

Abstract
The asymmetric allylic functionalization of unactivated internal alkenes is an emerging strategy for the conversion of simple unsaturated starting materials into a diverse range of enantioenriched products. This Perspective summarizes the development of reactions wherein a chiral catalyst facilitates the intermolecular stereoselective reaction between an achiral unactivated internal alkene and a reagent.

Keywords: allylic oxidation, allylic esterification, allylic amination, allylic alkylation, internal alkenes, enantioselective catalysis, enantioselective heteroene
1. INTRODUCTION
The stereo- and regioselective transformation of simple unsaturated hydrocarbons through allylic functionalization provides a direct path to enantioenriched building blocks from abundant and inexpensive petrochemical feedstocks. Another advantage to this method lies in its ability to generate these chiral synthons with preservation of the alkene as a handle for further elaboration. As a result, catalytic enantioselective allylic functionalization has emerged as a useful synthetic tool, streamlining the production of pharmaceuticals, natural products, fine chemicals, and other functional materials.1–5
The most developed and widely used approaches for catalytic asymmetric allylic functionalization fall within three distinct categories, which have been reviewed elsewhere: (1) intramolecular allylic rearrangement,6–8 (2) transition metalmediated allylic substitution/coupling,9–13 and (3) allylic C–H functionalization14–28 (Scheme 1). The first two categories rely on the presence of activating or directing groups embedded within the starting material. Often in these cases, the necessary insertion and deletion of these additional functional groups limits the economy and generality of the transformation.
Scheme 1.

Approaches to Catalytic Asymmetric Allylic Functionalization
The third method of allylic functionalization relies solely on the presence of an alkene moiety that lacks any activation at the allylic position. The vast majority of these reactions require the use of tethered substrates for intramolecular reactions or terminal alkene starting materials for intermolecular reactions.14–28 A general catalytic intermolecular enantioselective allylic C–H functionalization of unactivated internal alkenes remains a longstanding challenge. Recent advances in the development of this type of transformation are the subject of this Perspective.
The utility of allylic C–H functionalization for the synthesis of valuable commodities and materials relies heavily on the ability to access products with significant chemo-, regio-, and stereo-control. Selective reactions with terminal alkenes have been studied most extensively, presumably because this class of substrates contains only one set of enantiotopic allylic protons (Ha and Hb, Scheme 2A). Internal alkenes, on the other hand, possess two sets of protons on either side of the alkene functional group, which leads to the additional challenge of regioselectivity (Ha and Hb, Hc and Hd, Scheme 2B). In addition, since the product that results from allylic C–H functionalization of an internal alkene is often an internal alkene itself, the issue of E/Z selectivity in product formation must be addressed, along with over-reaction of the product. For this more challenging class of substrates, C–H functionalization in a substrate with many electronically and sterically similar allylic protons often leads to a mixture of constitutional isomers, E/Z olefin isomers, and enantiomers.
Scheme 2.

Allylic C–H Functionalization of Terminal and Internal Alkenes
The first examples of intermolecular asymmetric allylic C–H functionalization utilizing unfunctionalized internal alkenes began to appear nearly three decades ago. However, these transformations have been largely limited to symmetrical cycloalkenes. Nonetheless, these studies provided the first insights into the diffculty of tackling stereoselective allylic C–H activation without the challenges of E/Z- and regioselectivity.
2. ASYMMETRIC KHARASCH–SOSNOVSKY REACTION WITH UNACTIVATED CYCLOALKENES
One of the most notable advances in allylic C–H functionalization is the copper-catalyzed enantioselective Kharasch–Sosnovsky allylic oxidation.29,30 The initial discovery that copper(I) species catalyze the oxidation of alkenes with tert-butyl peroxybenzoate to yield allylic benzoates (e.g., 1 to 2, Scheme 3) laid the groundwork for one of the most common catalytic asymmetric allylic oxidations of unactivated alkenes.31,32
Scheme 3.

Early Kharasch–Sosnovsky Reaction with Cycloalkenes
Following two seminal attempts at rendering this radical process enantioselective (up to 16% ee),33,34 several groups reported enantioselective variants of this oxidation with unfunctionalized cycloalkenes.35–48 Enantioselectivity, yield, and reaction time varied substantially throughout these reports and are largely dependent on ligand identity.
The Muzart, Andersson, and Feringa laboratories independently explored the use of proline-based ligands for the stereoselective oxidation of cyclohexene 1 with catalytic Cu(OAc)2 (Scheme 4).37,39,44,46 Andersson’s bicyclic amino acid 5 appeared to be superior to the conventional l-proline 4, exhibiting the best enantioselectivity of the series (40% yield, 64% ee). Feringa and co-workers studied the effect of modifying 4, but efforts to further increase the enantioselectivity of this process were unsuccessful.48
Scheme 4.

Amino Acids as Chiral Ligands for the Asymmetric Kharasch–Sosnovsky Reaction
Several examples of the asymmetric Kharasch–Sosnovsky reaction have also been reported utilizing bisoxazoline-based ligands.36,40,41,43,45,49–53 One of the earliest examples originated from Pfaltz and co-workers who utilized catalytic copper(I) triflate benzene complex and bisoxazoline ligand 8 in the presence of tert-butyl perbenzoate to access benzoate 7 from cyclopentene 6 in 61% yield and 84% ee (Scheme 5A).36 Low temperatures were necessary to obtain higher selectivity but resulted in a slow reaction requiring 22 days to complete. Similar enantioselectivities were observed with a variety of C2-symmetric bisoxazoline ligands in studies conducted by numerous research groups with wide-ranging reaction times and yields. Katsuki later discovered that cyclopentene could be converted to allylic ester 7 in 93% ee using a modified C3-symmetric trisoxazoline ligand 9 (Scheme 5B).41 While various other ligand classes have been examined in the asymmetric Kharasch–Sosnovsky reaction, bisoxazoline ligands have shown the highest enantioselectivity to date.54–59 For example, Andrus utilized ligand 11 to form allylic ester 10 in 99% ee (Scheme 5C).50
Scheme 5.

Oxazolines as Chiral Ligands for the Asymmetric Kharasch–Sosnovsky Reaction
Trisubstituted alkenes 12 and 17 were also investigated under various reaction conditions (Scheme 6). In all cases, the secondary radical intermediate was preferentially formed over the tertiary, precluding any significant formation of 15 or 20. Still, regioselectivity between isomeric products 13 and 14 or 18 and 19 was poor, and the enantiomeric excess varied significantly. Interestingly, the most substituted allylic esters 15 and 20 were generated with the highest enantioselectivity. Acyclic alkenes surveyed in this chemistry were limited to terminal alkenes that displayed diminished enantioselectivities.35,41
Scheme 6.

Asymmetric Kharasch–Sosnovsky Reaction with Trisubstituted Alkenes
Andrus and Singh have both offered stereoinduction models for the copper-catalyzed enantioselective allylic oxidation (Scheme 7).35,43 Both authors assumed that the final step of the Kharasch–Sosnovsky reaction proceeds via a rearrangement of a copper(III)-allyl intermediate. In the model proposed by Andrus, the minimization of steric interactions between the substrate, ester functional group, and chiral ligand dictates the substrate’s approach (Scheme 7A). Singh’s model, employing a tridentate pyridyl bisoxazoline ligand, invokes an additional stabilizing π–π interaction between one of the phenyl groups of the chiral ligand and the aromatic ring of the benzoate (Scheme 7B). In both cases, stereochemical models account for the lowered selectivity exhibited by acyclic alkenes lacking in conformational rigidity.
Scheme 7.

Model for Stereoinduction in the Copper-Bisoxazoline-Catalyzed Asymmetric Kharasch–Sosnovsky Reaction
3. ASYMMETRIC ALLYLIC AMINATION OF UNACTIVATED ALKENES
Despite the plethora of reports that began emerging in the 1990s for catalytic asymmetric allylic C–H esterification, the first enantioselective C–H amination utilizing unactivated cycloalkenes did not appear until 2001. In the course of examining the aziridination chemistry between [N-(p-toluenesulfonyl)imino]-phenyliodinane and cyclohexene, Katsuki discovered that various electron-deficient salen-manganese(III) complexes catalyzed the formation of C–H amination product 21 instead (Scheme 8).60 While it is not uncommon for this reaction pathway to compete with aziridination, the electron-deficient cationic catalyst 22 was particularly well suited for asymmetric allylic amination, exclusively affording 21 in 44% yield and 67% ee (Scheme 8A). Additionally, cycloheptene 23 was assayed under the reaction conditions yielding 24 with lower enantioselectivity and as a mixture with the corresponding aziridine byproduct 25 (Scheme 8B). In 2003, Katsuki and co-workers reported a complementary enantioselective intermolecular benzylic and allylic C–H amination that is mediated by 2-(trimethylsilyl)ethanesulfonyl azide as an oxidant and a salen-based ruthenium complex.61
Scheme 8.

Catalytic Asymmetric C–H Amination with Manganese-Salen Complex
Four years later, the Clark lab reported a copper-catalyzed allylic C–H amination that proceeded with comparable levels of enantioselectivity.62 Inspired by a report on allylic C–H amination by Katsuki for the generation of racemic product, and their own work in the field of asymmetric allylic oxidation, Clark and co-workers were able to develop an enantioselective amination variant of the Kharasch–Sosnovsky reaction. Substoichiometric amounts of [Cu(CH3CN)4]PF6 and bisoxazoline 11 facilitated the amination of cyclohexene 1 with peroxycarbamate 26 to furnish allylic amination product 21 in up to 70% ee (Scheme 9A).
Scheme 9.

Amination Variant of Asymmetric Kharasch–Sosnovsky Reaction
Interestingly, the identity of the peroxycarbamate was critical for the formation of the amination product over that of the alternative oxidation product 27. The authors noted that the aryl carbamates of allylic alcohols undergo stereospecific rearrangement to yield the desired allylic amines. In some cases, the introduction of certain ligands biased reactivity toward esterification (Scheme 9B).
Dauban developed an elegant strategy for asymmetric C–H amination in which chiral rhodium complex 30 catalyzed the coupling of chiral sulfonimidamide 31 and various alkenes (Scheme 10).63–66 This amination is postulated to occur by the C–H insertion of a chiral metallanitrene, resulting from the oxidation of rhodium catalyst 30 by an iminoiodane intermediate generated in situ. Both cyclic and acyclic alkenes were viable substrates for this method. Most notably, acyclic unsymmetrical alkenes such as 29c and 29d exhibited high regioselectivity in allylic amination, as methylene C–H bonds underwent amination preferentially to methyl C–H bonds.
Scheme 10.

C–H Amination with a Chiral Rhodium Catalyst and Chiral Sulfonimidamide
4. ENANTIOSELECTIVE ALLYLIC ALKYLATION OF UNACTIVATED ALKENES
The asymmetric Mizoroki–Heck reaction has garnered considerable attention in recent years, due to the immense popularity of the robust and widely functional group tolerant racemic version of this reaction. Several reviews have been written to summarize the most important advances in enantioselective Mizoroki–Heck reactions (Scheme 11A),67–74 oxidative Mizoroki–Heck reactions (Scheme 11B),75 redox-relay Mizoroki–Heck reactions (Scheme 11C),76 and other variants of this venerable transformation. Examples have been reported for the asymmetric allylic arylation or alkylation of cyclic and acyclic internal alkenes with organohalides, organotriflates, organoboronic acids, or organodiazonium salts.
Scheme 11.
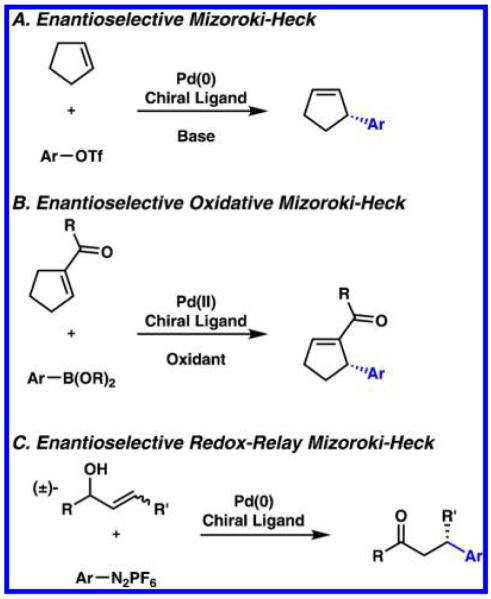
Asymmetric Mizoroki–Heck Reactions
Alternatively, site-selective carbenoid C–H functionalization has emerged as one of the most effective ways of accessing new C–C bonds in unactivated substrates with impressive regio- and stereoselectivity.77–79 Around the same time as Müller’s initial report of an acceptor/acceptor-substituted carbenoid for moderately enantioselective allylic C–H alkylations, Davies began to explore this mode of C–H functionalization with donor/acceptor stabilized carbenoids.80–85 These reports on the Rh2(DOSP)4-catalyzed decomposition of aryldiazoacetates have become one of the most selective methods for C(sp3)–H functionalization.
In 2001, Davies reported a catalytic asymmetric allylic C–H activation to access γ,δ-unsaturated esters, providing an alternative route to Claisen rearrangement products (Scheme 12).84 The donor/acceptor-stabilized carbenoids resulted in enhanced stability and chemoselectivity toward the C–H functionalization product over the alternative cyclopropanation pathway. A variety of substituted cyclohexenes were smoothly alkylated in the presence of catalytic D2-symmetric Rh2(S-DOSP)4 and diazoester 32 revealing high levels of regioselectivity as a result of steric and electronic bias (33a–d). Enantioselectivity was generally high, even when acyclic internal alkenes were used (33e–g). However, the diastereomeric ratios varied substantially with some dropping as low as 1.3:1 in response to altered steric environments.
Scheme 12.

Rhodium-Catalyzed Asymmetric Allylic C–H Alkylation with Stabilized Carbenoids
Recently, Davies reported a new method for accessing C–H functionalized products similar to 33e–g with increased selectivity.85 N-Sulfonyl-1,2,3-triazole 34 generates a donor/acceptor carbene with chiral rhodium(II) catalyst Rh2 (S-NTTL)4 that exhibits increased regio-, diastereo-, and enantioselectivity for allylic alkylation (Scheme 13). Though only four examples are described, the conversion of acyclic alkenes to 35a,b represents two of the most selective direct allylic alkylations of unactivated acyclic internal alkenes to date.
Scheme 13.

Rhodium-Catalyzed Asymmetric Allylic C–H Alkylation with Triazoles
5. ENANTIOSELECTIVE HETERO-ENE REACTIONS
The Lewis acid-catalyzed enantioselective heteroene reaction between glyoxylate esters and unfunctionalized alkenes, first examined by the Evans group, represents one of the earliest advances in the asymmetric functionalization of internal unactivated alkenes.86–89 Various cationic copper(II)-bisoxazoline catalysts were investigated for their ability to catalyze the formation of α-hydroxy esters with both 1,1- and 1,2-disubstituted alkenes. Copper complex 38 reacted smoothly with ethyl glyoxylate 36 and cyclohexene (1), for example, to provide α-hydroxy ester 37a in great yield and with exquisite enantioselectivity (Scheme 14).
Scheme 14.

Copper-Bisoxazoline-Catalyzed Asymmetric Hetero-Ene Reaction
X-ray crystallography and computational studies of [Cu((S,S)-t-Bu-BOX)(glyoxylate)]2+ indicate that the catalyst takes on a distorted square planar geometry with glyoxylate 36 (Figure 1). This conformation provides insight into the factors that govern stereoselectivity, as the flanking tert-butyl groups of the ligand both block the Re-face approach of the enophile and sterically disfavor the exo transition state.
Figure 1.
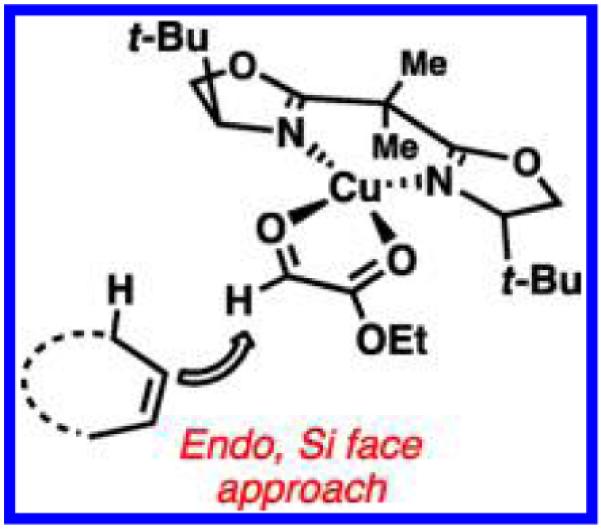
Model for stereoinduction of copper-bisoxazoline-catalyzed asymmetric hetero-ene reaction.
In noncoordinating solvents, various unfunctionalized internal alkenes furnished products in up to 98% ee and modest diastereoselectivity (37a–d). Even acyclic cis-2-butene produced α-hydroxy ester 37d with comparable enantioselectivity to the structurally rigid cycloalkenes. Interestingly, this trend did not extend to cycloheptene, which failed to afford synthetically useful yield of 37b under any of the conditions screened. Since this initial discovery by Evans, a palladium(II)-catalyzed version of this reaction has also been developed.90
Recently Tambar and co-workers developed a catalytic asymmetric heteroene reaction utilizing enophilic sulfurimide reagent 41 to convert a wide range of cis-alkenes 39 to allylic sulfinamides 40 utilizing a catalytic combination of SbCl5 and BINOL derivatives 42a or 42b (Scheme 15A).91 This chemistry, which accommodates a wide substrate scope including unactivated olefins, takes place with high enantioselectivity, diastereoselectivity, E-alkene selectivity, and regioselectivity. This reaction, thus, represents one of the earliest examples of asymmetric allylic C–S bond formation utilizing unactivated alkenes. A series of steric and electronic trends were identified to predict regio- and chemoselectivity in the enantioselective allylic oxidation of nonsymmetric internal alkenes (Scheme 15B).
Scheme 15.

Catalytic Asymmetric Allylic Oxidation of Internal Alkenes via a Hetero-Ene Reaction
Based on these reactivity trends, substrates accommodating multiple nonequivalent alkenes exhibited chemoselectivity, disfavoring the heteroene reaction with alkenes displaying diminished π-nucleophilicity (Scheme 16). It is important to note, however, that trans-alkenes and cycloalkenes provide the corresponding allylic sulfinamides with diminished enantioselectivities, presumably due to disfavored steric interactions.
Scheme 16.

Regio-, Enantio-, and Chemoselective Allylic Oxidation of Dienes with Multiple Internal Alkenes
The catalytic combination of Lewis acidic SbCl5 and BINOL derivatives 42a or 42b proved imperative, with diminished yields when either catalytic component was excluded. This observation, in combination with pioneering studies by Corey and co-workers,92,93 led the authors to further investigate and propose a Lewis acid-assisted Brønsted acidity-based mechanism wherein SbCl5 further acidifies a proton of BINOL to activate enophile 41.94–98 The regio-, diastereo-, and enantioselectivity are rationalized through an organized 6-membered closed transition state 43 with oxidant 41 favoring an exo-selective approach (Scheme 17).
Scheme 17.

Transition-State Model for SbCl5–BINOL-Catalyzed Allylic Oxidation of Internal Alkenes
The utility of Tambar’s stereoselective heteroene chemistry is showcased in the stereospecific elaboration of the allylic sulfinamide product 40a to a variety of derivatives containing stereocenters with C–S, C–C, C–Cl, C–N, and C–O bonds (Scheme 18). Copper-mediated alkylation chemistry affords allylic alkylation product 44.99 Simple reduction conditions yield allylic thiol 45. Rearrangements afford allylic alcohol 46, allylic amine 47, and allylic chloride 48. Thus, this chemistry represents a general strategy toward enantioenriched products from both unactivated and activated cis-alkene starting materials.
Scheme 18.

Synthetic Derivitization Products from Catalytic Regio- and Enantioselective Allylic Oxidation of Internal Alkenes
6. CONCLUSIONS
Recent advances in catalytic enantioselective intermolecular allylic C–H oxidation, amination, and alkylation of unactivated internal alkenes have begun to address several selectivity issues encountered when utilizing unsaturated hydrocarbons. These powerful strategies have provided chemists with the ability to directly access value-added products from inexpensive and abundant starting materials. While relatively few examples of selective intermolecular allylic C–H functionalization utilizing acyclic systems have been demonstrated, several recent discoveries have established a foundation to address this need. Future advances in the asymmetric allylic functionalization of internal alkenes will undoubtedly focus on selective transformations that can be performed in molecular settings that are amenable to the synthesis of complex target molecules.
ACKNOWLEDGMENTS
Financial support was provided by W. W. Caruth, Jr. Endowed Scholarship, Welch Foundation (I-1748), National Institutes of Health (R01GM102604), National Science Foundation (1150875), and Sloan Research Fellowship.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Farina V; Reeves JT; Senanayake CH; Song JJ Chem. Rev 2006, 106, 2734–2793. [DOI] [PubMed] [Google Scholar]
- (2).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (3).Hyneck M; Dent J; Hook JB In Chirality in Drug Design and Synthesis; Brown C, Ed.; Academic Press: San Diego, 1990; pp 1–28. [Google Scholar]
- (4).Wilson RM; Danishefsky SJ Angew. Chem., Int. Ed 2010, 49, 6032–6056. [DOI] [PubMed] [Google Scholar]
- (5).Brooks WH; Guida WC; Daniel KG Curr. Top. Med. Chem 2011, 11, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Overman LE; Carpenter NE In Organic Reactions; Overman LE, Ed.; Wiley: Hoboken, NJ, 2005; Vol. 66, pp 1–107. [Google Scholar]
- (7).Sweeney JB Chem. Soc. Rev 2009, 38, 1027–1038. [DOI] [PubMed] [Google Scholar]
- (8).Nasveschuk CG; Rovis T Org. Biomol. Chem 2008, 6, 240–254. [DOI] [PubMed] [Google Scholar]
- (9).Trost BM; Van Vranken DL Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- (10).Trost BM; Crawley ML Chem. Rev 2003, 103, 2921–2944. [DOI] [PubMed] [Google Scholar]
- (11).Alexakis A; Bäckvall JE; Krause N; Pàmies O; Diéguez M Chem. Rev 2008, 108, 2796–2823. [DOI] [PubMed] [Google Scholar]
- (12).Hartwig JF; Stanley LM Acc. Chem. Res 2010, 43, 1461–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hethcox JC; Shockley SE; Stoltz BM ACS Catal 2016, 6, 6207–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Collet F; Dodd RH; Dauban P Chem. Commun 20095061–5074. [DOI] [PubMed] [Google Scholar]
- (15).Liu G; Wu Y In C–H Activation; Yu J-Q, Ed.; Springer: Berlin, Heidelberg, 2010; pp 195–209. [Google Scholar]
- (16).Andrus MB In Stereoselective Synthesis; De Vries JG, Molander GA, Evans PA, Eds.; Thieme Verlag: Stuttgart, 2011; Vol. 3, 1st ed., pp 469–482. [Google Scholar]
- (17).Grennberg H; Bäckvall J-E In Transition Metals for Organic Synthesis; Wiley-VCH Verlag GmbH: Weinheim, 2008; pp 243–265. [Google Scholar]
- (18).Page PCB; McCarthy TJ In Comprehensive Organic Synthesis; Fleming I, Ed.; Pergamon: Oxford, 1991; pp 83–117. [Google Scholar]
- (19).Johannsen M; Jørgensen KA Chem. Rev 1998, 98, 1689–1708. [DOI] [PubMed] [Google Scholar]
- (20).Zalatan DN; Bois JD In C–H Activation; Yu J-Q, Shi Z, Eds.; Springer: Berlin, Heidelberg, 2010; pp 347–378. [Google Scholar]
- (21).Collet F; Lescot C; Dauban P Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]
- (22).Ramirez TA; Zhao B; Shi Y Chem. Soc. Rev 2012, 41, 931–942. [DOI] [PubMed] [Google Scholar]
- (23).Covell DJ; White MC Angew. Chem., Int. Ed 2008, 47, 6448–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ammann SE; Liu W; White MC Angew. Chem., Int. Ed 2016, 55, 9571–9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Race NJ; Hazelden IR; Faulkner A; Bower JF Chem. Sci 2017, 8, 5248–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vulovic B; Watson DA Eur. J. Org. Chem 2017, 2017, 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zalatan DN; Du Bois J J. Am. Chem. Soc 2008, 130, 9220–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Milczek E; Boudet N; Blakey S Angew. Chem., Int. Ed 2008, 47, 6825–6828. [DOI] [PubMed] [Google Scholar]
- (29).Eames J; Watkinson M Angew. Chem., Int. Ed 2001, 40, 3567–3571. [DOI] [PubMed] [Google Scholar]
- (30).Le Paih J; Schlingloff G; Bolm C; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2004; Vol. 2, pp 256–265. [Google Scholar]
- (31).Kharasch M; Fono A J. Org. Chem 1958, 23, 324–325. [Google Scholar]
- (32).Kharasch MS; Sosnovsky G J. Am. Chem. Soc 1958, 80, 756–756. [Google Scholar]
- (33).Araki M; Nagase T Chemical Abstracts 1977, 86, 120886r. [Google Scholar]
- (34).Denney DB; Napier R; Cammarata A J. Org. Chem 1965, 30, 3151–3153. [Google Scholar]
- (35).Andrus MB; Argade AB; Chen X; Pamment MG Tetrahedron Lett 1995, 36, 2945–2948. [Google Scholar]
- (36).Gokhale AS; Minidis ABE; Pfaltz A Tetrahedron Lett 1995, 36, 1831–1834. [Google Scholar]
- (37).Rispens MT; Zondervan C; Feringa BL Tetrahedron: Asymmetry 1995, 6, 661–664. [Google Scholar]
- (38).Loiseleur O; Meier P; Pfaltz A Angew. Chem., Int. Ed. Engl 1996, 35, 200–202. [Google Scholar]
- (39).Södergren MJ; Andersson PG Tetrahedron Lett 1996, 37, 7577–7580. [Google Scholar]
- (40).Andrus MB; Chen X Tetrahedron 1997, 53, 16229–16240. [Google Scholar]
- (41).Kawasaki K.-i.; Katsuki T Tetrahedron 1997, 53, 6337–6350. [Google Scholar]
- (42).Fahrni CJ Tetrahedron 1998, 54, 5465–5470. [Google Scholar]
- (43).Sekar G; DattaGupta A; Singh VK J. Org. Chem 1998, 63, 2961–2967. [Google Scholar]
- (44).Muzart J J. Mol. Catal 1991, 64, 381–384. [Google Scholar]
- (45).Kawasaki K; Tsumura S; Katsuki T Synlett 1995, 1995, 1245–1246. [Google Scholar]
- (46).Levina A; Muzart J Tetrahedron: Asymmetry 1995, 6, 147–156. [Google Scholar]
- (47).DattaGupta A; Singh VK Tetrahedron Lett 1996, 37, 2633–2636. [Google Scholar]
- (48).Zondervan C; Feringa BL Tetrahedron: Asymmetry 1996, 7, 1895–1898. [Google Scholar]
- (49).Aldea L; Delso I; Hager M; Glos M; García JI; Mayoral JA; Reiser O Tetrahedron 2012, 68, 3417–3422. [Google Scholar]
- (50).Andrus MB; Zhou Z J. Am. Chem. Soc 2002, 124, 8806–8807. [DOI] [PubMed] [Google Scholar]
- (51).Clark S; Tolhurst KF; Taylor M; Swallow S J. Chem. Soc., Perkin Trans 1 1998, 1167–1170. [Google Scholar]
- (52).Kohmura Y; Katsuki T Tetrahedron Lett 2000, 41, 3941–3945. [Google Scholar]
- (53).Ramalingam B; Neuburger M; Pfaltz A Synthesis 2007, 2007, 572–582. [Google Scholar]
- (54).Malkov AV; Bella M; Langer V; Kočovský P Org. Lett 2000, 2, 3047–3049. [DOI] [PubMed] [Google Scholar]
- (55).Hoang VDM; Reddy PAN; Kim T-J Organometallics 2008, 27, 1026–1027. [Google Scholar]
- (56).Zhou Z; Andrus MB Tetrahedron Lett 2012, 53, 4518–4521. [Google Scholar]
- (57).Malkov AV; Pernazza D; Bell M; Bella M; Massa A; Teplý F; Meghani P; Kočovský P J. Org. Chem 2003, 68, 4727–4742. [DOI] [PubMed] [Google Scholar]
- (58).Tan Q; Hayashi M Adv. Synth. Catal 2008, 350, 2639–2644. [Google Scholar]
- (59).Malkov AV; Stewart-Liddon AJP; Teplý F; Kobr L; Muir KW; Haigh D; Kočovský P Tetrahedron 2008, 64, 4011–4025. [Google Scholar]
- (60).Kohmura Y; Katsuki T Tetrahedron Lett 2001, 42, 3339–3342. [Google Scholar]
- (61).Nishioka Y; Uchida T; Katsuki T Angew. Chem., Int. Ed 2013, 52, 1739–1742. [DOI] [PubMed] [Google Scholar]
- (62).Clark JS; Roche C Chem. Commun 20055175–5177. [DOI] [PubMed] [Google Scholar]
- (63).Liang C; Robert-Peillard F; Fruit C; Mueller P; Dodd RH; Dauban P Angew. Chem., Int. Ed 2006, 45, 4641–4644. [DOI] [PubMed] [Google Scholar]
- (64).Liang C; Collet F; Robert-Peillard F; Mueller P; Dodd RH; Dauban P J. Am. Chem. Soc 2008, 130, 343–350. [DOI] [PubMed] [Google Scholar]
- (65).Collet F; Lescot C; Liang C; Dauban P Dalton Trans 2010, 39, 10401–10413. [DOI] [PubMed] [Google Scholar]
- (66).Lescot C; Darses B; Collet F; Retailleau P; Dauban P J. Org. Chem 2012, 77, 7232–7240. [DOI] [PubMed] [Google Scholar]
- (67).Guiry PJ; Hennessy AJ; Cahill JP Top. Catal 1997, 4, 311–326. [Google Scholar]
- (68).Link JT; Wada CK; John Wiley & Sons Ltd.: Hoboken, NJ, 2009; pp 433–462. [Google Scholar]
- (69).Loiseleur O; Hayashi M; Keenan M; Schmees N; Pfaltz A J. Organomet. Chem 1999, 576, 16–22. [Google Scholar]
- (70).McCartney D; Guiry PJ Chem. Soc. Rev 2011, 40, 5122–5150. [DOI] [PubMed] [Google Scholar]
- (71).Oestreich M Angew. Chem., Int. Ed 2014, 53, 2282–2285. [DOI] [PubMed] [Google Scholar]
- (72).Shibasaki M; Vogl EM J. Organomet. Chem 1999, 576, 1–15. [Google Scholar]
- (73).Shibasaki M; Vogl EM; Ohshima T Adv. Synth. Catal 2004, 346, 1533–1552. [Google Scholar]
- (74).Tietze LF; Lotz F; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2007; pp 147–152. [Google Scholar]
- (75).Lee AL Org. Biomol. Chem 2016, 14, 5357–5366. [DOI] [PubMed] [Google Scholar]
- (76).Werner EW; Mei T-S; Burckle AJ; Sigman MS Science 2012, 338, 1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Davies HML; Manning JR Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Davies HML; Morton D Chem. Soc. Rev 2011, 40, 1857–1869. [DOI] [PubMed] [Google Scholar]
- (79).Müller P; Fruit C Chem. Rev 2003, 103, 2905–2920. [DOI] [PubMed] [Google Scholar]
- (80).Müller P; Tohill S Tetrahedron 2000, 56, 1725–1731. [Google Scholar]
- (81).Davies HML; Coleman MG; Ventura DL Org. Lett 2007, 9, 4971–4974. [DOI] [PubMed] [Google Scholar]
- (82).Davies HML; Dai X; Long MS J. Am. Chem. Soc 2006, 128, 2485–2490. [DOI] [PubMed] [Google Scholar]
- (83).Davies HML; Gregg TM Tetrahedron Lett 2002, 43, 4951–4953. [Google Scholar]
- (84).Davies HML; Ren P; Jin Q Org. Lett 2001, 3, 3587–3590. [DOI] [PubMed] [Google Scholar]
- (85).Kubiak RW; Mighion JD; Wilkerson-Hill SM; Alford JS; Yoshidomi T; Davies HML Org. Lett 2016, 18, 3118–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Mikami K; Shimizu M Chem. Rev 1992, 92, 1021–1050. [Google Scholar]
- (87).Snider BB; Ron E J. Am. Chem. Soc 1985, 107, 8160–8164. [Google Scholar]
- (88).Evans DA; Burgey CS; Paras NA; Vojkovsky T; Tregay SW J. Am. Chem. Soc 1998, 120, 5824–5825. [Google Scholar]
- (89).Evans DA; Tregay SW; Burgey CS; Paras NA; Vojkovsky T J. Am. Chem. Soc 2000, 122, 7936–7943. [Google Scholar]
- (90).Hao J; Hatano M; Mikami K Org. Lett 2000, 2, 4059–4062. [DOI] [PubMed] [Google Scholar]
- (91).Bayeh L; Le PQ; Tambar UK Nature 2017, 547, 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Surendra K; Corey EJ J. Am. Chem. Soc 2012, 134, 11992–11994. [DOI] [PubMed] [Google Scholar]
- (93).Surendra K; Rajendar G; Corey EJ J. Am. Chem. Soc 2014, 136, 642–645. [DOI] [PubMed] [Google Scholar]
- (94).Huang Y; Unni AK; Thadani AN; Rawal VH Nature 2003, 424, 146–146. [DOI] [PubMed] [Google Scholar]
- (95).Yamamoto H; Futatsugi K Angew. Chem., Int. Ed 2005, 44, 1924–1942. [DOI] [PubMed] [Google Scholar]
- (96).Taylor MS; Jacobsen EN Angew. Chem., Int. Ed 2006, 45, 1520–1543. [DOI] [PubMed] [Google Scholar]
- (97).Akiyama T Chem. Rev 2007, 107, 5744–5758. [DOI] [PubMed] [Google Scholar]
- (98).Johnston JN; Muchalski H; Troyer TL Angew. Chem., Int. Ed 2010, 49, 2290–2298. [DOI] [PubMed] [Google Scholar]
- (99).Bao H; Bayeh L; Tambar UK Angew. Chem., Int. Ed 2014, 53, 1664–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]