

Abstract
There is limited data describing endothelial cell (EC) gene expression between aneurysms and arteries partly because of risks associated with surgical tissue collection. Endovascular biopsy (EB) is a lower risk alternative to conventional surgical methods, though no such efforts have been attempted for aneurysms. We sought (1) to establish the feasibility of EB to isolate viable ECs by fluorescence-activated cell sorting (FACS), (2) to characterize the differences in gene expression by anatomic location and rupture status using single-cell qPCR, and (3) to demonstrate the utility of unsupervised clustering algorithms to identify cell subpopulations. EB was performed in 10 patients (5 ruptured, 5 non-ruptured). FACS was used to isolate the ECs and single-cell qPCR was used to quantify the expression of 48 genes. Linear mixed models and exploratory multilevel component analysis (MCA) and self-organizing maps (SOMs) were performed to identify possible subpopulations of cells. ECs were collected from all aneurysms and there were no adverse events. A total of 437 ECs was collected, 94 (22%) of which were aneurysmal cells and 319 (73%) demonstrated EC-specific gene expression. Ruptured aneurysm cells, relative controls, yielded a median p value of 0.40 with five genes (10%) with p values < 0.05. The five genes (TIE1, ENG, VEGFA, MMP2, and VWF) demonstrated uniformly reduced expression relative the remaining ECs. MCA and SOM analyses identified a population of outlying cells characterized by cell marker gene expression profiles different from endothelial cells. After removal of these cells, no cell clustering based on genetic co-expressivity was found to differentiate aneurysm cells from control cells. Endovascular sampling is a reliable method for cell collection for brain aneurysm gene analysis and may serve as a technique to further vascular molecular research. There is utility in combining mixed and clustering methods, despite no specific subpopulation identified in this trial.
Keywords: Cerebral aneurysm, Vascular biology, Gene expression, Cerebrovascular procedures, Endothelium
Introduction
Cerebral aneurysms are common, affecting 1–6% of the population, and their rupture carries significant morbidity and mortality. Consequently, the clinical and aneurysm morphological features associated with rupture have been extensively studied [1, 2]. There are also numerous in vitro and animal studies characterizing the cellular and molecular events involved in aneurysm formation, growth, and rupture [3–12]. Despite these efforts, there remain only a few series describing the histopathological features of aneurysms as well as the differences in gene expressivity between aneurysmal and non-aneurysmal arteries and non-ruptured and ruptured aneurysms [11, 13–19]. This paucity of data stems in part from the risks associated with aneurysm tissue collection. Furthermore, these bulk genetic studies represent the collective expression of all cell types that compose the aneurysm wall limiting conclusions as to genetic changes within the respective cell layers affected in aneurysm formation and rupture.
The concept of endovascular tissue collection is known and has been performed using a range of different devices in many anatomic locations [20, 21]. The technique is less injurious to the vessels it targets for study relative to classical open surgical tissue collection and, given the expanse of endovascular techniques in most surgical disciplines, can be applied to a much wider array of patients and diseases. It also has the benefit of isolating the EC population more efficiently than ex vivo tissue digestion methods. This is important, as the ECs are the primary biomechanical interface translating chemical and fluid dynamic forces to the vessel wall as a whole. Despite its expanding use, endovascular cell sampling has not been used in the cerebral vasculature.
The endovascular treatment of cerebral aneurysms is widely employed and commonly performed using detachable coils. At times during embolization, a coil may not fit within the aneurysm and needs to be removed. That coil, normally discarded, may collect cells during its exposure to the aneurysm and those cells may be harvested from the device and studied, a concept established using pigs and the rabbit-elastase aneurysm model [22, 23]. The benefit of coil-based sampling is the specificity and safety the technique provide, though the cell yields are often small (< 20 cells) compared to stents. As such, we established methods using fluorescence-activated cell sorting (FACS) analysis to optimize cell collection as well as single-cell qPCR techniques to better confirm cell identity using the genetic expression patterns of cells in combination with their cell surface markers [24, 25].
Using the combination of endovascular coil-based cell sampling, FACS, and single-cell qPCR analysis, we studied a cohort of 10 patients undergoing cerebral aneurysm treatment (5 non-ruptured and 5 ruptured). The aims of this study were threefold: (1) to establish the feasibility of the coil-based technique to isolate viable endothelial cells; (2) to characterize gene expression profiles of ECs collected from ruptured and non-ruptured aneurysms and the iliac arteries; and (3) to demonstrate effective bioinformatics analysis methods, such as unsupervised, machine-learning analytical techniques of gene expression at the individual cell level, to help identify clusters in the data that might represent distinct subpopulations.
Methods
Patient Selection
Patients were screened from outpatient clinics and in-hospital census lists. If a patient was to undergo treatment, he or she and/or their family was questioned about their interest in participating in the study. In addition to the standard surgical consent, written informed consent (CHR No. 10-03924) was collected permitting collection of an additional coil during treatment to be used for the express purpose of cell collection. All patients were adults (> 17 years) and all aneurysms had not been previously treated.
As part of CHR approval, formal stopping rules were put in place. In the event of a single adverse event, defined as symptomatic periprocedural stroke (hemorrhagic or ischemic), the study was to stop for formal review by IRB-appointed members. The other risks of the study, including additional anesthesia, additional ionizing radiation, potential exposure of patient health information, and potential disease discovery through genetic analysis, were considered and discussed as part of informed consent. These secondary risks, however, were not felt to be as potentially harmful as that of the added stroke risk and not considered significant adverse events.
Endovascular Cell Sampling
To address the feasibility of endovascular coil-based EC sampling, we used three criteria: (1) safety, (2) cell yield and viability for purposes of gene expression analysis, and (3) veracity of the EC population as defined by a combination of surface markers and gene expression profile. Safety was defined as no unintended hemorrhagic or thromboembolic complications either during deployment or retrieval of the sampling coil. Cell yield was quantified using FACS (described subsequently) and cell viability for purposes of gene expression was defined by the success of quantitative PCR methods (described subsequently).
Cell sampling procedures were carried out as previously described [22]. Routine surgical practice was followed for all procedures. Patients were administered general endotracheal anesthesia and prepped and draped in the usual fashion. A transfemoral approach was performed for all procedures. Following control femoral angiography, a 0.035-in. guidewire (Benson or “J” curved wire) was advanced into the sheath permitting contact with the iliac arterial segment. The wire was removed and the distal 7-cm cut and immediately placed into a 50-ml Falcon tube containing dissociation buffer. At this time, a 5-ml arterial blood sample was also collected. This sample served as the internal control for the study subject.
All patients received an intravenous bolus of heparin with target activated clotting time (ACT) double of baseline or > 250 s. We recognized systemic heparinization might affect cell collection yields, but given our standard use of anticoagulation during aneurysm treatment, we opted to maintain the practice for thromboembolic risk reduction. Aneurysm treatment was performed using telescoping guide and microcatheters as each patient’s anatomy required. Once the microcatheter was positioned within the aneurysm fundus, a coil was selected for the purpose of aneurysm treatment, not to maximize cell yield (e.g., coil with a diameter and/or length oversized for the aneurysm). The primary goal of the procedure was treatment of the aneurysm and if the operator thought removal of the coil posed a significant risk to the patient, it was left in place and the procedure continued without cell collection. Only the first coil was used for cell collection in each case. We considered the approach whereby only those coils that were discarded for clinical reasons would be used for sampling. One major issue with this approach is that if the coil to be discarded was not the first coil in contact with the aneurysm, then cells were too often absent all together. During our preliminary investigations in animals and humans, we noted this effect and posit that the inability of the coil to have multiple direct points of contact with the aneurysm, as opposed to other coils, limits cell collection. Once the coil was fully deployed with in the aneurysm, it was left in place for ~ 30 s after which time it was pulled back into the microcatheter and then removed (coil only, not the microcatheter). The coil was then placed and cut into a separate 50-ml Falcon tube containing dissociation buffer. A new coil was selected and the procedure carried out as per routine practice.
EC Enrichment by FACS
Workflow of the post-processing steps is outlined in Fig. 1. EC enrichment methods were described in our previous report [22–24]. Briefly, after dislodging the attached cells on the endovascular devices with vortexing and centrifuging, RBCs were first removed by ACK Lysing Buffer (Gibco, Grand Island, NY). The nucleated cells were then stained with seven fluorescently conjugated monoclonal antibodies for further single EC sorting on FACS. Four EC markers, CD31, CD34, CD105, and CD146, were used for selecting ECs; three WBC markers, CD45, CD42b, and CD11b, for removing contaminated leucocytes, myeloid cells, and platelets. Each of the sorted EC candidates was sorted to individual wells of 96-well plate that were filled with reverse transcription-specific target amplification buffer containing 5 μl Cells Direct 2X Reaction Mix (Life Technologies, Carlsbad, CA), 0.2 μl SuperScript III RT Platinum Taq Mix (Life Technologies, Carlsbad, CA), 2.8 μl nuclease-free water, and 1 μl 10× primer mixture (500 nM).
Fig. 1.
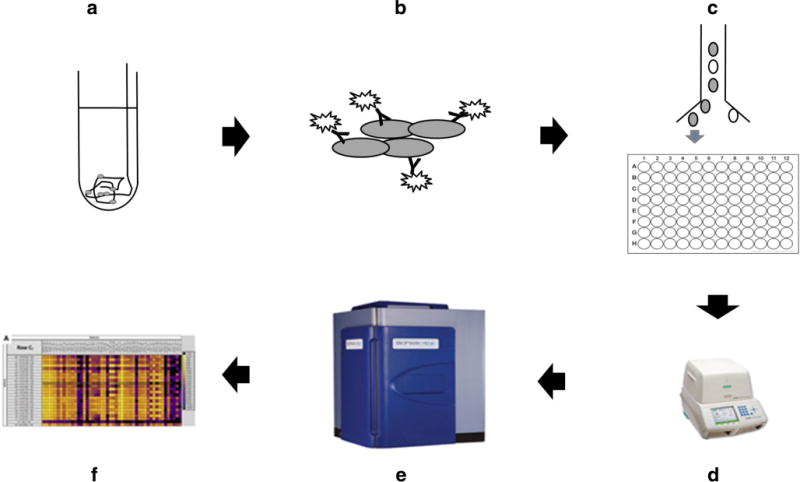
Flow chart for experiments. Cells collected by guide wire and aneurysm coil were dislodged (a) and stained by antibodies specific to different cell surface markers (b). Individual ECs were sorted into a 96-well plate by FACS (c). Reverse transcription and cDNA pre-amplification of the 48 target genes specific gene cDNA were then finished on a thermocycler (d). Microfluidic qPCRs were performed on Biomark HD system (Fluidigm, South San Francisco, CA) (e). qPCR results were collected and analyzed by qPCR analysis software (f; Fluidigm, South San Francisco, CA)
Single-Cell Reverse Transcription Quantitative PCR
Single-cell RT qPCRs were carried out following the protocol in our previous report [24]. Briefly, a PCR thermocycler was used to finish reverse transcription and cDNA pre-amplification, and BioMark system (Fluidigm, South San Francisco, CA) and 48.48 nanofluidic chips were used for microfluidic single-cell qPCR. The detected 48 genes were selected (Table 1) from prior works implicating their involvement in aneurysm pathophysiology that was summarized in our previous study [24]. We grouped these genes into functional groups: angiogenesis, inflammation, and extracellular matrix maintenance to assist in final analysis [26, 27]. Additional genes for cell identification were also selected to identify the triple-positive EC gene expression (CD31, CD34, and CD105) cohort (triple-positive cells) to best ensure the identity of the studied cells was both EC by surface antigens and genetic functionality [28, 29]. The reverse transcription was carried on at 50 °C for 15 min then 95 °C for 2 min to inactivate reverse transcriptase and activate Taq polymerase. Eighteen PCR cycles were then used (95 °C 15 s then 60 °C for 4 min for each cycle) for specific target amplification (STA) or cDNA pre-amplification. Exonuclease I (New England BioLabs, Ipswich, MA) incubation was then used to remove the unincorporated primers at 37 °C for 30 min. PCR was carried on through 35 cycles of 5 s at 96 °C and 20 s at 60 °C after a hot start phase of 60 s at 95 °C. The output qPCR data were processed by Fluidigm quantitative RT-PCR Analysis software (Fluidigm, South San Francisco, CA) to calculate Ct values for further statistical analysis. Normalization was performed by an assumed detection Ct level of 28.
Table 1.
48 genes selected for single-cell gene expression profile analysis
| Cell marker [14] | Angiogenesis [12] | Inflammation [10] | ECM remodeling [12] | ||||
|---|---|---|---|---|---|---|---|
| Symbol | Gene name | Symbol | Gene name | Symbol | Gene name | Symbol | Gene name |
| PTPRC | CD45 | VEGFA | VEGF-A | IL6 | – | MMP2 | MMP-2 |
| PECAM1a | CD31 | TGFB1 | TGF-β1 | IL8 | – | MMP9 | MMP-9 |
| CD34a | CD34 | PCNA | PCNA | VCAM1 | VCAM-1 | MMP14 | MMP-14 |
| ENGa | CD105 | CAT | – | ICAM1 | ICAM-1 | SERPINE1 | PAI-1 |
| MCAMa | CD146 | SGK1 | SGK | TBXAS1 | THA-2 | TNF | TNF-α |
| KDR | Flk1 | ANGPT1 | – | NOS3 | eNOS | ITGA7 | – |
| FLT1 | VEGFR1 | ANGPT2 | – | CCL2 | MCP-1 | TIMP1 | TIMP-1 |
| TIE1 | Tie1 | HIF1A | HIF-1α | SELP | – | TIMP2 | TIMP-2 |
| THBD | – | NR4A1 | TR3 | PTGS1 | COX-1 | FN1 | – |
| VWF | vWF | ALOX5 | 5-LO | PTGS2 | COX-2 | TNC | – |
| TEK | Tie2 | CD44 | – | – | – | SCEL | – |
| ACTG2 | α-actin | ACE | – | – | – | PPL | – |
| EPHB2 | EphB2 | – | – | – | – | – | – |
| EPHB4 | EphB4 | – | – | – | – | – | – |
Genes used for EC sorting on FACS
Statistical Analysis
Patient characteristics, aneurysm size and location, coil specifications, and cell yields were summarized. We tested whether aneurysm cell yields were associated with rupture status with a Wilcoxon rank-sum test. Using mixed-effects logistic regression, we tested whether there was an association between triple-positive status and cell location, i.e., iliac or aneurysmal; we included patient-specific random intercepts in the models to account for clustering.
Linear Mixed Models
To assess gene expression differences between anatomic sites (iliac artery vs. aneurysm) and disease status (ruptured vs. non-ruptured), we ran a series of linear mixed models each with a single-gene expression level as the outcome and multiple predictors that included (1) a history of smoking, as smoking can confound the relationship between rupture status and gene expression levels, (2) patient rupture status, (3) cell location (iliac vs. aneurysm), and (4) the interaction of rupture and location. This last analysis was the primary one because our main goal was to assess the effect of rupture on gene expression in aneurysm cells over and above any effect rupture has on other types of cells. Since individual cells were the observational units, we included patient-specific random intercepts in the models to account for clustering. We used this framework for each of the 48 pre-selected genes. For our results, we report the coefficient for predictor (1) which measures the effect of smoking, the coefficient for predictor (2) which quantifies the difference between ruptured and unruptured aneurysmal cells, the linear contrast (3) comparing aneurysm cells to iliac cells irrespective of patient rupture status, and the coefficient for predictor (4) which measures whether the difference between aneurysm and iliac cells is more or less pronounced in ruptured patients than in unruptured patients. These analyses were performed using cell data from the triple-positive cohort to best ensure a true EC identity [28, 29]. Results were oriented such that unruptured status, iliac location, and no history of smoking were the reference groups. The gene expression levels were analyzed as Log2Ex units, which was defined as the limit of detection (set to 28) minus the gene expression CT value (negative Log2Ex values were set to 0). Thus, each unit increase is interpreted as a twofold increase in gene expression; results were presented as fold change values. These data analyses were performed using Stata 13.1 (StataCorp. 2013. Stata Statistical Software: Release 13. College Station, TX: StataCorp LP).
Unsupervised Clustering
Because the data represent the functionality of individual cells, we expected a wide diversity of gene co-expression profiles among all cells studied. More specifically, we expected cells sampled from aneurysm sites to have both pathological and normal expressivity; likewise for cells sampled from the iliac site. Given there is ample evidence demonstrating the significant genetic expressivity variation among cells of a singular phenotype [30–37], we performed an unsupervised, machine-learning analysis to help identify potential cell subpopulations based on gene expressivity. Namely, we employed multilevel component analysis, self-organizing maps and hierarchical agglomerative clustering using data restricted to the within-subject deviation after split-variation decomposition.
Split-Variation Decomposition Pre-Processing
Due to the multilevel nature of the qPCR data (multiple cells sampled from within patients) the correlations between cells sampled within patients must be taken into account during unsupervised clustering analysis. Similar to mixed effects modeling where variation is partitioned into random effects (between-patient variation) and residual (within-patient variation), gene expression data must be decomposed as a pre-processing step. This approach has been used in multilevel sparse partial least-squares discriminant analysis (sPLS-DA) of genetics data [38, 39]. With this technique, the gene expression for cell j for patient s for gene k can be defined as:
where, is the overall gene k expression across all patients (offset), is the average gene k expression for patient s minus the overall gene k expression across all patients (between-subject deviation) and is the actual gene k expression for cell j of patient s minus the average gene k expression across all cells of patient s. By separating the sources of variation and removing the between-subject variability, the genetic expression within patients can be focused on, specifically the differences in genetic expression for cells sampled from the aneurysm vs. iliac sites. Therefore, the within-subject variation χw data, with between-subject variability removed, was used in unsupervised clustering algorithms.
Hierarchical Agglomerative Clustering (HAC)
To visually determine the effects of intra-patient gene expressivity correlation, we used HAC on the raw triple-positive data (not χw). HAC of the 48 gene expressions from triple-positive cells (n = 319) was conducted by scaling the data (cell-wise) and using the hierarchical cluster function “hclust” from base package stats in R [40]. The Pearson method was used to develop the covariance matrix for cells and Spearman’s method for genes. Genes and cells with similar co-expression relationships were grouped using the complete linkage method to create a topological heat map. Each cell was arranged on the map according to the strength of covariance of cells and genes. Cells were colorized according to their z-score, or expression value in reference to the mean gene expression for all genes for each cell. A color bar was created which represents the patient wherein each cell was sampled. Blocks of similar color indicate collections of cells grouped together for the same patient. The same analysis was conducted using χw data to inspect the differences in clustering with and without compensating for patient-specific correlations.
Multilevel Component Analysis (MCA)
MCAwas used to visualize clustering of the aneurysm vs. iliac cells. MCA consists of the following: (i) isometric log ratio transformation of the genetics data, (ii) split-variation decomposition to produce the χw within-subject variation matrix, and (iii) principal component χw analysis on the data. The MCA process was conducted in R using the “mixOmics” package [41]. PCA was used to visualize how cells cluster in the high-dimensional data space. In PCA, each cell is represented as a point within the 48-dimensional data space; in this case, each dimension is a χw gene expression value. New axes are defined, known as principal components; the number of which is equal to the number of variables [48]. The first component is a vector that explains the most variance in the data; subsequent components are orthogonal vectors to the preceding component and explains the highest remaining variance. Principal component 1 (PC1) is a linear combination of the 48 genes that explains the most variance between cells. Likewise, principal component 2 (PC2) is the orthogonal rotation from PC1 and explains the remaining variance, and so on. Principal component 1 and 2 projections of the triple-positive cells were colored by cell type. Loadings for each gene on PC1 were plotted as a bar chart. Scree plotting the variance explained by each component was used to determine the optimum number of components.
Self-Organizing Maps (SOMs)
SOM networks of the χw gene data were also used to identify clusters of cells based on gene co-expression profiles. Briefly, SOM provides a means to cluster cells on the basis of similar gene expression. The technique uses pre-set mathematical relationships to simultaneously explore heterogeneous data and minimize bias of classical supervised tools. Unsupervised methods have emerged as single-cell genomic study has expanded in use [42]. It is customary for studies investigating single cells from a common phenotypic source to find heterogeneous lineages as it relates to the molecular profile [30, 32, 35–37, 42–45]. In the oncological field, such results speak to subpopulations that may be more and less susceptible to various therapies, while others have noted subpopulations demonstrating greater pluripotency than others. Beyond disease-specific states, others have pointed out some of the heterogeneity may be related to cell cycle and others the spatial arrangement of the cells themselves [31, 33]. Regardless of the cause, it is expected that gene expression variation would exist even among a cell group taken from a discrete anatomic site.
Each cell in the SOM is stored as an n-dimensional vector (where n is the number of variables characterizing the cell), which represents a point in an n-dimensional data space. A pre-defined number of nodes are set into the data space. This unsupervised system uses a competitive learning strategy wherein, during training, the Euclidian distance for each node to all weight vectors is calculated as nodes move incrementally closer to cells they are similar to based on n features. After a pre-determined minimum change in mean distance for all nodes has been reached, permutations of the training end and the node that is most similar to collections of vectors, dubbed the best matching unit (BMU), represent a cluster in the SOM network. Therefore, nodes in the SOM represent a collection of statistically similar vectors. In the current analysis, initial map weights for each node were based on loadings on the first principal component. From initialization, in each epoch of the training process, nodes move towards cells in the n-dimensional data space that they are most similar to (minimizing the Euclidian distance) and eventually “lock-in.” Patterns in the data, at this point, can be represented by a pre-defined number of nodes with genetically similar cells linked to them. In the current analysis, the SOM creates a discretized low-dimensional representation of the triple-positive cells as a smaller number of nodes. The gene weights on each node represent the weighted gene expression for cells linked to the respective node. Neighboring nodes in the network are most similar in terms of genetic expressivity profiles. To visually inspect the pattern of gene expressivity weights across the network, each node was plotted as a circle with fan plots in the circle measuring the specific gene weight on that node based on the genetically similar cells linked to the node. SOM was chosen for its ability to cluster cells on the bases of similar gene expression data and for its ability to be plotted for visual inspection. For more information on the mathematical features of SOMs, see [46–48]. Each stage of the SOM analysis applied to the triple-positive data is illustrated. The features of this analysis are described in detail in “Supplementary Materials,” “Methods” section.
K-means clustering of nodes in the SOM network was used to identify groups in the data. Node background colors (gray levels) were used to show nodes in each cluster after k-means clustering. The number of clusters identified using SOM was determined using the elbow rule of the within-sum of squares plot.
Results
Feasibility: Patient Characteristics and EC Enrichment Efficiency
Patient-level characteristics are summarized in Table 2. Four of the 10 patients were male. The average patient age was 62 years old (± 13 years). Six patients were hypertensive and 4 patients were smokers. The average aneurysm size was 6.5 mm (± 2 mm) in the greatest dimension. Viable endothelial cells were collected from all aneurysms and the iliac arteries. A total of 437 FACS-sorted cells was collected. Of these, 94 (22%) were from aneurysms. On average, 9.4 (the median was 6.5) cells were collected from the aneurysm of each patient; the minimum value was 1 and the maximum was 24. There was a trend, albeit not a statistically significant one, for higher cell yields from ruptured aneurysms (13.8 ± 8.6 vs. 5.0 ± 1.9, p = 0.116). Three hundred nineteen of 437 cells (73%) were classified as CD31, CD34, and CD105 triple-positive. Iliac cells were more likely to be triple-positive than aneurysmal cells, but not significantly so (76 vs. 62%, p = 0.114); the proportion of cells classified as triple-positive varied from patient to patient (p < 0.001).
Table 2.
Patient-level characteristics
| Patient characteristics | Aneurysm characteristics | Coil parameters | FACS-sorted | Triple-positive | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| ID | Age | Sex | HTN | Smoker | Ruptured | Size (mm) | Location | Manufacturer | Diameter (mm) | Length (mm) | Soft | Aneurysm | Iliac | Aneurysm | Iliac |
| 1 | 46 | Male | No | No | Yes | 7.5 | Acorn | Penumbra | 6 | 15 | Yes | 19 | 26 | 4 | 5 |
| 2 | 84 | Female | Yes | No | No | 7.2 | ICA | Codrnan | 6 | 20 | Yes | 2 | 27 | 1 | 18 |
| 3 | 72 | Male | Yes | Yes | No | 5.2 | Acorn | Target | 5 | 15 | No | 5 | 22 | 3 | 19 |
| 4 | 70 | Male | No | Yes | No | 5.8 | ICA | Target (XL) | 6 | 20 | Yes | 7 | 41 | 6 | 32 |
| 5 | 59 | Female | Yes | No | Yes | 6.5 | PeriCal | Target (XL) | 5 | 15 | Yes | 1 | 47 | 0 | 35 |
| 6 | 69 | Female | Yes | No | No | 7.2 | Acorn | Target | 5 | 15 | No | 5 | 43 | 5 | 42 |
| 7 | 45 | Female | No | Yes | No | 4.8 | SCA | Target | 4 | 10 | No | 6 | 42 | 6 | 40 |
| 8 | 60 | Male | Yes | Yes | Yes | 5.0 | Basiliar | Target | 4 | 6 | Yes | 24 | 24 | 21 | 20 |
| 9 | 49 | Female | No | No | Yes | 4.4 | Pcom | Target | 3 | 6 | Yes | 12 | 36 | 11 | 29 |
| 10 | 63 | Female | Yes | No | Yes | 11.0 | ICA | Target | 6 | 20 | Yes | 13 | 35 | 1 | 21 |
| 62 ± 13 | 40% M | 60% Y | 40% Y | 50% Y | 6.5 ± 1.9 | 5.0 ± 1.1 | 14.2 ± 5.3 | 70% Y | 9.4 ±7.5 | 34.3 ± 9.0 | 5.8 ±6.2 | 26.1 ± 11.5 | |||
Summaries provided on the bottom row are mean ± sd or a percentage
Abbreviations: HTN hypertension, Acorn anterior communicating artery, ICA internal carotid artery, PeriCal pericallosal artery, SBA superior cerebellar artery, Pcom posterior communicating artery
Gene Expression Analysis
Quality of Single-Cell Analysis
Before microfluidic qPCR assay, quality control of single-cell samples by PCR of housekeeping gene GAPDH was carried out after cDNA pre-amplification step, and all the single-cell samples gave positive PCR results that justified the quality of cDNA samples.
Statistical Outcomes
Linear Mixed Models
Results from linear mixed model analyses are presented in Supplementary Table 1, with p value distributions presented in Fig. 2 as histograms. Using the triple-positive EC cohort, we found a history of smoking had the strongest association with overall gene expression level in both aneurysm and iliac cells. The median p value for the set of 48 gene expression analyses was 0.10 for the predictor measuring the effect of smoking (Fig. 2a), with 11 analyses (23%) yielding a p value of less than 0.05 (bolded p values in Supplementary Table 1). The median p value for the effect of rupture status on gene expression levels of aneurysm cells was 0.36, with five instances (10%) of p values less than 0.05 (Fig. 2b). The median p value for the linear contrast comparing aneurysmal and iliac cells was 0.56, with one instance (2%) of a p value less than 0.05 (Fig. 2c). The median p value of the interaction of location and rupture was 0.40, with five instances (10%) of p values less than 0.05 (Fig. 2d). The expressions of TIE1, ENG, VEGFA, MMP2, and VWF were reduced in the ECs collected from ruptured aneurysms were relative compared to ECs collected from unruptured aneurysms and the iliac arteries. Given the potential for more limited accessibility of the Fluidigm microfluidic system at other institutions, we performed the same statistical analyses using the FACS EC cohort to help inform other groups interested in using the technique. We do not report the FACS data, though the results were similar to the triple-positive cohort analysis and are presented in Supplementary Table 2.
Fig. 2.
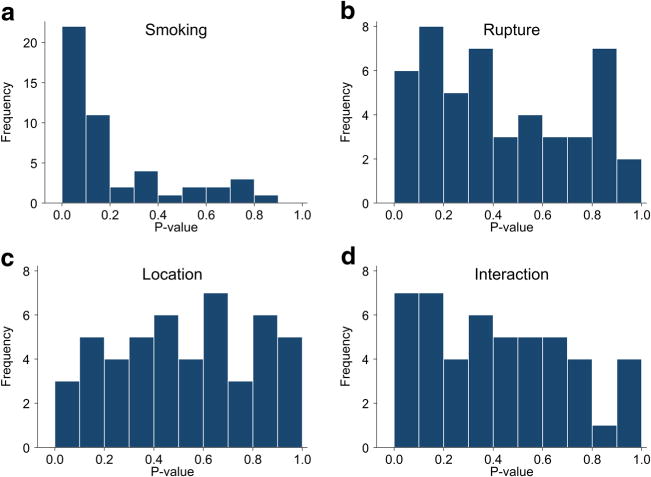
Histograms of p values from all multipredictor linear mixed models with gene expression as the outcome. “Smoking” is the comparison of patients with a history of smoking to those without one. “Rupture” is the comparison of ruptured and unruptured aneurysm cells. “Location” is the comparison of the aneurysmal to iliac cells, irrespective of rupture status. “Interaction” compares whether the difference between the aneurysmal and iliac cells is more or less pronounced in ruptured patients than in unruptured patients
HAC
Heat maps from HAC are presented in Fig. 3a (using χw data) and Fig. 3b (using raw triple-positive data). Visual inspection of the color bars for each plot shows that there is significant clustering of cells due to patient (color bar, Fig. 3a) compared to data limited to within-subject variation (color bar, Fig. 3b). This cell clustering due to intra-patient genetic correlations validates the need to account for the nested structure of the data when doing unsupervised clustering analysis.
Fig. 3.
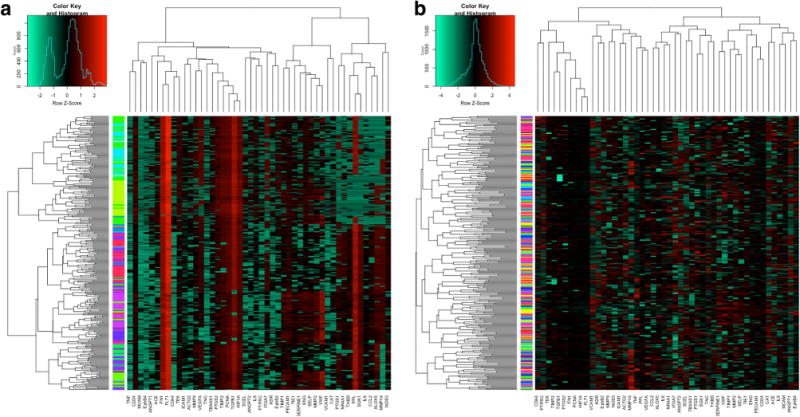
HAC heat maps. Hierarchical heat map plotted with genes clustered as column-wise dendrogram and cells clustered as row-wise dendrogram using triple-positive data (a). Color bars indicate the patient χw ID by which cells were sampled. The same analysis done using the data (b)
MCA
MCA applied to the triple-positive data resulted in PC1 explaining 6.4% of the variance and PC2 explaining 4.4%. Figure 4a shows the proportion of variance explained by each principal component (scree plot). Figure 4b shows the distribution of cells projected on PC1 and PC2, colorized by cell type (aneurysm cells blue; iliac cells orange). The bar chart (Fig. 4c) shows the loadings of each gene on PC1. As can be seen in Fig. 4b, there is a cluster of seven cells (five control, two aneurysm) from different patients that are separated by PC1. Genes which had the highest loading on PC1 were CD34, ENG, MCAM, and TIE1; all EC marker genes. The negative loading on PC1 for these genes suggests that there is an inverse relationship between the genetic expression and PC score; therefore, these cells have very low expressivity for these four genes. Because these seven cells separated on genes in the cell marker family, they were treated as outliers (non-endothelial cells) and removed from subsequent analysis. Figure 4d–e show the same PCA using data with these seven cells removed. It is apparent that, after removal of these outliers, no inherent clustering of cells is seen on the PC1 and PC2 axes for cell sampling location or otherwise. Subsequent SOM analysis was done using χw data with these outliers removed.
Fig. 4.
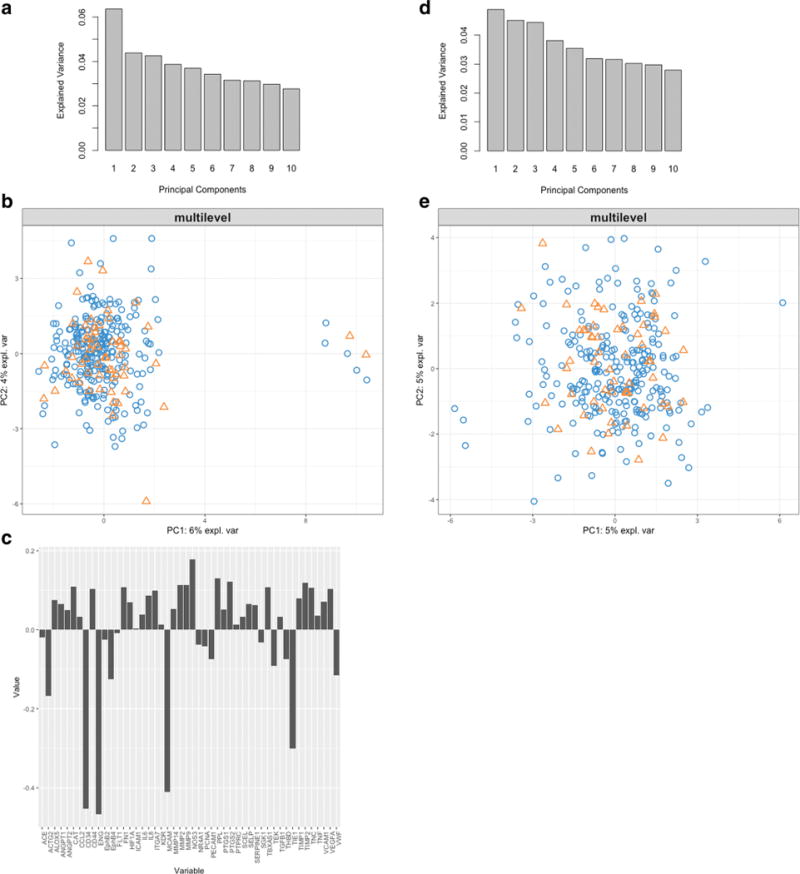
Variance explained by each PC component using the decomposed data (a). Cells projected onto the PC1 and PC2 axes, labels are the cell ids (labeled as coil-aneurysm vs. wire-iliac sampling. Patient ID. Cell number) (b). Labels are colorized by sampling location (coil-aneurysm vs. wire-iliac). Gene loadings on the PC1 component which separates the outlying seven cells (c). Variance explained by each PC component with outliers removed identified in b (d). Cells projected onto PC1 and PC2 axes with outliers removed (e)
SOM
The SOM demonstrated clustering of four groups based on aberrant gene family marker profiles of four cells. Figure 5a shows the distribution of gene family marker weights across the SOM network. Background colors in variegations of gray show the groups. Only two cells were linked to group 1 (dark gray), one cell to group 2 (light gray), and one cell to group 3 (white); the remaining cells (308) clustered into one group (group 4—in black). Similar to MCA, the application of SOM on the χw -MCA filtered data was able to identify additional outlying cells with a very different family marker gene expressivity. These cells were considered non-endothelial cells and were removed. A second SOM was conducted to determine if cell clustering could be seen without outliers. Figure 5b shows the distribution of gene weights throughout the network using χw data with MCA- and SOM-identified outliers removed. Figure 5c shows the within-sum of squares for each number of clusters using k-means clustering of the SOM nodes. From this plot, no specific number of clusters drastically reduces the within-sum of squares, and therefore, there did not appear to be any inherent clustering in the data. Clustering results were statistically validated using the “clvalid” package in R [49–52]. Internal and stability scores were reported in Fig. 6. Using internal validity based on connectivity, Dunn index, and silhouette scores, hierarchical clustering at two groups of cells scored the best. For stability validation, hierarchical clustering with two groups was identified for the average proportion of non-overlap and average distance between means indices. Difference in average distance using full data vs. iterative single-gene removal identified 10 groups using PAM in the data and similarly using, figure of merit (FOM) SOM was chosen using nine clusters. Although, based on the majority rule of these indices, hierarchical clustering indicates two clusters in the data; the overall internal and stability scores are inconsistent across clustering algorithms. Additionally, HAC heat map modeling of the data and visual inspection of z-scores do not indicate two clusters based on gene co-expression profiles. This is further corroborated by the fact that within-sum of squares of SOM nodes and MCA both showed no inherent clustering of the data.
Fig. 5.
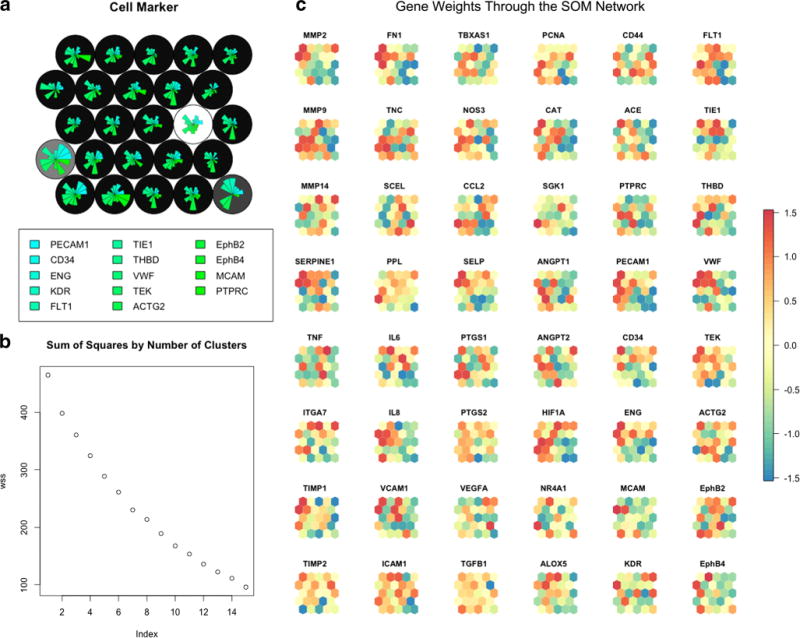
SOM network using 25 nodes (circles) colorized with background colors (grays) clustered based on k-means clustering of the nodes (a). Fan plots are gene weights for genes in the family marker family. SOM node network showing the distribution of each gene’s weights throughout the network (b). The color of each node (ranging from − 1.5 (red) to 1.5 (blue), 0 = yellow) indicates the gene weight on each node. Groups of clusters with identical colors, or a dichotomy of colors, indicate clustering of gene weights in the network. Genes showing homogenous colorization throughout the network have no impact on differentiating cell groups
Fig. 6.
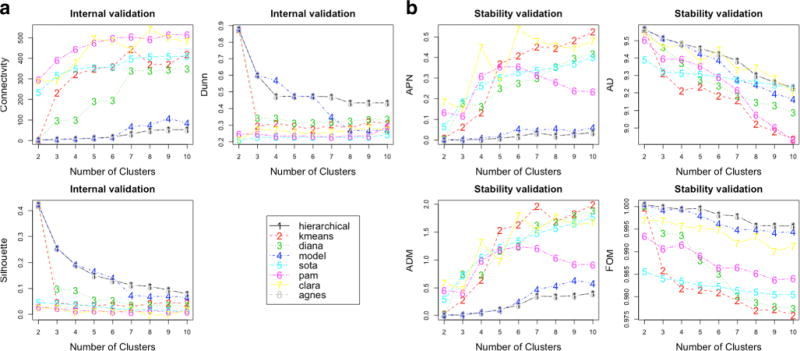
Internal validation scores for each clustering method for each number of clusters (a). Stability validation scores for each clustering method for each number of clusters (b)
Discussion
In this study, we accomplished three goals: (1) demonstrated the feasibility of the coil-based technique to isolate viable ECs from human aneurysms; (2) characterized the differences in gene expression between ECs collected from the iliac arteries and aneurysms and ECs from non-ruptured and ruptured aneurysms; and (3) demonstrated that an unsupervised, machine-learning analytical method can be used for analysis of single cell-gene expression while adjusting for the correlation of gene expression profile for cells sampled within the same patient. That we were unable to identify EC gene expression profiles definitive of aneurysms, ruptured or non-ruptured, is disappointing, though not surprising given the small sample size and limited number of genes analyzed. Each step of the overall method was, however, successful in its prescribed scope and collectively promise to be a powerful approach in the study of vascular disorders.
Aneurysms are now more commonly treated using endovascular methods than open, microsurgical techniques; a trend that parallels many other vascular surgical fields whereby less invasive treatments can translate into better patient outcomes. What is lost, however, in such widespread adoption of endovascular methods is the ability to gather tissue to better understand the treated diseases themselves. Relative to oncology, we have a much more limited appreciation for the molecular heterogeneity of vascular disorders, largely binning them into those that cause ischemia or hemorrhage. We use simple angioarchitectural details, e.g., degree of stenosis or diameter of an aneurysm, to homogenize these likely genetically variegated diseases into those amenable to treatment and those that are not. There is growing interest in more nuanced imaging-based metrics (e.g., vessel wall imaging or computational fluid dynamics), though we still lack a safe, consistent, and valued means to collect vascular tissue for genetic study.
Just as the study of vascular disorders might learn from oncology the value of systematically collecting tissue for genetic study, it then would also benefit from adopting the statistical methods used to analyze such single-cell data. We opted to use an ensemble combination of traditional linear mixed modeling and more exploratory unsupervised methods. Overall, the linear mixed modeling approach is well established and accounts for patient identity, which may affect gene expression. This approach was used to determine whether pre-specified factors were associated with gene expression levels, though no finding from the statistical models was significant even after a Bonferroni correction for multiple comparisons. Consequently, we focused on the distribution of p values, as detailed previously. As it relates to the second aim, our analysis revealed little evidence that the expressions of the suite of the 48 genes were driven by anatomic location or rupture status. That is not to say that all 48 genes selected were useless markers, but, overall, the set of 48 chosen genes performed poorly and good markers may exist that simply were not chosen. We did note greater differential gene expression through the interaction of the rupture and aneurysm location, as there was a slight preponderance of smaller p values. This hints that supervised binary groupings may lack the necessary nuance to meaningfully capture such differential gene expression in complex, multifactorial diseases such as aneurysms. Interestingly, smoking was strongly associated with differential gene expression levels, which is in agreement with previous findings [53–56].
As it relates to unsupervised clustering, the techniques (MCA and SOM) shed light on the utility of gene expressivity groups to identify small groups of non-endothelial cells based on gene family expressivity profiles. These 7 false-positive cells were missed during FACS and triple-positive filtering. After removing these outlying cells, we were unable to show clustering of iliac vs. aneurysmal cohorts although the SOM methods revealed some asymmetrical distributions along the network (Fig. 4b). Despite focusing on genes implicated in aneurysmal pathogenesis, 48 targets still remain a limited sampling of the genetic possibilities to understand the varied molecular events between endothelial cells in different locations and functional states. Nonetheless, within the respective cohorts, we noted a small number of genes that were significantly differentially expressed with respect to cell location and cell rupture status.
The unsupervised clustering approaches should be seen as a first step in exploring the gene expression landscape of endothelial cell data when cells are sampled from a number of patients, creating nested data. We, however, emphasize the limitations of the analysis given the number of patients, genes, and cells studied. Consequently, making definitive statements of endothelial cell expressivity as it relates to cell location and/or rupture status are ill considered. Instead, we wish to highlight the value of the entire approach, particularly unsupervised MCA and SOM methods, to reveal data trends otherwise occult using more traditional, supervised methods. The range of gene expression profiles provides evidence of cell heterogeneity related to a spectrum of vascular endothelial cell function. It is reasonable to assume the entire endothelial cell population of any vascular segment need not have a uniform gene expression pattern to manifest disease, whether it be an aneurysm or an atherosclerotic plaque, but instead that there is a specific fraction of cells, as function of surface area and/or environmental forces, that must be dysfunctional for macroscopic pathology to arise. More importantly is that the identification of such critical cell cohorts could reveal those disease states that would respond more favorably to lifestyle modifications and medical therapies rather than to invasive treatments.
As noted, there are multiple limitations of this analysis; the most significant being the small number of patients and cells studied. As it relates to safety, the absence of an adverse event within 10 patients is encouraging, but does guarantee similar results if widely used in general practice. Endovascular aneurysm cell collection is experimental and should only be carried out under full IRB approval and with informed consent. As it relates to statistical shortcomings, mixed models are relatively robust in the presence of low numbers of observations per cluster. The low number of clusters used in the model was the greater source of bias in the mixed models, specifically when estimating the within- and between-subject variations. However, in the current analysis, our goal was to estimate the genetic expressivity differences between sampling sites while controlling for the variance component. These point estimates are unlikely to be biased severely due to the low number of cells per cluster, but the precision around these estimates is likely worse, making it more difficult to detect significance at the 0.05 level. Subsequently, because between-subject variation was removed in the unsupervised clustering approach, the low cell yield makes it more difficult to detect clusters based on cell co-expressivity profiles. Certainly, a larger study including more patients and cells per patient will allow better estimates of the precision around the findings estimated here.
Despite such paucity of data, there is promise of a larger scale study. Given the number of aneurysms treated in an endovascular manner worldwide, the potential of collecting more cells is feasible. Furthermore, recent methods of single-cell RNA sequencing have been established and would enable full-transcriptome analysis rather than a select number of genes. Additional limitations include training bias (no additional data outside of this cohort to test model with), the supervised method relying on sampling site to label cell identify and thereby missing single-cell heterogeneity, and an inability to control for confounders such as the cell cycle.
Overall, aneurysms remain an incompletely understood and heterogeneous disorder. Despite some degree of an inheritable component, the majority of aneurysms are considered sporadic. As such, we have a very limited ability to predict who will form an aneurysm, where that aneurysm might arise within the cerebral vasculature, and most importantly, which aneurysms will rupture. There are a handful of gene microarray studies based on aneurysmal dome samples collected during surgical clipping [10, 11]. These studies are informative as they relate to directing research of certain genes or functional gene families, but limited in that they are a homogenization of multiple-cell types (endothelial, smooth muscle, monocytes, fibroblasts, etc.); each group of which may contribute to aneurysm formation, and potentially rupture, in a different way. Furthermore, the technique of tissue collection is limited by the safety of resecting aneurysmal tissue in surgery, something that is not routinely performed, and in that the majority of aneurysms are now treated in an endovascular means. The methods put forth permit a safe and reliable approach to collect tissue for genetic study with the bonus of its ability to isolate the endothelial cell population as well as its potential for more widespread use given the continued expansion of endovascular techniques for all vascular disorders, whether in the brain or not.
Conclusion
In summary, we have demonstrated the proof-of-principle of human in vivo aneurysm endovascular-based endothelial cell collection. Additionally, we have demonstrated methods to purify those viable cells for quantitative single-cell gene expression as well as both supervised and unsupervised statistical methodologies for their analysis. These results are valuable as the ability to safely and reliably collect endothelial cells in vivo for molecular characterization, particularly within the central nervous system, represents an opportunity to further our understanding of vascular pathophysiology. The combination of single-cell genomic analysis and the use of unsupervised data analysis methods, such as multilevel component analysis and self-organizing maps, offer powerful tools in identifying sub-populations of cells sharing common gene expression profiles involved in vascular diseases, aneurysms, or otherwise.
Supplementary Material
Acknowledgments
The authors would like to thank the UCSF Neurointerventional Radiology service, particularly its fellows, for their help in the sample collection.
Sources of Funding
The project was funded in part by the Society for Neurointerventional Surgery, the UCSF Department of Radiology, and by a grant from the National Cancer Institute R01 CA056721 to Z.W.
Abbreviations
- EB
Endovascular biopsy
- FACS
Fluorescence-activated cell sorting
- EC
Endothelial cell
- qPCR
Quantitative polymerase chain reaction
- MCA
Multilevel component analysis
- SOMs
Self-organizing maps
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s12975-017-0560-4) contains supplementary material, which is available to authorized users.
Compliance with Ethical Standards
Conflict of Interest The authors declare that they have no conflict of interest.
Ethical Approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
References
- 1.Backes D, Vergouwen MD, Tiel Groenestege AT, Bor AS, Velthuis BK, Greving JP, et al. PHASES score for prediction of intracranial aneurysm growth. Stroke. 2015;46(5):1221–6. doi: 10.1161/STROKEAHA.114.008198. [DOI] [PubMed] [Google Scholar]
- 2.Greving JP, Wermer MJ, Brown RD, Morita A, Juvela S, Yonekura M, et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol. 2014;13(1):59–66. doi: 10.1016/S1474-4422(13)70263-1. [DOI] [PubMed] [Google Scholar]
- 3.Frösen J. Smooth muscle cells and the formation, degeneration, and rupture of saccular intracranial aneurysm wall—a review of current pathophysiological knowledge. Transl Stroke Res. 2014;5(3):347–56. doi: 10.1007/s12975-014-0340-3. [DOI] [PubMed] [Google Scholar]
- 4.Marbacher S, Marjamaa J, Bradacova K, von Gunten M, Honkanen P, Abo-Ramadan U, et al. Loss of mural cells leads to wall degeneration, aneurysm growth, and eventual rupture in a rat aneurysm model. Stroke. 2014;45(1):248–54. doi: 10.1161/STROKEAHA.113.002745. [DOI] [PubMed] [Google Scholar]
- 5.Frösen J, Tulamo R, Heikura T, Sammalkorpi S, Niemelä M, Hernesniemi J, et al. Lipid accumulation, lipid oxidation, and low plasma levels of acquired antibodies against oxidized lipids associate with degeneration and rupture of the intracranial aneurysm wall. Acta Neuropathol Commun. 2013;1(1):71. doi: 10.1186/2051-5960-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, et al. Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathol. 2012;123(6):773–86. doi: 10.1007/s00401-011-0939-3. [DOI] [PubMed] [Google Scholar]
- 7.Frösen J, Marjamaa J, Myllärniemi M, Abo-Ramadan U, Tulamo R, Niemelä M, et al. Contribution of mural and bone marrow-derived neointimal cells to thrombus organization and wall remodeling in a microsurgical murine saccular aneurysm model. Neurosurgery. 2006;58(5):936–44. doi: 10.1227/01.NEU.0000210260.55124.A4. discussion–44. [DOI] [PubMed] [Google Scholar]
- 8.Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, et al. Growth factor receptor expression and remodeling of saccular cerebral artery aneurysm walls: implications for biological therapy preventing rupture. Neurosurgery. 2006;58(3):534–41. doi: 10.1227/01.NEU.0000197332.55054.C8. discussion–41. [DOI] [PubMed] [Google Scholar]
- 9.Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35(10):2287–93. doi: 10.1161/01.STR.0000140636.30204.da. [DOI] [PubMed] [Google Scholar]
- 10.Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T, et al. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke. 2014;45(8):2239–45. doi: 10.1161/STROKEAHA.114.005851. [DOI] [PubMed] [Google Scholar]
- 11.Marchese E, Vignati A, Albanese A, Nucci CG, Sabatino G, Tirpakova B, et al. Comparative evaluation of genome-wide gene expression profiles in ruptured and unruptured human intracranial aneurysms. J Biol Regul Homeost Agents. 2010;24(2):185–95. [PubMed] [Google Scholar]
- 12.Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Gene expression profile of the intima and media of experimentally induced cerebral aneurysms in rats by laser-microdissection and microarray techniques. Int J Mol Med. 2008;22(5):595–603. [PubMed] [Google Scholar]
- 13.Yong-Zhong G, van Alphen HA. Pathogenesis and histopathology of saccular aneurysms: review of the literature. Neurol Res. 1990;12(4):249–55. doi: 10.1080/01616412.1990.11739952. [DOI] [PubMed] [Google Scholar]
- 14.Hasan D, Hashimoto T, Kung D, Macdonald RL, Winn HR, Heistad D. Upregulation of cyclooxygenase-2 (COX-2) and microsomal prostaglandin E2 synthase-1 (mPGES-1) in wall of ruptured human cerebral aneurysms: preliminary results. Stroke. 2012 doi: 10.1161/STROKEAHA.112.655829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bygglin H, Laaksamo E, Myllärniemi M, Tulamo R, Hernesniemi J, Niemelä M, et al. Isolation, culture, and characterization of smooth muscle cells from human intracranial aneurysms. Acta Neurochir (Wien) 2011;153(2):311–8. doi: 10.1007/s00701-010-0836-x. [DOI] [PubMed] [Google Scholar]
- 16.Jia W, Wang R, Zhao J, Liu IY, Zhang D, Wang X, et al. E-selectin expression increased in human ruptured cerebral aneurysm tissues. Can J Neurol Sci. 2011;38(6):858–62. doi: 10.1017/s0317167100012439. [DOI] [PubMed] [Google Scholar]
- 17.Pera J, Korostynski M, Krzyszkowski T, Czopek J, Slowik A, Dziedzic T, et al. Gene expression profiles in human ruptured and unruptured intracranial aneurysms: what is the role of inflammation. Stroke. 2010;41(2):224–31. doi: 10.1161/STROKEAHA.109.562009. [DOI] [PubMed] [Google Scholar]
- 18.Jin D, Sheng J, Yang X, Gao B. Matrix metalloproteinases and tissue inhibitors of metalloproteinases expression in human cerebral ruptured and unruptured aneurysm. Surg Neurol. 2007;68(Suppl 2):S11–6. doi: 10.1016/j.surneu.2007.02.060. discussion S6. [DOI] [PubMed] [Google Scholar]
- 19.Peters D, Kassam A, Feingold E, Heidrich-O’Hare E, Yonas H, Ferrell R, et al. Molecular anatomy of an intracranial aneurysm: coordinated expression of genes involved in wound healing and tissue remodeling. Stroke. 2001;32(4):1036–42. doi: 10.1161/01.str.32.4.1036. [DOI] [PubMed] [Google Scholar]
- 20.Feng L, Stern D, Pile-Spellman J. Human endothelium: endovascular biopsy and molecular analysis. Radiology. 1999;212(3):655–64. doi: 10.1148/radiology.212.3.r99au28655. [DOI] [PubMed] [Google Scholar]
- 21.Yu S, Huang L, Song Y, Li A, Qin J, Yu X, et al. Identification of human coronary artery endothelial cells obtained by coronary endovascular biopsy. Zhonghua Xin Xue Guan Bing Za Zhi. 2008;36(3):240–2. [PubMed] [Google Scholar]
- 22.Cooke DL, Bauer D, Sun Z, Stillson C, Nelson J, Barry D, et al. Endovascular biopsy: technical feasibility of novel endothelial cell harvesting devices assessed in a rabbit aneurysm model. Interv Neuroradiol. 2015;21(1):120–8. doi: 10.15274/INR-2014-10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooke DLSH, Sun Z, Guo Y, Guo D, Saeed MM, Hetts SW, Higashida RT, Dowd CF, Young WL, Halbach VV. Evaluating the feasibility of harvesting endothelial cells using detachable coils. Interv Neuroradiol. 2013 doi: 10.1177/159101991301900401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Z, Lawson DA, Sinclair E, Wang CY, Lai MD, Hetts SW, et al. Endovascular biopsy: strategy for analyzing gene expression profiles of individual endothelial cells obtained from human vessels(✩) Biotechnol Rep (Amst) 2015;7:157–65. doi: 10.1016/j.btre.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Z, Su H, Long B, Sinclair E, Hetts SW, Higashida RT, et al. Endothelial cell high-enrichment from endovascular biopsy sample by laser capture microdissection and fluorescence activated cell sorting. J Biotechnol. 2014;192(Pt A):34–9. doi: 10.1016/j.jbiotec.2014.07.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehoux S, Tedgui A. Cellular mechanics and gene expression in blood vessels. J Biomech. 2003;36(5):631–43. doi: 10.1016/s0021-9290(02)00441-4. [DOI] [PubMed] [Google Scholar]
- 27.Anwar MA, Shalhoub J, Lim CS, Gohel MS, Davies AH. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J Vasc Res. 2012;49(6):463–78. doi: 10.1159/000339151. [DOI] [PubMed] [Google Scholar]
- 28.Navone SE, Marfia G, Invernici G, Cristini S, Nava S, Balbi S, et al. Isolation and expansion of human and mouse brain microvascular endothelial cells. Nat Protoc. 2013;8(9):1680–93. doi: 10.1038/nprot.2013.107. [DOI] [PubMed] [Google Scholar]
- 29.van Beijnum JR, Rousch M, Castermans K, van der Linden E, Griffioen AW. Isolation of endothelial cells from fresh tissues. Nat Protoc. 2008;3(6):1085–91. doi: 10.1038/nprot.2008.71. [DOI] [PubMed] [Google Scholar]
- 30.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–96. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33(2):155–60. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 32.Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163(6):1400–12. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scialdone A, Natarajan KN, Saraiva LR, Proserpio V, Teichmann SA, Stegle O, et al. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods. 2015;85:54–61. doi: 10.1016/j.ymeth.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 34.Szulwach KE, Chen P, Wang X, Wang J, Weaver LS, Gonzales ML, et al. Single-cell genetic analysis using automated microfluidics to resolve somatic mosaicism. PLoS One. 2015;10(8):e0135007. doi: 10.1371/journal.pone.0135007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar RM, Cahan P, Shalek AK, Satija R, DaleyKeyser AJ, Li H, et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516(7529):56–61. doi: 10.1038/nature13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498(7453):236–40. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liquet B, Lê Cao KA, Hocini H, Thiébaut R. A novel approach for biomarker selection and the integration of repeated measures experiments from two assays. BMC Bioinf. 2012;13:325. doi: 10.1186/1471-2105-13-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westerhuis JA, van Velzen EJ, Hoefsloot HC, Smilde AK. Multivariate paired data analysis: multilevel PLSDA versus OPLSDA. Metabolomics. 2010;6(1):119–28. doi: 10.1007/s11306-009-0185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Team RC. R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2016. [Google Scholar]
- 41.Liquet K-ALCFRGDBGBPMCZY. mixOmics: omics. Data integration project. R package version 6.1.2. 2017 ed: https://CRAN.R-project.org/package=mixOmics.
- 42.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 2016;166(5):1308–23. doi: 10.1016/j.cell.2016.07.054. e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202–14. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods. 2013;10(7):623–9. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saarinen J, Kohonen T. Self-organized formation of colour maps in a model cortex. Perception. 1985;14(6):711–9. doi: 10.1068/p140711. [DOI] [PubMed] [Google Scholar]
- 47.Kohonen T. Adaptive, associative, and self-organizing functions in neural computing. Appl Opt. 1987;26(23):4910–8. doi: 10.1364/AO.26.004910. [DOI] [PubMed] [Google Scholar]
- 48.Kohonen T. Self-organized formation of topologically correct feature maps. Biol Cybern. 1982;43:59–69. [Google Scholar]
- 49.Datta S. Comparisons and validation of statistical clustering techniques for microarray gene expression data. Bioinformatics. 2003;19(4):459–66. doi: 10.1093/bioinformatics/btg025. [DOI] [PubMed] [Google Scholar]
- 50.Datta S. Methods for evaluating clustering algorithms for gene expression data using a reference set of functional classes. BMC Bioinf. 2006;7:397. doi: 10.1186/1471-2105-7-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brock G, Pihur V, Datta S, Datta S. clValid: an R package for cluster validation. J Stat Softw. 2008;25(4) http://www.jstatsoft.org/v25/i04. [Google Scholar]
- 52.Handl J, Knowles J, Kell DB. Computational cluster validation in post-genomic data analysis. Bioinformatics. 2005;21(15):3201–12. doi: 10.1093/bioinformatics/bti517. [DOI] [PubMed] [Google Scholar]
- 53.Kim TG, Kim NK, Baek MJ, Huh R, Chung SS, Choi JU, et al. The relationships between endothelial nitric oxide synthase polymorphisms and the formation of intracranial aneurysms in the Korean population. Neurosurg Focus. 2011;30(6):E23. doi: 10.3171/2011.2.FOCUS10227. [DOI] [PubMed] [Google Scholar]
- 54.Foroud T, Sauerbeck L, Brown R, Anderson C, Woo D, Kleindorfer D, et al. Genome screen in familial intracranial aneurysm. BMC Med Genet. 2009;10:3. doi: 10.1186/1471-2350-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Foroud T, Sauerbeck L, Brown R, Anderson C, Woo D, Kleindorfer D, et al. Genome screen to detect linkage to intracranial aneurysm susceptibility genes: the Familial Intracranial Aneurysm (FIA) study. Stroke. 2008;39(5):1434–40. doi: 10.1161/STROKEAHA.107.502930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krischek B, Inoue I. The genetics of intracranial aneurysms. J Hum Genet. 2006;51(7):587–94. doi: 10.1007/s10038-006-0407-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.