

Abstract
Growing evidence demonstrates that the highly conserved serine/threonine kinase CK2 promotes Th17 cell differentiation while suppressing the generation of Foxp3+ Tregs; however, the exact mechanism by which CK2 regulates the Th17/Treg axis remains unclear. CK2 can be comprised of 3 distinct subunits: 2 catalytic subunits, CK2α and CK2α′, and the regulatory subunit, CK2β. We generated mice that lack the major catalytic subunit of CK2, CK2α, specifically in mature T cells utilizing the distal Lck-Cre (CK2α−/−). Importantly, CK2α deficiency resulted in a significant decrease in the overall kinase activity of CK2. Further, CK2α deficiency resulted in a significant defect in Th17 cell polarization and a reciprocal increase in Tregs both in vitro and in vivo in the context of autoimmune neuroinflammation. The transcription factor FoxO1 directly inhibits Th17 cell differentiation and is essential for the generation of Tregs. CK2α−/− CD4+ T cells exhibit less phosphorylated FoxO1 and a corresponding increase in the transcription of FoxO1-regulated genes. Treatment of CK2α−/− CD4+ T cells with the FoxO1 inhibitor AS1842856 or shRNA knockdown of Foxo1 is sufficient to rescue Th17 cell polarization. Through utilization of a genetic approach to target CK2 kinase activity, the current study provides evidence of a major mechanism by which CK2 regulates the Th17/Treg axis through the inhibition of FoxO1.
INTRODUCTION
Protein kinase CK2 is a highly conserved and constitutively active serine/threonine kinase that promotes survival and proliferation in many tumor cells (1, 2). The CK2 holoenzyme is comprised of two catalytic subunits, CK2α and/or CK2α′, associated with two regulatory subunits, CK2β, each encoded by separate genes. The regulatory subunit is not essential for the catalytic activity of CK2α/α′, but confers specificity and therefore can direct the phosphorylation of certain substrates in a cell and environment specific manner (1). With over 500 identified target proteins, CK2 is capable of promoting the activation of numerous signaling pathways, ultimately leading to increased cell survival, proliferation and inflammation (2–4). One key signaling network sensitive to CK2 activity is PI3K/Akt/mTOR (5). CK2 phosphorylation of PTEN results in the inhibition of phosphatase activity and relief of PTEN-mediated inhibition of PI3K (6). In addition, direct phosphorylation of S129 Akt by CK2 leads to enhanced kinase activity of Akt compared to that of S473 phosphorylation alone (7).
The PI3K/Akt/mTOR pathway is essential for the function of CD4+ T cells. Strong signals through the pathway downstream of activation drive the differentiation of effector CD4+ T cells, while inhibition promotes the generation of Foxp3+ regulatory T cells (Tregs) (8–10). One mechanism by which Akt regulates T cell differentiation is through the negative regulation of the transcription factor forkhead box protein O1 (FoxO1) (11). When phosphorylated by Akt, FoxO1 is sequestered in the cytosol, and therefore unable to promote the transcription of FoxO1-regulated genes (12). In the context of CD4+ T cell differentiation, FoxO proteins promote the expression of Foxp3 and additional genes essential for the generation and function of Tregs (13, 14). In addition, FoxO1 can directly bind and inhibit the activity of the T helper 17 (Th17)-promoting transcription factor RORγt (15).
Utilizing the pharmacologic inhibitor, CX-4945, which targets both CK2α and CK2α′, we previously demonstrated a critical role for CK2 in promoting Th17 cell differentiation at the expense of Tregs (16); however, the molecular basis by which CK2 regulates the Th17/Treg axis remains unclear. Here we demonstrate that deletion of CK2α in CD4+ T cells is sufficient to significantly decrease overall kinase activity and target the Th17/Treg axis both in vitro and during experimental autoimmune encephalomyelitis (EAE), a murine model of Multiple Sclerosis (MS). In addition we demonstrate that regulation of the Th17/Treg axis by CK2 is dependent on the activity of FoxO1. Inhibition of FoxO1 in CK2α−/− CD4+ T cells resulted in a near complete rescue of Th17 cell differentiation, accompanied by significant inhibition of Foxp3 expression. Our findings provide mechanistic evidence that CK2 promotes Th17 cell differentiation and suppresses Tregs through the negative regulation of the transcription factor FoxO1.
MATERIALS AND METHODS
Mice
Csnk2a1fl/fl mice were obtained from Dr. Heike Rebholz (City College of New York, New York, NY) (17). Distal Lck-Cre (dLckCre) mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were crossed to create offspring that were homozygous for the floxed Csnk2a1 gene and heterozygous for dLckCre, allowing for the generation of CK2αfl/fl and CK2α−/− littermates. CK2αfl/fl, CK2α−/− and Rag1−/− mice were maintained in the animal facility at UAB. Male and female mice between 8 and 12 weeks old were used for all experiments. All experiments using animals were reviewed and approved by the Institutional Animal Care and Use Committee of UAB.
Inhibitors
The FoxO1 inhibitor AS1842856 was purchased from Calbiochem and reconstituted in DMSO for in vitro studies.
Cytokines, Antibodies and Dyes
Murine cytokines used for in vitro polarizations were purchased from Biolegend. Anti-mouse CD3, anti-mouse CD28 and neutralizing antibodies to murine IL-4 and IFN-γ were purchased from BioXCell. Antibodies to CK2α, phosphorylated S129 Akt, total Akt, phosphorylated T24 FoxO1 and total FoxO1 were purchased from Cell Signaling. Antibody to mouse β-actin was purchased from Abcam. Flow cytometry antibodies against mouse CD3, CD4, CD8, CD25, IL-17A and IFN-γ were purchased from Biolegend. Flow cytometry antibodies against mouse CD44, CD62L, CD69, Foxp3 and GM-CSF were purchased from eBioscience. Flow cytometry antibodies for phosphorylated STAT3, STAT5, S6 and Akt were purchased from Cell Signaling. Aqua Live/Dead Viability dye and CFSE proliferation dye were purchased from ThermoFisher Scientific.
Naïve CD4+ T cell Enrichment, Activation and Polarization
Naive CD4+ T cells were enriched from the spleens of 8-12 week old mice using the Naïve CD4+ T cell Kit purchased from Stem Cell Technologies. Cells were cultured at a density of 0.75 – 1 × 106 cells/ml in RPMI supplemented with 10% FBS, L-glutamine, Hepes buffer, sodium pyruvate, β-mercaptoethanol and penicillin/streptomycin (R10). Cells were activated with plate-bound anti-CD3 (10 μg/ml) and soluble anti-CD28 (1 μg/ml) for activation and polarization experiments. Th1 polarizing conditions were IL-12 (10 ng/ml) and anti-IL-4 (10 μg/ml); Th2 polarizing conditions were IL-4 (10 ng/ml) and anti-IFN-γ (10 μg/ml); Th17 conditions were IL-6 (20 ng/ml), TGFβ1 (2.5 ng/ml), IL-23 (10 ng/ml), anti-IL-4 (10 μg/ml), and anti-IFN-γ (10 μg/ml); and Treg polarizing conditions were TGFβ1 (5 ng/ml), IL-2 (5 ng/ml), anti-IL-4 (10 μg/ml), and anti-IFN-γ (10 μg/ml). Cells were polarized for 72 h unless otherwise noted.
Flow Cytometry
For surface protein detection, cells were incubated with Fc Block (2.4G2) for 15 min, washed and incubated with viability dye and anti-surface protein antibodies. For intracellular CK2 subunit and phosphorylated protein detection, cells were permeabilized with 70% and 90% MeOH, respectively. For cytokine detection, cells were stimulated with PMA (25 ng/ml) and Ionomycin (1 μg/ml) in the presence of GolgiStop (BD Biosciences) for 4 h and permeabilized using the Foxp3 Staining Buffer Kit (eBioscience). For in vitro proliferation assays, naïve CD4+ T cells were incubated in CFSE dye, washed and activated with plate-bound anti-CD3 and soluble CD28 antibodies for 72 h. Stained cells were run on an LSRII flow cytometer (BD Biosciences), and data was analyzed using FlowJo Software (TreeStar). Representative flow plots are gated on live, CD4+ cells, unless otherwise noted.
Immunoblotting
CD4+ T cells were lysed in buffer containing 1% Triton X-100 (Sigma-Aldrich), and protein lysate was separated by electrophoresis, transferred to a nitrocellulose membrane and blotted with immunoblotting antibodies, as previously described (18). Quantification of immunoblots were performed using ImageJ.
CK2 Kinase Activity
The Casein Kinase 2 Assay Kit (Millipore) was used to assess CK2 kinase activity., as previously described (19). Normalized cell numbers were lysed and both catalytic subunits, CK2α and CK2α′, were immunoprecipitated. Resulting lysates were incubated with CK2-specific peptide sequence and P32-labeled ATP. The reaction was quenched, and the release of P32 measured with a scintillation counter.
Genomic DNA Isolation and PCR
CD3− and CD3+ cells were sorted from the spleens of CK2α fl/fl and CK2α−/− mice using a FACS Aria cell sorter. Genomic DNA was extracted from sorted cells using the Wizard SV Genomic Purification System (Promega). PCR was performed using a Csnk2a1 primer (Forward: GAGTCATCTGTCATCTGTAGC; Reverse: GACCACATCCTAACTATCC), and the resulting product was separated on an agarose gel.
RNA Isolation and qRT-PCR
Total RNA was isolated from cells using Trizol extraction. For quantitative RT-PCR analysis, 500 to 1000 ng of RNA was used to reverse transcribe into cDNA. cDNA was subjected to qRT-PCR using TaqMan primers purchased from ThermoFisher Scientific. The data were analyzed using the comparative cycle threshold method to obtain relative quantitation values.
RNA Interference
Naïve CD4+ T cells from CK2αfl/fl and CK2α−/− mice were activated under Th0 conditions [anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), anti-IFN-γ (10 μg/ml) and anti-IL-4 (10 μg/ml)]. At days 1 and 2 post activation, cells were infected with retrovirus containing shRNA targeting CD8 (control) or shRNA targeting FoxO1 and containing an Ametrine reporter. CD4+ T cells were then removed from CD3 and CD28 stimulation and expanded for another 2 days in the presence of IL-2 (2 ng/ml). Ametrine+ CD4+ T cells were sorted at day 4 for assessment of knockdown efficiency and Th17 cell polarization.
EAE Induction, Assessment and Cell Isolation
Active EAE was induced in CK2αfl/fl and CK2α−/− mice using MOG35-55 emulsified in complete Freund’s adjuvant purchased from Hooke Laboratories, as previously described (18). For Th17 adoptive transfer EAE, donor mice were immunized with MOG35-55 emulsified in CFA and on day 10-14, lymph node cells were extracted and cultured in Th17 polarized media containing MOG35-55 (50 μg/ml), IL-6 (10 ng/ml), IL-23 (10 ng/ml), IL-1β (10 ng/ml) and neutralizing antibodies to IFN-γ (10 μg/ml) and IL-4 (10 μg/ml). After 4 days in culture, CD4+ cells were isolated using dynabeads, and 2-5 × 106 cells transferred using intraperitoneal injection into Rag1−/− recipients. All EAE mice were monitored daily and scored on a scale of 0 to 5: 0, no disease; 1, tail paralysis; 2, hind limb paresis; 3, complete hind limb paralysis; 4, front limb weakness and hind limb paralysis; and 5, moribund state. Mononuclear cells were isolated from the spinal cords of immunized mice using a Percoll gradient, as previously described (18, 20), and assessed by flow cytometry.
Statistics
Levels of significance for comparison between two groups were determined by one-sided two-sample Mann–Whitney rank sum test, in the case of EAE scores, and the Student t test distribution. A p value of <0.05 was considered statistically significant. All error bars represent SEM.
RESULTS
Generation of mice with T cell-specific CK2α deficiency
Global deletion of CK2α is embryonic lethal (21). In order to target CK2 activity utilizing a genetic approach, we chose to delete the major catalytic subunit of CK2, CK2α, specifically in mature T cells. Csnk2a1fl/fl mice (CK2αfl/fl), in which exon 2, containing the ATP binding site, can be excised by homologous recombination (17) were crossed with mice expressing Cre recombinase controlled by the distal Lck promoter (22). The resulting Csnk2a1fl/fl dLckCre (CK2α−/−) mice experienced T cell specific deletion of Csnk2a1 (Figure 1A). CK2α−/− mice appeared phenotypically normal out to 12 weeks of age, with no signs of systemic or organ-specific inflammation. CK2α−/− mice had comparable numbers of cells and distributions of CD4+ and CD8+ T cells in the spleen and lymph nodes compared to CK2αfl/fl littermates (Figures 1B and 1C). In addition, CK2α−/− mice had a comparable distribution of peripheral naïve and effector T cells, as detected by CD44 and CD62L expression (Figure 1D), and Tregs, as assessed by CD25 and Foxp3 expression (Figure 1E).
Figure 1. Characterization of CK2α−/− Mice.
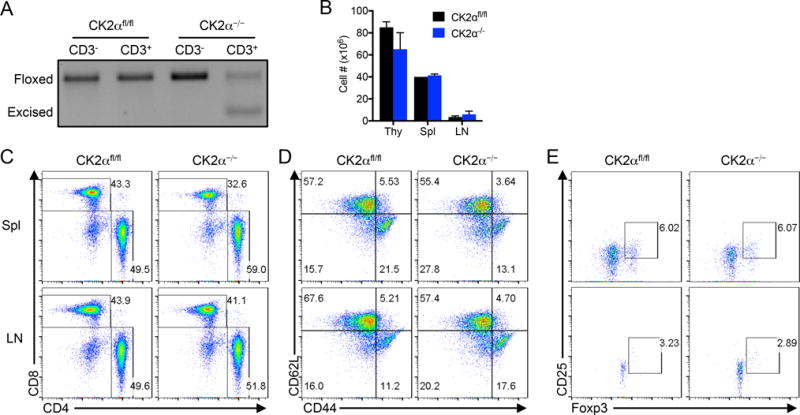
(A) CD3− and CD3+ cells were sorted from the spleens of CK2αfl/fl and CK2α−/− mice, genomic DNA isolated, and Csnk2a1 detected by PCR. (B) Cells from the thymus, spleen and lymph nodes of 12 week old CK2αfl/fl and CK2α−/− mice were counted; n=2. (C) Cells from the spleen and lymph nodes of 12-week old CK2αfl/fl and CK2α−/− mice were stained with anti-CD4 and anti-CD8 antibodies for detection by flow cytometry. (D) Cells from the spleen and lymph nodes of 12-week old CK2αfl/fl and CK2α−/− mice were stained with anti-CD62L and anti-CD44 antibodies for detection by flow cytometry. Plots are gated on CD4+ cells. (E) Cells from the spleens and lymph nodes of 12-week old CK2αfl/fl and CK2α−/− mice were stained with anti-CD25 and anti-Foxp3 antibodies for detection by flow cytometry. Plots are gated on CD4+ cells.
CK2α deficiency in CD4+ T cells results in decreased overall CK2 kinase activity in response to activation
Naïve CD4+ T cells from CK2α−/− mice lacked Csnk2a1 mRNA and expressed comparable mRNA levels of the other CK2 subunits, Csnk2a2 (CK2α′) and Csnk2b (CK2β), as cells from CK2αfl/fl littermates (Figure 2A). We previously demonstrated that expression of the CK2 subunits is induced upon CD4+ T cell activation with anti-CD3 and anti-CD28 antibodies (16). Interestingly, deletion of CK2α resulted in a significant reduction in Csnk2a2 and Csnk2b mRNA expression upon activation, demonstrating a dependence of the expression of these subunits on the activity of CK2α. This finding provides evidence that CK2 may regulate its own expression in T cells, as demonstrated previously in a cancer cell line (23). Further, CK2α protein was undetectable in CK2α−/− CD4+ T cells. CK2α deletion resulted in a significant decrease in overall CK2 kinase activity, as demonstrated by decreased phosphorylation of Akt1 at the CK2-specific site S129 (Figure 2B), and a kinase activity assay utilizing radio-labeled ATP (Figure 2C). Importantly, these findings validate our genetic approach to target CK2.
Figure 2. CK2α Deficiency in CD4+ T Cells Results in Decreased CK2 Kinase Activity and Akt/mTOR Signaling Upon Activation.
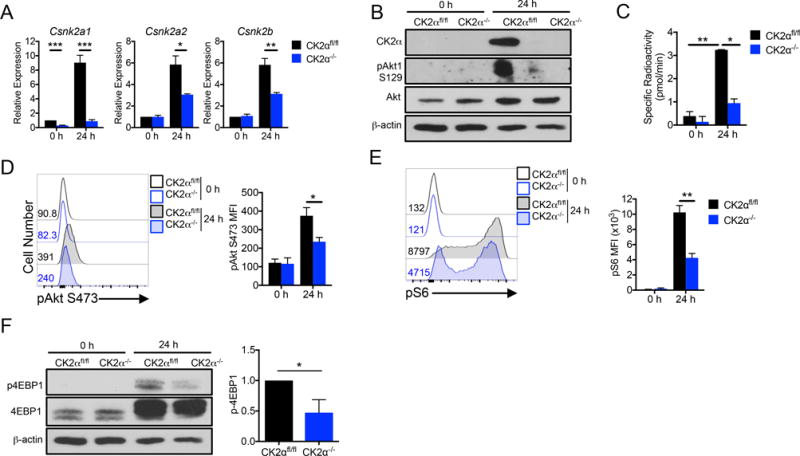
Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were activated with plate-bound anti-CD3 (10 μg/ml) and soluble anti-CD28 (1 μg/ml) antibodies for 24 h. (A) RNA was extracted and qRT-PCR performed using primers for Csnk2a1 (CK2α), Csnk2a2 (CK2α′) and Csnk2b (CK2β); n=4. (B) Cells were lysed and immunoblotted for CK2α, phosphorylated Akt S129, total Akt and β-actin. (C) Cells were assayed for CK2 kinase activity; n=2 technical replicates. Representative experiment of 3 individual experiments is shown. (D, E) Phosphorylated Akt S473 and S6 ribosomal protein S235/236 were detected by flow cytometry; pAkt, n=3; pS6, n=4. (F) Cells were lysed and immunoblotted for phosphorylated 4EBP1 T37/46, total 4EBP1 and β-actin. *p < 0.05, **p < 0.01.
CK2α deficiency in CD4+ T cells results in decreased Akt/mTOR signaling upon activation
The Akt/mTOR signaling pathway is activated in response to CD4+ T cell activation and plays a key role in regulating CD4+ T cell function (8–10). In addition, we have previously shown this pathway to be sensitive to CK2 inhibition in CD4+ T cells (16). CK2α−/− CD4+ T cells exhibited a significant decrease in phosphorylation of Akt at S473, essential for Akt activity (Figure 2D). In addition, CK2α−/− cells exhibited decreases in the phosphorylation of the downstream targets of mTOR complex 1 such as ribosomal protein S6 (Figure 2E) and 4EBP1 (Figure 2F) compared to CK2αfl/fl cells upon activation. These findings demonstrate a specific role of CK2α in promoting Akt/mTOR signaling in CD4+ T cells upon activation.
CK2α is not required for CD4+ T cell survival, activation or proliferation upon activation
A major function of CK2 activity is the promotion of cell survival (1). Therefore, before moving forward with functional experiments, it was important to determine the effect of CK2α deficiency on CD4+ T cell viability in vitro. CK2αfl/fl and CK2α−/− CD4+ T cells did not experience significantly altered cell survival in vitro, as measured by viability dye uptake 24 and 72 h post-activation (Figure 3A).
Figure 3. CK2α Expression is Not Required for CD4+ T Cell Survival, Activation or Proliferation.

Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were activated with plate-bound anti-CD3 anti-CD28 antibodies. (A) At 24 and 72 h, cells were incubated with viability dye, and the frequency of live cells was detected by flow cytometry. (B, C) At 24 h, CD25 and CD69 expression were detected by flow cytometry; n=7. (D) At 24 h, IL-2 in the supernatants of activated CK2αfl/fl and CK2α−/− cells was measured by ELISA; n=5. (E) At 24 h, Phosphorylated STAT5 Y694 was detected by flow cytometry. MFI of activated cells was normalized to corresponding naïve controls; n=5 (F) Cells were incubated in CFSE dye, and activated with anti-CD3 and anti-CD28 antibodies for 72 h. % Proliferation represents the frequency of cells undergoing at least 2 divisions as measured by CFSE dilution; n=3. *p < 0.05.
We next sought to determine the potential contribution of CK2α to CD4+ T cell activation, and first examined the effect of CK2α deficiency on the expression of activation markers. CK2α−/− CD4+ T cells activated with anti-CD3 and anti-CD28 antibodies exhibited no significant difference in CD25 or CD69 expression compared to CK2αfl/fl cells after 24 h (Figures 3B and 3C). In addition, CK2α−/− cells exhibited no defect in IL-2 production or signaling through STAT5 (Figures 3D and 3E). To determine the potential effect of CK2α deficiency on proliferation, CK2αfl/fl and CK2α−/− cells were incubated with CFSE dye prior to activation. CK2α−/− cells exhibited a small but significant decrease in proliferation, as determined by the frequency of cells undergoing more than 2 divisions in 72 h (Figure 3F). Although small differences were detected, together these data demonstrate that CK2α is largely dispensable for CD4+ T cell survival, activation and proliferation.
CK2α deletion impairs Th17 cell differentiation and promotes the generation of Foxp3+ Tregs in vitro
We next examined if CK2α played a critical role in regulating CD4+ T cell differentiation. CK2α−/− cells had no significant defect in polarization to the IFN-γ-producing Th1 or IL-4-producing Th2 phenotypes (Figures 4A and 4B). Alternatively, CK2α−/− cells had a significantly enhanced capacity to differentiate into Foxp3+ Tregs (Figure 4C). Most strikingly, CK2α−/− CD4+ T cells had a significantly impaired capacity to differentiate into the Th17 phenotype and exhibited a higher frequency of Foxp3+ cells than CK2αfl/fl cells in Th17 conditions (Figure 4D). Inhibition of IL-17A production was associated with suppressed basal and IL-6-induced STAT3 phosphorylation (Figure 4E) and significant inhibition of the expression of a panel of Th17 effector genes, as detected by qRT-PCR (Figure 4F). The robust increase in Foxp3 also occurred at the level of Foxp3 gene expression (Figure 4G). These data demonstrate a critical role of CK2α in regulating early signaling events that ultimately determine the fate of the Th17 and Treg transcriptional programs.
Figure 4. CK2α Deficiency Selectively Suppresses Th17 Cell Differentiation and Promotes the Generation of Foxp3+ Tregs.
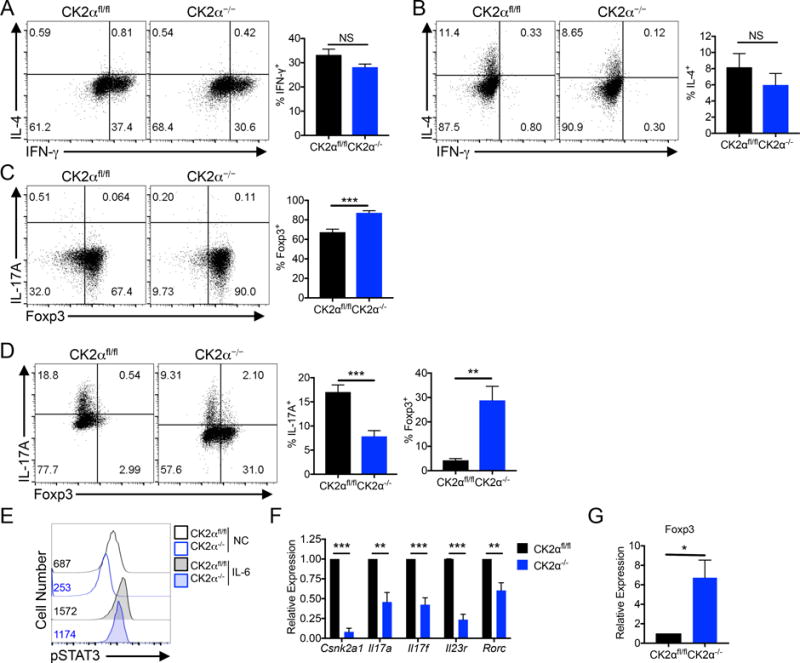
(A) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Th1 phenotype for 72 h, and IFN-γ production measured by flow cytometry; n=3. (B) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Th2 phenotype for 72 h, and IL-4 production measured by flow cytometry; n=3. (C) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Treg phenotype for 72 h, and Foxp3 expression measured by flow cytometry; n=6. (D) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Th17 phenotype for 72 h. IL-17A production and Foxp3 expression was measured by flow cytometry; n=6. (E) CK2αfl/fl and CK2α−/− CD4+ T cells were activated for 24 h in the absence or presence of IL-6 (10 ng/ml), and phosphorylated STAT3 Y705 detected by flow cytometry. Representative experiment of 3 individual experiments is shown; numbers represent MFI. (F, G) RNA was extracted from Th17 cultures and expression of Th17-associated genes (F) and Foxp3 (G) detected by qRT-PCR. *p < 0.05, **p < 0.01, ***p < 0.001.
CK2α deletion results in impaired Th17 cell and enhanced Treg differentiation during EAE
To determine if CK2α is critical for regulating the differentiation of Th17 and Treg cells in vivo, we utilized a murine model of MS, EAE. CK2α−/− mice exhibited significantly lower clinical scores during the peak and resolution phases of the disease course compared to CK2αfl/fl littermates (Figure 5A). The total number of infiltrating mononuclear cells into the spinal cord at the peak of disease did not differ between groups (Figure 5B). However, protection observed at the peak of disease in CK2α−/− mice was associated with a decrease in CD4+ T numbers and a significant decrease in frequencies of CD4+ T cells present in the spinal cord (Figure 5B), potentially reflecting a defect in either migration or proliferation. Further, CK2α−/− mice had significantly decreased frequencies of IL-17A+ cells present in the inflamed spinal cord (Figures 5C and 5D). Pathogenic Th17 cells that co-produce IFN-γ and GM-CSF were also significantly reduced compared to CK2αfl/fl littermates (Figures 5C and 5E). Consistent with our in vitro findings, the defect was specific to Th17 cells, as the CK2α deficiency had no significant affect on frequencies of IL-17A−IFN-γ+ and IL-17A−GM-CSF+ T cells (Figure 5E). In addition, protection observed during disease resolution in CK2α−/− mice was associated with significantly higher frequencies of CD25+Foxp3+ Tregs in the spinal cord than CK2α−/− littermates. (Figures 5F and 5G). Importantly, these data demonstrate a critical role for CK2α in regulating Th17 and Treg differentiation in vivo.
Figure 5. CK2α Deletion Results in Impaired Th17 Cell and Enhanced Treg Differentiation During EAE.
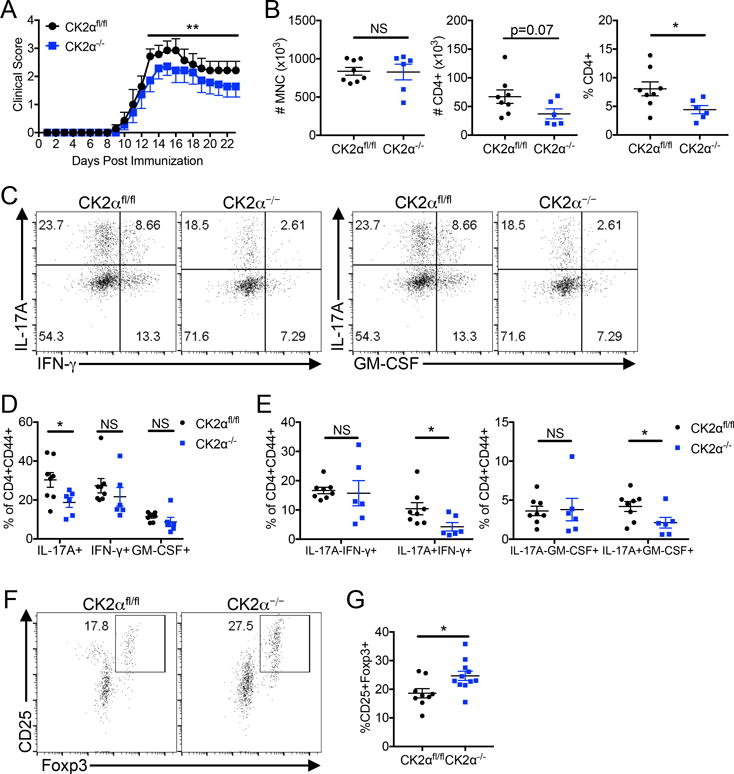
(A) CK2αfl/fl and CK2α−/− were immunized with MOG35-55 in CFA, and scored daily for signs of clinical disease; n=7 mice/group. Data is combined from 3 separate experiments. (B) At disease peak, between days 14 and 16 post immunization (P.I.), mononuclear cells (MNC) were enriched from the spinal cords, and CD4 was expression detected by flow cytometry. Total numbers of enriched MNCs, and numbers and frequencies of CD4+ cells are shown. (C) Cytokine production from CD4+ T cells in the spinal cords was detected by flow cytometry on MNCs. Cells are gated on live, CD4+ CD44+ cells. (D, E) Frequencies of total IL-17A+, IFN-γ+, and GM-CSF+ cells (D) and single and double cytokine-producing cells (E) are quantified; CK2αfl/fl, n=8 mice; CK2α−/−, n=6 mice. Data is combined from 3 separate experiments. (F) During disease resolution, between days 22 and 24 P.I., MNCs were enriched from the spinal cords and Tregs detected by expression of CD25 and Foxp3 by flow cytometry. (G) Frequencies of Tregs in the spinal cord during resolution are quantified; n=7 mice/group. Data is combined from 3 separate experiments. *p < 0.05, ***p < 0.001.
CK2α deletion impairs the pathogenicity of Th17 cells during adoptive transfer EAE
To determine the intrinsic ability of CK2α−/− CD4+ T cells to become pathogenic effector cells capable of mediating neuroinflammation, we utilized an adoptive transfer approach. MOG-reactive CD4+ T cells from the lymph nodes of previously immunized CK2αfl/fl and CK2α−/− mice were expanded in Th17 conditions and transferred into Rag1−/− recipients. Although the donor cells were similar in phenotype upon transfer (Figure 6A), CK2α−/− cells exhibited a significantly impaired ability to facilitate EAE (Figure 6B), demonstrating an essential role for CK2α in promoting the functional pathogenicity of Th17 cells. After transfer, recipients of CK2α−/− Th17 cells exhibited significantly fewer mononuclear cells and a significant decrease in the frequency of CD4+ T cells present in the spinal cord (Figure 6C). In addition, CK2α−/− cells transferred into recipient mice displayed lower frequencies of Th17 cells and enhanced Foxp3 expression during active disease (Figures 6D and 6E). These data further demonstrate that CK2α regulates the Th17/Treg axis. In addition, these data provide evidence that CK2α is essential for Th17 cell maintenance, recruitment to the spinal cord, and overall pathogenicity in the context of EAE.
Figure 6. CK2α promotes Th17 Cell Pathogenicity in Adoptive Transfer EAE.
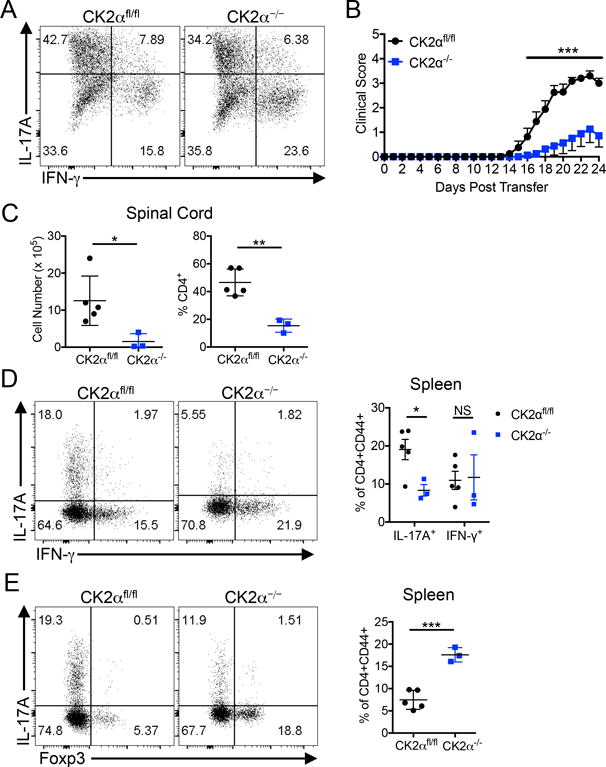
(A) CK2αfl/fl and CK2α−/− mice were immunized, and on days 10-14 P.I, lymph nodes cultured in Th17 conditions. Cytokine production was determined by flow cytometry. A representative plot from 3 separate experiments is shown. (B) Cells were adoptively transferred into Rag1−/− recipients, and recipient mice scored daily; n=8 mice/group. (C) At disease peak, mononuclear cells were enriched from the spinal cords, counted, and CD4 expression detected by flow cytometry. (D) At disease peak, effector T cells from the spleens were stained for IL-17A and IFN-g. Representative flow plots and combined frequencies of total IL-17A+ and IFN- g+ CD4+CD44+ T cells are shown. (E) Tregs in the spleens of recipient mice were identified by Foxp3. Representative flow plots and combined frequencies of Foxp3+ cells are shown; CK2afl/fl, n = 5; CK2a−/−, n = 3. * p<0.05, ** p<0.01, *** p<0.001.
CK2α promotes Akt-mediated phosphorylation of FoxO1, thereby inhibiting its transcriptional activity
One downstream target of Akt that has a major role in regulating the differentiation of Th17 cells and Tregs is the transcription factor FoxO1 (13, 14). When phosphorylated by Akt, FoxO1 remains sequestered in the cytosol where it is unable to promote expression of target genes such as Foxp3, Klf2 and Eomes. Consistent with the observed decreased in Akt phosphorylation (Figures 2B, 2D and 2E), CD4+ T cells that lack CK2α had substantially less phosphorylated FoxO1 after activation (Figure 7A). In addition, CK2α−/− cells polarized to the Th17 phenotype exhibited substantially reduced phosphorylation of FoxO1 than CK2αfl/fl cells (Figure 7B). Decreased FoxO1 phosphorylation in Th17 cultures resulted in an increased expression of the FoxO1-regulated genes Eomes and Klf2 (Figure 7C), and as shown previously, expression of Foxp3 (Figure 4G). These data demonstrate that CK2α promotes phosphorylation of FoxO1 in CD4+ T cells, therefore inhibiting its transcriptional activity.
Figure 7. Impaired Th17 Cell Differentiation and Enhanced Foxp3+ Expression in CK2α−/− Cells is Dependent on FoxO1.
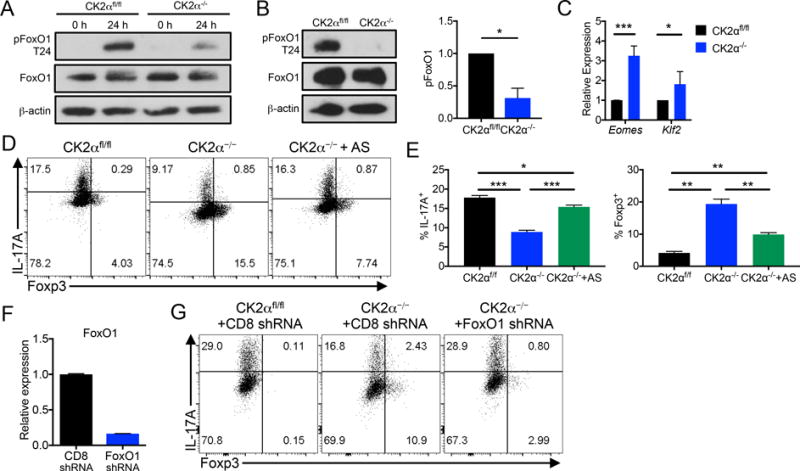
(A) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were activated with anti-CD3 and anti-CD28 antibodies for 24 h, lysed and immunoblotted for phosphorylated FoxO1 T24, total FoxO1 and β-actin. (B) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Th17 phenotype for 72 h, lysed and immunoblotted for phosphorylated FoxO1 T24, total FoxO1 and β-actin. Relative quantifications are normalized to total FoxO1; n=3. (C) RNA was extracted from Th17 cultures and expression of FoxO1-regulated genes was detected by qRT-PCR; n=3 (D, E) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were polarized to the Th17 phenotype for 72 h in the presence of DMSO or 25 nM AS1842856 (AS). IL-17A and Foxp3 expression were detected by flow cytometry; n=7. (F) Naïve CK2αfl/fl and CK2α−/− CD4+ T cells were activated and infected with retrovirus containing shRNA targeted to either CD8 or FoxO1 and expressing the reporter Ametrine. Ametrine+ cells were sorted and FoxO1 gene expression detected by qRT-PCR. (G) Ametrine+ cells were sorted and polarized to the Th17 phenotype for 72 h. IL-17A and Foxp3 expression were detected by flow cytometry. *p < 0.05, **p < 0.01, ***p < 0.001.
CK2α promotes Th17 cell differentiation via inhibition of FoxO1
In addition to being essential for the generation of Tregs, studies have shown that FoxO1 directly suppresses the generation of Th17 cells (15). We next sought to determine if the impaired Th17 cell differentiation and enhanced Treg differentiation in CK2α−/− cells was dependent on increased FoxO1 transcriptional activity utilizing the FoxO1 inhibitor AS1842856 (AS; 25 nM). FoxO1 inhibition in CK2α−/− cells resulted in the near complete rescue of IL-17A expression and significant inhibition of Foxp3 (Figures 7D and 7E). In addition to pharmacologic inhibtion, we targeted FoxO1 expression utilizing shRNA. CD4+ T cells from CK2αfl/fl and CK2α−/− mice were infected with retrovirus containing shRNA targeted to either CD8 or FoxO1, and expressing the reporter Ametrine. After infection, Ametrine+ cells infected with FoxO1 shRNA had markedly reduced FoxO1 gene expression compared to those infected with CD8 shRNA (Figure 7F). Importantly, knockdown of FoxO1 was sufficient to rescue Th17 cell differentiation in CK2α−/− cells (Figure 7G). Together, the data from these two approaches support that CK2α promotes Th17 cell differentiation and suppresses Tregs through the inhibition of the transcription factor FoxO1.
DISCUSSION
We previously demonstrated that targeting of CK2 with the pharmacologic inhibitor CX-4945, which inhibits both CK2α and CK2α′, inhibited Th17 cell differentiation and promoted the generation of Foxp3+ Tregs (16). Here we employ a genetic approach to target CK2 activity by specifically deleting CK2α, the major catalytic subunit of CK2, in mature T cells, which proved sufficient to significantly decrease overall kinase activity (Figure 2C). In addition to validating our previous findings with CX-4945, our current findings demonstrate a dominant role for CK2α in regulating the Th17/Treg axis and further, a lack of ability of CK2α′ to compensate in its absence. Interestingly, Ulges and colleagues described a similar effect on Th17 and Treg polarization with the deletion of the regulatory subunit CK2β (24), demonstrating that the catalytic activity of CK2 alone is not sufficient to drive Th17 cell differentiation, but regulation of substrate specificity conferred by CK2β is also essential. Although many studies have elucidated the role of CK2 in regulating numerous signaling pathways, the regulation of CK2 itself is not well understood (1). Together, our findings highlight a need to further understand the understudied mechanisms that regulate CK2 activity and specificity in the context of CD4+ T differentiation.
Here, we also demonstrate that regulation of the Th17/Treg axis by deletion of CK2α occurs in vivo in the context of EAE (Figure 5). In addition to a decrease in total IL-17+ CD4+ T cells infiltrating the spinal cord during the peak of disease symptoms, CK2α−/− mice had significantly decreased frequencies of Th17 cells that co-produce IFN-γ and GM-CSF (Figure 4E). These data support a role for CK2 in promoting the maturation of Th17 cells to the particularly pathogenic, double producing phenotype (25, 26). We also demonstrated regulation of Th17 cell maturation by CK2 utilizing pharmacologic inhibition of CK2 kinase activity (16). The more specific targeting of CK2α in Th17 cells will provide the tools necessary to define mechanisms by which CK2 regulates the pathogenic functions of lineage-committed Th17 cells, with further implications on disease outcome.
Although CK2α−/− mice experience significant protection at later stages of EAE, the protection seen with the T cell specific deletion was not as profound as protection previously demonstrated with global inhibition utilizing CX-4945 treatment (16). One explanation for this discrepancy is an overlooked role for CK2 activity in other cell types relevant to EAE. De Bourayne and colleagues recently described a role for CK2 activity in monocyte-derived dendritic cells to promote inflammatory cytokine production and their capacity to polarize Th1 and Th17 cells (27), demonstrating a potential role for CK2 in the inflammatory function of myeloid cells in EAE. In addition, Ulges and colleagues demonstrated a more complete protection from EAE when CK2β was deleted from CD4+ cells in the periphery utilizing a tamoxifen inducible Cre, which may suggest subtle differences in CK2 function between the late stages of thymic maturation and activation in the periphery (24). Lastly, when interpreting the EAE data, it is important to consider that the CK2α−/− mice also lack CK2α in CD8+ T cells, a subset relevant to EAE in which the role of CK2 has yet to be examined (28).
Ultimately, employing a genetic approach to target CK2 activity enabled us to perform mechanistic studies without the confounding potential off-target effects of utilizing a pharmacologic inhibitor. Inhibition of FoxO1 was able to nearly completely reverse the effects of CK2α deletion on the Th17/Treg axis, demonstrating a CK2/FoxO1 axis governing CD4+ T cell differentiation. We demonstrate that CK2α deletion results in decreased Akt phosphorylation at S473 (Figure 2E). Phosphorylated by mTOR complex 2, this site reflects an overall decrease in pathway activation in the absence of CK2α. In addition, targeting CK2α results in the absence of phosphorylation of the CK2-specific site S129 on Akt1. Although we primarily use phosphorylation at S129 as a read-out for CK2 kinase activity, this isoform-specific phosphorylation site is known to enhance Akt activity above S473 phosphorylation alone (5). Consequently, the Akt-mediated inhibitory phosphorylation of FoxO1, leading to decreased transcriptional activity, was decreased in CK2α−/− cells (Figures 7A and 7B). Although we propose this indirect regulation of Foxo1 by CK2 to be important for the regulation of CD4+ T cell differentiation, we cannot rule out the possibility of a direct interaction between CK2 and FoxO1, considering its immense pleiotropy (4). Further, important roles for FoxO1 outside of regulation of the Th17/Treg axis in CD4+ T cells, such as the regulation of the T-follicular helper subset and naïve cell homeostasis, have been well documented (29, 30). Utilizing additional in vivo models to test these T cell functions in CK2α−/− mice may reveal additional critical roles for CK2 in CD4+ T cell biology.
Acknowledgments
We thank Dr. Heike Rebholz for generously providing the Csnk2a1fl/fl mice and Drs. Chander Raman and Laurie Harrington at UAB for helpful discussions. We would also like to thank Drs. Hui Hu and Yinhu Wang for their aid in the shRNA knockdown experiments.
This work was funded by NIH T32AI007051 (SAG), R01NS057563 (ENB), R01CA194414 (ENB), and NMSS RG-1606-24794 (HQ). The Comprehensive Flow Cytometry Core at UAB is supported by NIH P30 AR048311 and P30 AI27667.
ABBREVIATIONS
- EAE
experimental autoimmune encephalomyelitis
- FoxO1
forkhead box protein O1
- MS
Multiple Sclerosis
- Th17
T helper 17
- Treg
regulatory T cell
Footnotes
CK2 Controls the Th17/Treg Axis via FoxO1
References
- 1.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nunez de Villavicencio-Diaz T, Rabalski AJ, Litchfield DW. Protein kinase CK2: intricate relationships within regulatory cellular networks. Pharmaceuticals (Basel) 2017;10:pii: E27. doi: 10.3390/ph10010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 4.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta. 2009;1793:847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Ruzzene M, Bertacchini J, Toker A, Marmiroli S. Cross-talk between the CK2 and AKT signaling pathways in cancer. Adv Biol Regul. 2017;64:1–8. doi: 10.1016/j.jbior.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- 7.Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F, Pinna LA, Ruzzene M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005;12:668–677. doi: 10.1038/sj.cdd.4401604. [DOI] [PubMed] [Google Scholar]
- 8.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, Shokat KM, Fisher AG, Merkenschlager M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawse WF, Sheehan RP, Miskov-Zivanov N, Menk AV, Kane LP, Faeder JR, Morel PA. Cutting Edge: Differential regulation of PTEN by TCR, Akt, and FoxO1 controls CD4+ T cell fate decisions. J Immunol. 2015;194:4615–4619. doi: 10.4049/jimmunol.1402554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12:649–661. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 13.Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 14.Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, Meijer D, Zhao K, Rudensky AY, Atwal G, Zhang MQ, Li MO. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laine A, Martin B, Luka M, Mir L, Auffray C, Lucas B, Bismuth G, Charvet C. Foxo1 is a T cell-intrinsic inhibitor of the RORgammat-Th17 program. J Immunol. 2015;195:1791–1803. doi: 10.4049/jimmunol.1500849. [DOI] [PubMed] [Google Scholar]
- 16.Gibson SA, Yang W, Yan Z, Liu Y, Rowse AL, Weinmann AS, Qin H, Benveniste EN. Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J Immunol. 2017;198:4244–4254. doi: 10.4049/jimmunol.1601912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rebholz H, Zhou M, Nairn AC, Greengard P, Flajolet M. Selective knockout of the casein kinase 2 in d1 medium spiny neurons controls dopaminergic function. Biol Psychiatry. 2013;74:113–121. doi: 10.1016/j.biopsych.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox THI, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Qin H, Stuart F, Deng L, Litchfield DW, Terfferi A, Pardanani A, Lin FT, Li J, Sha B, Benveniste EN. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood. 2011;118:156–166. doi: 10.1182/blood-2010-01-266320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Holdbrooks AT, De Sarno P, Rowse AL, Yanagisawa LL, McFarland BC, Harrington LE, Raman C, Sabbaj S, Benveniste EN, Qin H. Therapeutic efficacy of suppressing the JAK/STAT pathway in multiple models of experimental autoimmune encephalomyelitis. J Immunol. 2014;192:59–72. doi: 10.4049/jimmunol.1301513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28:131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, Killeen N. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- 23.Lupp S, Gumhold C, Ampofo E, Montenarh M, Rother K. CK2 kinase activity but not its binding to CK2 promoter regions is implicated in the regulation of CK2α and CK2β gene expressions. Mol Cell Biochem. 2013;384:71–82. doi: 10.1007/s11010-013-1782-8. [DOI] [PubMed] [Google Scholar]
- 24.Ulges A, Witsch EJ, Pramanik G, Klein M, Birkner K, Buhler U, Wasser B, Luessi F, Stergiou N, Dietzen S, Bruhl TJ, Bohn T, Bundgen G, Kunz H, Waisman A, Schild H, Schmitt E, Zipp F, Bopp T. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc Natl Acad Sci U S A. 2016;113:10145–50. doi: 10.1073/pnas.1523869113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, Duquette P, Prat A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66:390–402. doi: 10.1002/ana.21748. [DOI] [PubMed] [Google Scholar]
- 26.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Bourayne M, Gallais Y, Ali Z El, Rousseau P, Damiens MH, Cochet C, Filhol O, Chollet-Martin S, Pallardy M, Kerdine-Romer S. Protein kinase CK2 controls T-cell polarization through dendritic cell activation in response to contact sensitizers. J Leukoc Biol. 2016;101(3):703–715. doi: 10.1189/jlb.3A0715-320RR. [DOI] [PubMed] [Google Scholar]
- 28.Steinman L. Myelin-specific CD8 T cells in the pathogenesis of experimental allergic encephalitis and multiple sclerosis. J Exp Med. 2001;194:F27–F30. doi: 10.1084/jem.194.5.f27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, Lin YC, Yang E, Goldrath AW, Li MO, Cantrell DA, Hedrick SM. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity. 2015;42:239–251. doi: 10.1016/j.immuni.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]