kit (Life Technologies), using the rigorous DNase treatment protocol. Sequencing libraries were prepared from 3.4 μg of RNA of each sample using the ScriptSeq Complete kit (Epicentre, Madison, WI), following the protocol from the manufacturer. Samples were barcoded using the Epicentre ScriptSeq Index PCR primers 1 to 12 and combined into a single sample. Sequencing was performed at the Tufts University Core Genomics Facility using a single lane in an Illumina HiSeq 2500 sequencer run in rapid mode with single-end 100-nucleotide reads. Sequences were aligned, quantified, and tested for differential expression using Rockhopper (18).
Quantitative real-time PCR.
Samples were grown as indicated for RNA-seq experiments. Three independent cultures were grown in mineral salts medium on two separate days (AKD915 average OD600, 0.88 ± 0.02; AKD916 average OD600, 0.72 ± 0.01). One-milliliter samples were harvested 6 h after treatment with the nitric oxide generator DPTA-NONOate and treated with RNA Protect Bacteria Reagent (Qiagen) following the manufacturer's protocol, and the resultant pellets were resuspended in 2 ml of TRIzol reagent (Life Technologies). RNA was isolated using the Zymo Direct-zol RNA miniprep kit, following the manufacturer's protocol (Zymo Research, Irvine, CA). To remove contaminating DNA, RNA samples were treated with the Turbo DNA-free kit (Life Technologies), using the rigorous DNase treatment protocol. cDNA was prepared using the Applied Biosystems high-capacity cDNA RT kit using 2 μg RNA as a template following the manufacturer's protocol. Quantitative PCR was performed using an Applied Biosystems Step One Plus PCR machine in combination with GoTaq qPCR master mix (Promega). Cycling conditions were 95°C for 1 min followed by 40 cycles of 95°C for 15 s, 55°C for 1 min, and 72°C for 1 min. The relative standard curve method was used to calculate relative expression levels using ftsK as an endogenous control. Data presented are averages from three separate PCR runs, one for each independent replicate. Primers are listed in Table 4.
Fluorescence-based assays for reactive oxygen species and membrane integrity.
Three replicate cultures of each strain were grown in glucose mineral salts medium as described for flask-based growth. Strain AKD916 containing pAKD601Baox was grown in medium both with and without 0.1 mM IPTG. Six hours after addition of DPTA-NONOate, 0.5-ml samples were collected for analysis (AKD915 average OD600, 0.72 ± 0.01; AKD916 average OD600, 0.61 ± 0.01). For assays for reactive oxygen species, 1 μl of aminophenyl fluorescein (APF) (Invitrogen) was added to each sample with incubation in the dark at 28°C for 20 min. Samples were pelleted and washed twice with filter-sterilized artificial seawater (35 ppt; Instant Ocean, Blacksburg, VA). Two hundred microliters was loaded into a 96-well white microtiter plate (Corning, Corning, NY), and fluorescence was measured in a Bio-Tek Synergy H1 microplate multimode reader with excitation at 485 nm and emission at 515 nm, with gain set at 100. Data are reported as fluorescence units. The LIVE/DEAD BacLight bacterial viability kit (Invitrogen) was used for assays for membrane integrity. Components A and B were mixed in a 1:1 ratio, and 1.5 μl of the mixture was added to 0.5 ml of cell culture. Samples were incubated at room temperature for 20 min, and 200 μl was loaded per well of a 96-well white microtiter plate (Corning). Fluorescence was measured using a Bio-Tek Synergy H1 microplate multimode reader with excitation at 485 nm and emission at 530 and 630 nm, with gain set at 100. Data are reported as a ratio of green fluorescence (intact membrane) divided by red fluorescence (disrupted membrane). Each experiment was repeated three times, with results from one representative experiment shown. Significance was measured using Student's t test.
NAD+/NADH assays.
One-milliliter samples were collected during the flask-based growth curve assays in mineral salts medium at 6 h after addition of DPTA-NONOate to analyze NAD+ and NADH levels (AKD915 average OD600, 0.80 ± 0.01; AKD916 average OD600, 0.69 ± 0.02). Samples were processed and assays performed as previously described using an enzyme cycling-based colorimetric assay (30). Absorbance data were collected using a Bio-Tek Synergy H1 Microplate Multi-Mode reader. Each experiment included three independent replicates for each strain and was performed on three separate days. Data are presented as combined averaged results from the three experiments (with error bars indicating standard error), and significance was measured using Student's t test.
Accession number(s).
Raw Illumina reads have been deposited in the Sequence Read Archives (SRA) under SRA accession numbers SRS2800744 and SRS2800745.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the National Science foundation awarded to A.K.D. (grant MCB-1050687).
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00797-17.
REFERENCES
- 1.McDonald AE, Vanlerberghe GC. 2006. Origins, evolutionary history, and taxonomic distribution of alternative oxidase and plastoquinol terminal oxidase. Comp Biochem Physiol D Genomics Proteomics 1:357–364. doi: 10.1016/j.cbd.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Vanlerberghe GC, McIntosh L. 1997. Alternative oxidase: from gene to function. Annu Rev Plant Physiol Plant Mol Biol 48:703–734. doi: 10.1146/annurev.arplant.48.1.703. [DOI] [PubMed] [Google Scholar]
- 3.Meeuse BJD. 1975. Thermogenic respiration in aroids. Annu Rev Plant Physiol 26:117–126. doi: 10.1146/annurev.pp.26.060175.001001. [DOI] [Google Scholar]
- 4.Vanlerberghe G. 2013. Alternative oxidase: a mitochondrial respiratory pathway to maintain metabolic and signaling homeostasis during abiotic and biotic stress in plants. Int J Mol Sci 14:6805–6847. doi: 10.3390/ijms14046805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del-Saz NF, Ribas-Carbo M, McDonald AE, Lambers H, Fernie AR, Florez-Sarasa I. 2018. An in vivo perspective of the role(s) of the alternative oxidase pathway. Trends Plant Sci 23:206–219. doi: 10.1016/j.tplants.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 6.McDonald AE, Amirsadeghi S, Vanlerberghe GC. 2003. Prokaryotic orthologues of mitochondrial alternative oxidase and plastid terminal oxidase. Plant Mol Biol 53:865–876. doi: 10.1023/B:PLAN.0000023669.79465.d2. [DOI] [PubMed] [Google Scholar]
- 7.McDonald AE, Vanlerberghe GC. 2005. Alternative oxidase and plastoquinol terminal oxidase in marine prokaryotes of the Sargasso Sea. Gene 349:15–24. doi: 10.1016/j.gene.2004.12.049. [DOI] [PubMed] [Google Scholar]
- 8.Stenmark P, Nordlund P. 2003. A prokaryotic alternative oxidase present in the bacterium Novosphingobium aromaticivorans. FEBS Lett 552:189–192. doi: 10.1016/S0014-5793(03)00920-7. [DOI] [PubMed] [Google Scholar]
- 9.Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcon LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC. 2007. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris RM, Rappe MS, Connon SA, Vergin KL, Siebold WA, Carlson CA, Giovannoni SJ. 2002. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420:806–810. doi: 10.1038/nature01240. [DOI] [PubMed] [Google Scholar]
- 11.May B, Young L, Moore AL. 2017. Structural insights into the alternative oxidases: are all oxidases made equal? Biochem Soc Trans 45:731–740. doi: 10.1042/BST20160178. [DOI] [PubMed] [Google Scholar]
- 12.Dunn AK, Karr EA, Wang Y, Batton AR, Ruby EG, Stabb EV. 2010. The alternative oxidase (AOX) gene in Vibrio fischeri is controlled by NsrR and upregulated in response to nitric oxide. Mol Microbiol 77:44–55. doi: 10.1111/j.1365-2958.2010.07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boettcher KJ, Ruby EG. 1990. Depressed light emission by symbiotic Vibrio fischeri of the sepiolid squid Euprymna scolopes. J Bacteriol 172:3701–3706. doi: 10.1128/jb.172.7.3701-3706.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visick KL, Ruby EG. 2006. Vibrio fischeri and its host: it takes two to tango. Curr Opin Microbiol 9:632–638. doi: 10.1016/j.mib.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Davidson SK, Koropatnick TA, Kossmehl R, Sycuro L, McFall-Ngai MJ. 2004. NO means ‘yes’ in the squid-vibrio symbiosis: nitric oxide (NO) during the initial stages of a beneficial association. Cell Microbiol 6:1139–1151. doi: 10.1111/j.1462-5822.2004.00429.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Dunn AK, Wilneff J, McFall-Ngai MJ, Spiro S, Ruby EG. 2010. Vibrio fischeri flavohaemoglobin protects against nitric oxide during initiation of the squid-Vibrio symbiosis. Mol Microbiol 78:903–915. doi: 10.1111/j.1365-2958.2010.07376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuny C, Lesbats M, Dukan S. 2007. Induction of a global stress response during the first step of Escherichia coli plate growth. Appl Environ Microbiol 73:885–889. doi: 10.1128/AEM.01874-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woodmansee AN, Imlay JA. 2002. Reduced flavins promote oxidative DNA damage in non-respiring Escherichia coli by delivering electrons to intracellular free iron. J Biol Chem 277:34055–34066. doi: 10.1074/jbc.M203977200. [DOI] [PubMed] [Google Scholar]
- 20.Ruby EG, Urbanowski M, Campbell J, Dunn A, Faini M, Gunsalus R, Lostroh P, Lupp C, McCann J, Millikan D, Schaefer A, Stabb E, Stevens A, Visick K, Whistler C, Greenberg EP. 2005. Complete genome sequence of Vibrio fischeri: a symbiotic bacterium with pathogenic congeners. Proc Natl Acad Sci U S A 102:3004–3009. doi: 10.1073/pnas.0409900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engl C, Jovanovic G, Lloyd LJ, Murray H, Spitaler M, Ying L, Errington J, Buck M. 2009. In vivo localizations of membrane stress controllers PspA and PspG in Escherichia coli. Mol Microbiol 73:382–396. doi: 10.1111/j.1365-2958.2009.06776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi PS, Naal Z, Moore C, Casado-Rivera E, Abruna HD, Helmann JD, Shapleigh JP. 2006. Assessing the impact of denitrifier-produced nitric oxide on other bacteria. Appl Environ Microbiol 72:2200–2205. doi: 10.1128/AEM.72.3.2200-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millenaar FF, Lambers H. 2003. The alternative oxidase: in vivo regulation and function. Plant Biol 5:2–15. doi: 10.1055/s-2003-37974. [DOI] [Google Scholar]
- 24.Vanlerberghe GC, Cvetkovska M, Wang J. 2009. Is the maintenance of homeostatic mitochondrial signaling during stress a physiological role for alternative oxidase? Physiol Plant 137:392–406. doi: 10.1111/j.1399-3054.2009.01254.x. [DOI] [PubMed] [Google Scholar]
- 25.Miller JH. 1992. A short course in bacterial genetics, p 456. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.Stabb EV, Reich KA, Ruby EG. 2001. Vibrio fischeri genes hvnA and hvnB encode secreted NAD(+)-glycohydrolases. J Bacteriol 183:309–317. doi: 10.1128/JB.183.1.309-317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stabb EV, Ruby EG. 2002. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol 358:413–426. doi: 10.1016/S0076-6879(02)58106-4. [DOI] [PubMed] [Google Scholar]
- 28.Bose JL, Rosenberg CS, Stabb EV. 2008. Effects of luxCDABEG induction in Vibrio fischeri: enhancement of symbiotic colonization and conditional attenuation of growth in culture. Arch Microbiol 190:169–183. doi: 10.1007/s00203-008-0387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaudy AF Jr, Abu-Niaaj F, Gaudy ET. 1963. Statistical study of the spot-plate technique for viable-cell counts. Appl Microbiol 11:305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kern SE, Price-Whelan A, Newman D. 2014. Extraction and measurement of NAD(P)+ and NAD(P)H. Methods Mol Biol 1149:311–323. doi: 10.1007/978-1-4939-0473-0_26. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 32.Dunn AK, Martin MO, Stabb EV. 2005. Characterization of pES213, a small mobilizable plasmid from Vibrio fischeri. Plasmid 54:114–134. doi: 10.1016/j.plasmid.2005.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.