

Abstract
Rhomboids are intramembrane serine proteases and belong to the group of structurally and biochemically most comprehensively characterized membrane proteins. They are highly conserved and ubiquitously distributed in all kingdoms of life and function in a wide range of biological processes, including epidermal growth factor signaling, mitochondrial dynamics, and apoptosis. Importantly, rhomboids have been associated with multiple diseases, including Parkinson’s disease, type 2 diabetes, and malaria. However, despite a thorough understanding of many structural and functional aspects of rhomboids, potent and selective inhibitors of these intramembrane proteases are still not available. In this study, we describe the computer-based rational design, chemical synthesis, and biological evaluation of novel N-methylene saccharin-based rhomboid protease inhibitors. Saccharin inhibitors displayed inhibitory potency in the submicromolar range, effectiveness against rhomboids both in vitro and in live Escherichia coli cells, and substantially improved selectivity against human serine hydrolases compared to those of previously known rhomboid inhibitors. Consequently, N-methylene saccharins are promising new templates for the development of rhomboid inhibitors, providing novel tools for probing rhomboid functions in physiology and disease.
Graphical Abstract

At least 600 protease-encoding genes have been identified in the human genome. Proteases regulate diverse biological processes, including blood coagulation, antigen presentation, cell motility, apoptosis, and neuronal development. 1,2 Dysregulation of protease activity can lead to severe diseases like arthritis, cancer, and cardiovascular diseases, which makes proteases attractive targets for the development of new pharmaceuticals.3–9 Soluble, globular proteases have been studied intensively for decades and now represent the best characterized and understood enzymes.2 In contrast, intra-membrane proteases were identified only recently. The founding member of the rhomboid superfamily of intramembrane serine proteases was discovered in Drosophila in 2001.10 Rhomboid intramembrane proteases can be found in all forms of life ranging from archaea and bacteria to fungi, plants, and animals.11,12 In contrast to classical serine proteases that use a catalytic triad (Ser/His/Asp),13,14 rhomboids use a catalytic dyad (Ser/His) within their active site15,16 and their substrates are transmembrane proteins.
Because of their broad phylogenetic distribution, their implication in various diseases, and the availability of high-resolution crystal structures, rhomboid proteases are potential targets for drug development, yet potent and selective inhibitors are not available.16–20 Isocoumarins were the first inhibitors observed to be effective against rhomboids, including dichloroisocoumarin (DCI),10,21 JLK-6 (IC50 ~ 6 μM), and IC16 (IC50 ~ 0.7 μM).22,23 However, isocoumarins are in general highly reactive and lack selectivity.24,25 In fact, DCI was later established as a pan-rhomboid inhibitor.26 Rational synthesis approaches and the screening of small molecule libraries have identified other classes of rhomboid inhibitors. N-Sulfonylated β-lactams inhibited bacterial rhomboids but showed limited potency in vivo (EC50 ~ 5–10 μM) and poor selectivity over soluble serine proteases such as chymotrypsin.27 Fluorophosphonates (IC50 ~ 50 μM)28 and β-lactone-based inhibitors (IC50 ~ 26–44 μM)29 displayed poor potency toward rhomboids. Recently, peptidyl chloromethylketones30 and peptidyl aldehydes31 were reported as peptidomimetic inhibitors of rhomboids, but these compounds also lacked sufficient potency and selectivity.
To fill the apparent gap between a comprehensive biochemical understanding of rhomboid enzymes and the absence of selective inhibitors, we applied a computer-aided rational approach with a starting template based on N-methylene-substituted saccharins with a leaving group (LG). This scaffold has several advantages as (i) it is a well-characterized sweetener with a safe toxicological profile,32 (ii) it can act as a suicide inhibitor, resulting in a double-bonded enzyme–inhibitor complex,24,33,34 and (iii) it has substantial potential for derivatization. A computational algorithm was used to virtually screen suitable candidate molecules prior to chemical synthesis and compound testing during in vitro enzyme activity assays (Figure 1A),35 which yielded a subset of N-methylene-substituted saccharins as potential rhomboid inhibitors. Mass spectrometry and mutagenesis were used to establish the reaction mechanism of rhomboid inhibition by saccharins, and the in vivo potency was determined in Escherichia coli. The selectivity of the compounds for a variety of rhomboids and off-target effects against human serine hydrolases were tested using activity-based probe approaches. 36,37 This effort provided submicromolar, partially reversible, and selective rhomboid protease inhibitors based on a novel and modifiable scaffold.
Figure 1.

(A) Workflow for the development of novel rhomboid inhibitors. (B) Proposed reaction mechanism of a saccharin-based inhibitor with the catalytic residues in the active site of the E. coli rhomboid protease GlpG. The mechanism involves the enzyme-induced formation of a Michael acceptor and results in an end product that is covalently cross-linked to the enzyme.
MATERIALS AND METHODS
Computational Chemistry
Various methods of Molecular Operating Environment (MOE) of the Chemical Computing group38 were used for the docking of saccharins into the rhomboid protease pocket. See the Supporting Information for details.
Chemical Synthesis
For details of the chemical synthesis of the saccharin inhibitors and the chemical data, see the Supporting Information.
Protein Expression and Purification
Bacterial expression constructs for N-terminally His-tagged E. coli GlpG,16 N-terminally GST-tagged Aquifex aeolicus AaROM,21 and C-terminally His-tagged Gurken chimeric substrate (MBP-GurkenTMD-Trx-His6)39 were kindly provided by Y. Ha (Yale School of Medicine, New Haven, CT), S. Urban (Johns Hopkins University, Baltimore, MD), and M. Freeman (University of Oxford, Oxford, U.K.). Both rhomboids and the substrate Gurken were expressed in E. coli C43(DE3) cells. After induction with isopropyl β-D-1-thiogalactopyranoside at an OD600 of 0.8, GlpG and AaROM were expressed overnight at 18 °C and Gurken was expressed for 3 h at 37 °C. The cells were broken by three passages through a nitrogen cavitation bomb; bacterial membranes were isolated by differential centrifugation, and proteins were solubilized from the membrane preparations with 1.5% dodecyl β-maltoside (DDM) (Glycon Biochemicals). GlpG and Gurken were purified via immobilized metal ion affinity chromatography (IMAC) using a TalonCrude 1 mL HiTrap column and an Akta prime plus chromatography system (GE Healthcare). The GST-tagged AaRom was incubated with glutathione sepharose resin overnight at 4 °C, and the rhomboid was eluted with PreScission (Amersham Pharmacia Biotech) protease cleavage according to the manufacturer’s instructions. All recombinant proteins were further concentrated using 10 or 30 kDa molecular weight cutoff Amicon Ultra Centrifugal filters (Millipore). Protein purity was estimated by Coomassie Brilliant Blue (Merck)-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE).
GlpG and AaROM in Vitro Activity Assays
Activity assays were performed in a volume of 20 μL of reaction buffer [50 mM HEPES-NaOH (pH 7.5), 5 mM EDTA, 0.4 M NaCl, 10% (v/v) glycerol, and 0.05% DDM] containing either GlpG (0.35 μM) or AaROM (0.094 μM) and Gurken substrate (1.8 μM). Saccharin inhibitors were preincubated with the rhomboids in reaction buffer for 30 min at 37 °C while being gently shaken. The substrate was added, and the reaction was continued for an additional 90 min at 37 °C. Subsequently, the reaction mixture was separated by SDS–PAGE and stained with Coomassie Brilliant Blue. The N-terminal Gurken cleavage fragment was quantified using ImageJ, normalized to the dimethyl sulfoxide (DMSO) vehicle control condition, and the values were plotted in GraphPad Prism. A nonlinear regression curve fit of the log(inhibitor concentration) versus percent N-terminal Gurken product was used to determine IC50 values.
GlpG Activity in Live E. coli Cells
The inhibitory potency of saccharin inbitiors against GlpG in live E. coli was determined as described previously.40
Cell-Based RHBDL2 Activity Assay
Human HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 1 mM sodium pyruvate, and 100 units/mL penicillin/streptomycin (Thermo Fisher Scientific). Expression constructs for HA-tagged mouse rhomboid RHBDL2 and FLAG-tagged Gurken substrate (kind gifts of M. Freeman27) were transiently co-transfected into HEK293T cells using GeneJuice transfection reagent (Merck). Two hours after the transfection, cells were treated with the saccharin inhibitors or DMSO as a control. After 48 h, cells lysates were prepared with standard 1% NP40 lysis buffer and N-terminal Gurken cleavage fragments were analyzed by Western blotting with the anti-FLAG BioM2 monoclonal antibody (1:1000, Merck).
Mass Spectrometry
The rhomboid GlpG at 0.2 mg/mL in 25 mM HEPES, 150 mM NaCl, and 0.2% (w/v) DDM (pH 8.0) was preincubated with 50 μM inhibitor or DMSO solvent control at 23 °C overnight. The sample was then diluted with 0.1% formic acid to a concentration of 0.1 mg/mL. Five microliters of the sample was injected onto a desalting column (MassPREP desalting, Waters) and desalted with a fast gradient (4 min) of acetonitrile in water with 0.1% formic acid. The separation was performed by a liquid chromatography system (I-class, Waters), which was coupled online to a mass spectrometer (Synapt G2, Waters) to acquire the intact mass of the protein by electrospray ionization. The raw spectra were subtracted and deconvoluted (MaxEnt 1, Waters) to produce the final spectra shown in panels B and C of Figure 4.
Figure 4.

Mechanism of saccharin-based inhibitors. (A) The reaction of N-methylated saccharins with the rhomboid involves the release of the acidic LG. A saccharin derivative with anthranilic acid as the LG (BSc5193) was incubated with recombinantly expressed GlpG. According to the proposed mechanism, nucleophilic attack of Ser201 should lead to the formation of a Michael acceptor and release of the anthranilic acid LG, which becomes fluorescent. Incubation of GlpG with BSc5193 resulted in an increase in fluorescence as compared to those of reaction mixtures containing only the enzyme or the compound. However, no increase in fluorescence was observed for a different potent saccharin-based inhibitor (BSc5195) with a nonfluorescent LG. Average values of two independent experiments are shown. Error bars represent the standard deviation. (B) Purified wild-type GlpG was reacted in vitro with five different saccharins (BSc5205, BSc5156, BSc5195, BSc5196, and BSc5198), and the mass difference between free GlpG and the inhibitor–GlpG adduct was determined by electrospray mass spectrometry. The shift in the m/z value was the same after incubation with all five compounds. This indicated that all compounds inhibited GlpG via the same mechanism, which was expected because the covalently bound core structure was the same and only the LG differed between the compounds. Three independent experiments were performed. (C) Upon incubation of wild-type GlpG with the saccharin BSc5196, a shift in the m/z value corresponding to a covalent double-bonded end product after reaction with the protease was observed. Mutation of the catalytically active Ser201 to threonine or His254 to alanine abolished the difference in m/z observed with the wild-type protein. In contrast, mutation of His150 had no effect and produced a difference in m/z as seen with wild-type GlpG. Three independent experiments were performed, and one representative experiment is shown. (D) Reversibility of the saccharin reaction mechanism was investigated by the rapid dilution method. Saccharin BSc5195 (5 μM), irreversible isocoumarin inhibitor JLK-6 (10 μM), or reversible β-lactam inhibitor L29 (1 μM) was preincubated with GlpG for 1 h at 10 times their IC50 concentrations. The reaction mixtures were then rapidly diluted 100-fold, and a fluorogenic rhomboid substrate peptide (10 μM) was added. No recovery of enzyme activity was observed with JLK-6, while substantial recovery was seen with L29 as expected. With the saccharin inhibitor BSc5195, less but noticeable recovery of GlpG activity was observed over the time period of 100 min, demonstrating partial reversibility of the inhibition mechanism. No recovery of activity was apparent when the saccharin inhibitor concentration was maintained at 5 μM after the dilution step. Three independent experiments were performed, and one representative experiment is shown.
GlpG Enzyme Recovery Assay
Rhomboid activity recovery assays were performed in black 96-well plates. Saccharin inhibitor Bsc5195 (5 μM), β-lactam L29 (1 μM), or isocoumarin JLK-6 (10 μM) was preincubated with 0.4 μM GlpG in reaction buffer [20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, and 0.05% (w/v) DDM] for 1 h. Subsequently, the reaction mixture was rapidly diluted 100-fold into reaction buffer containing 10 μM fluorogenic substrate KSp76.40 This quenched fluorescent peptide is cleaved by the rhomboid, leading to activation of a red fluorophore. Substrate cleavage and enzyme activity recovery were monitored continuously by measuring the fluorescence over 100 min with excitation at 553 nm and emission at 583 nm using a microplate reader (Tecan Infinite M1000).
α-Chymotrypsin in Vitro Activity Assay
To determine the inhibitory potency of saccharin inhibitors against α-chymotrypsin, the compounds were preincubated in a defined range of concentrations (from 0 μM to 1 mM) with 4 μM bovine chymotrypsin (PanReac AppliChem) in reaction buffer [50 mM HEPES-NaOH (pH 7.5), 5 mM EDTA, 0.4 M NaCl, 10% (v/v) glycerol, and 10% DMSO] for 30 min at 25 °C. Then, 2.5 μL of the preincubated reaction mixture was added to 100 μL of 0.12 mM N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Suc-AAPF-pNA, Merck) substrate in reaction buffer. Enzymatic cleavage of the 4-nitroanilide substrate resulted in 4-nitroaniline, which has a yellow color under alkaline conditions.41 The increase in absorbance at 410 nm over 5 min at 25 °C was measured with a microtiter plate reader (Beckman Coulter). IC50 values were calculated by fitting a nonlinear regression curve to a plot of log(compound concentration) versus percent α-chymotrypsin activity using GraphPad Prism.
Rhomboid Activity Panel
Selectivity profiling of rhomboid inhibitors using activity-based probes was performed as described previously.26
Soluble Serine Hydrolase Panel
Selectivity profiles of the saccharin-based inhibitors against human soluble serine hydrolases were determined as described previously.37
RESULTS AND DISCUSSION
Chemical Rationale
To synthesize a panel of enzyme-activated heterocyclic compounds with inhibitory activity against rhomboid proteases, we considered the saccharin (1,2-benzisothiazol-3-one 1,1-dioxide) template. The basic structure of the saccharin heterocyclic ring system consists of an aromatic ring and a sulfimide ring with lactam and sulfonamide moieties. Saccharin-based inhibitors were first described in 1980 as irreversible, acylating compounds of human leukocyte elastase and chymotrypsin.42 The protease’s active-site serine initiates attack at the carbonyl group of the saccharin, which leads to formation of an acyl–enzyme complex and enzyme-induced ring opening (Figure 1B). An important advantage of saccharin-based inhibitors is their ability to differentiate between different serine proteases, which allows a rather broad usage. A possible explanation for this observation is the variable stability of the acyl–enzyme complex that is formed when a specific saccharin inhibitor reacts with the active site of a particular serine protease.24,34
We investigated saccharins with a leaving group (LG) attached to the nitrogen atom of the heterocyclic ring system (so-called next-generation N-methylene-substituted saccharin inhibitors).43–46 Attack of the active-site serine is followed by a prototropic shift, and the release of the leaving group to form a reactive intermediate that cross-links to the active-site nucleophilic histidine residue. Overall, this leads to a cross-linked structure and an irreversible enzyme–inhibitor complex (Figure 1B).24
Modifications to the saccharin scaffold can be introduced as different N-functional LGs or in the heterocyclic ring of the saccharin core. Because derivatization of the N-functional group can be achieved much easier than at the core, we started to identify the most favorable LGs at this position. A consequence of this strategy is that the reaction between the rhomboid protease and the saccharin-based inhibitors with different LGs could result in the same double-bonded enzyme/inhibitor end product (provided that the reaction mechanism with the rhomboid is the same as with the classical, triad-containing serine proteases). We anticipated that binding of a putative inhibitor to the active site of rhomboid proteases would involve the formation of covalent bonds as well as hydrogen bonds and hydrophobic interactions. The final reaction product should feature two covalent bonds, one toward the catalytic Ser and one toward the catalytic His, and would result in the release of the N-functional LG (Figures 1B and 2C).
Figure 2.
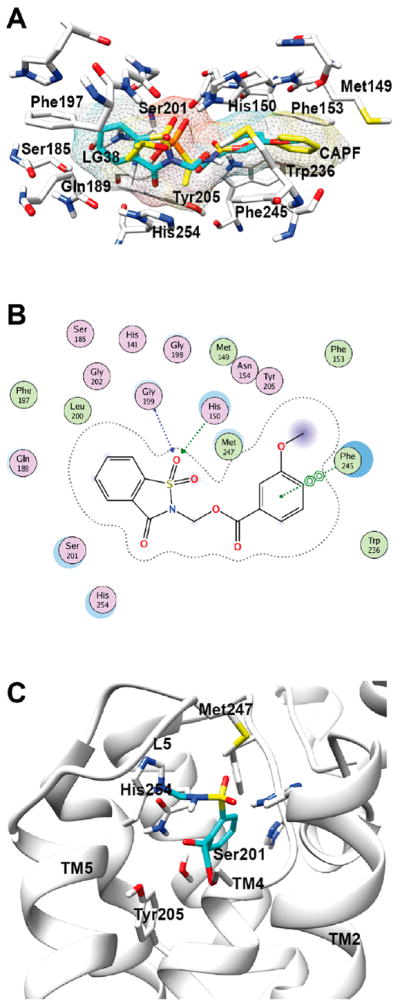
Docking of saccharin-based inhibitors into the active site of the rhomboid protease GlpG. (A) The model shows the initial interactions of a known rhomboid inhibitor (CAPF, yellow) and a saccharin-based inhibitor (with LG 38, turquoise) with amino acid side chains in the active site of GlpG. (B) Ligand interaction map of GlpG and the saccharin with LG38. The map shows the formation of two H-bonds of the sulfoxide moiety of the saccharin with His150 and Gly199. In addition, a π–π interaction between the aryl ring of the saccharin and Phe245 is visible. (C) Molecular model of N-methylated saccharin within the GlpG active site after the enzymatic reaction between the inhibitor (blue) and the rhomboid protease (gray). The model shows the end product based on the proposed reaction mechanism (see Figure 1B). Two covalent bonds have been formed between the saccharin and Ser201 and His254.
Docking Studies
Because our focus was on the effect of different LGs attached to the saccharin core structure, we considered in the docking studies the initial interactions between the inhibitor and the active site of the rhomboid protease rather than the final reaction product. Therefore, a molecular database of 50 saccharin-based serine protease targeting ligands with favorable LGs (Supplementary Table 1) was docked into the active-site cleft of the crystal structure of the E. coli rhomboid protease GlpG.
We used a co-crystal structure of GlpG and the inhibitor CAPF (Protein Data Bank entry 3UBB)47 that is covalently bound to the active-site Ser201 as a template for the docking studies. The receptor (GlpG, 3UBB) was prepared for docking, and the modeling experiments were performed using the DOCK module with the Amber12:EHT force field in the Molecular Operating Environment (MOE).38 The resulting poses for all 50 saccharin inhibitors were scored by London dG and Affinity dG followed by energy minimization within the enzyme active-site cleft. The output database contained the 10 best poses for each inhibitor along with their scoring function (S).
Subsequently, the molecules were ranked on the basis of the calculated ligand efficiencies (cLEs) followed by their scoring function and ligand interactions (Supplementary Table 2). As a result, saccharins with an aryl carboxylic acid LG were identified as the best-suited ligand and termed “hit molecules” (Supplementary Figure 2). Another observation of the docking studies was that the saccharin derivatives interacted with the rhomboid active site similar to CAPF. The sulfoxide moiety of the saccharin scaffold formed hydrogen bonds to His150 and Gly199, which is depicted in the ligand interaction map of GlpG and the saccharin scaffold with LG38 (Figure 2B; see Supplementary Figure 3 for a ligand interaction map of GlpG with CAPF). Another striking similarity between CAPF and the saccharin derivatives was the occupancy of the S region by the saccharin core ring and the LG extending toward the S′ region of GlpG. This observation together with significant π–π interactions between the aryl ring of LG38 and Phe245 of the rhomboid GlpG led us to propose that the LG might play a major role in the recognition process of the inhibitor and enzyme (Figure 2A).
Synthesis and Test of the First Compound Batch
Calculations from the docking experiments revealed that saccharins with aryl carboxylic acid LGs showed the highest cLE along with saccharins that had aryl sulfides and sulfones as LGs (Supplementary Figure 2 and Supplementary Table 2). Subsequently, we synthesized a preliminary batch of 10 N-methylene saccharin derivatives with different LGs (Supplementary Table 3; for details of the synthesis, see Supplementary Methods and Supplementary Figure 1) to evaluate their inhibitory potential in an established in vitro rhomboid activity assay.21,27,39
The rhomboid protease GlpG from E. coli is an extremely well characterized serine intramembrane protease, both biochemically and structurally.16,28,48–50 GlpG also served as a standard model for the development of new rhomboid inhibitors and provided new insights into substrate recognition processes and related mechanistic details.23,27,30,51 Interestingly, no natural substrates have been described for GlpG, but its sequence preferences for cleavage are well-known.30 We therefore used transmembrane domain 2 of the Drosophila protein Gurken as a substrate for the cleavage assays.39,52 GlpG and Gurken were expressed and purified as described elsewhere (see the Supporting Information and ref 39). To evaluate the preliminary batch of saccharin inhibitors, GlpG was preincubated with each compound at concentrations ranging from 1 to 250 μM for 30 min at 37 °C prior to the addition of the Gurken substrate. Saccharins with aryl carboxylic acid LGs were able to inhibit GlpG at a concentration of 10 μM, while saccharins with aryl sulfide and sulfone LGs were inactive at 250 μM. These results were consistent with our findings from the docking experiments, which indicated that carboxylic acid LGs might optimally target the rhomboid protease GlpG. The docking studies showed that the aryl ring contacted Phe245 via π–π interactions and fitted nicely into a hydrophobic region within the GlpG active-site cleft (Figure 2A). Analysis of electrostatic interactions revealed that hydrophobic extensions of a LG in the para position might be favorable, whereas a LG substituted at the ortho position was engaged in proton donor/acceptor interactions. In addition, strong electron-withdrawing groups could activate the saccharin core structure for an attack by the active-site Ser201. Substitutions with small extensions were possible without steric clashes or external exposure at the para and ortho positions to optimize the contact between the inhibitor and enzyme as well as activating the inhibitor. Consequently, N-methylated saccharins with an aryl carboxylic acid LG were selected as “lead molecules”.
Synthesis and Test of the Second Compound Batch
To further examine the most suitable extensions on the aryl ring of the LG and the influence on the electron-withdrawing properties of the LG, we performed additional docking experiments with a database of 103 saccharin derivatives. Interestingly, the docking experiments did not reveal any significantly improved interactions between the aryl ring substituents and their neighboring residues within the active site of GlpG. However, to analyze how substitutions of the aryl acid moiety with different electron donor/acceptor groups would affect the inhibitory potency, 20 N-methylated saccharin derivatives (Supplementary Table 4) were synthesized using synthesis scheme 3 (for details, see Supplementary Methods and Supplementary Figure 1), and their biological activity was determined via the in vitro GlpG cleavage assay.
All aryl acid saccharin derivatives were able to inhibit GlpG in the in vitro activity assay at 250 μM, and compounds that showed activity at 10 μM were further selected for determination of IC50 values. First, to benchmark the GlpG in vitro activity assay with known rhomboid inhibitors, the IC50 values for the β-lactam inhibitor L16 and the isocoumarin DCI were determined and found to be 0.9 and 19 μM, respectively (Table 1), which were comparable to literature data.21,27 Subsequent experiments identified 12 saccharin derivatives with IC50 values of <1 μM (Figure 3A and Table 1). The aryl acid derivatives substituted with electron-withdrawing groups such as halogens at the ortho position (BSc5188, BSc5195, and BSc5197) had IC50 values of <1 μM, while an inhibitor substituted with an electron-donating group such as -NH2 (BSc5193) was unable to completely inhibit GlpG at 10 μM. Therefore, electron-withdrawing groups at the ortho position of the aryl carboxylic acid LG appeared to increase the inhibitor potency. However, with this limited range of derivatives, it was not possible to definitively conclude how the electronwithdrawing properties of the LG affected the inhibitor potency. The IC50 value of the most potent compound that evolved from our screen was determined to be 200 nM (Figure 3A and Table 1).
Table 1.
Overview of Selected Saccharin-Based Inhibitors with Different Leaving Groups and IC50 Values of <10 μM in the E. coli GlpG in Vitro Activity Assaya
| ID | Structure | IC50 [μM] GlpG |
IC50 [μM] α- chymo- trypsin |
EC50 [μM] E. coli in vivo |
ID | Structure | IC50 [μM] GlpG |
IC50 [μM] α- chymo- trypsin |
EC50 [μM] E. coli in vivo |
|---|---|---|---|---|---|---|---|---|---|
| DCI |

|
19 | 3.5 | n.d. | BSc5195 |

|
0.20 | 10 | 0.28 |
| L16 |

|
0.9 | n.d. | n.d. | BSc5196 |

|
0.56 | 208 | 1.2 |
| BSc5156 |
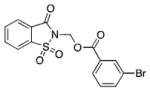
|
0.61 | 33 | 0.99 | BSc5197 |
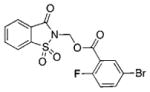
|
0.64 | 68 | n.d. |
| BSc5187 |

|
0.54 | 50 | n.d. | BSc5198 |

|
0.80 | 56 | 0.27 |
| BSc5188 |

|
0.36 | 9 | n.d. | BSc5199 |

|
>10 | 39 | n.d. |
| BSc5189 |

|
>10 | 29 | n.d. | BSc5200 |

|
>10 | 120 | n.d. |
| BSc5190 |

|
>10 | 27 | n.d. | BSc5202 |

|
1.26 | n.d. | n.d. |
| BSc5191 |

|
>10 | 14 | n.d. | BSc5203 |

|
0.33 | 40 | n.d. |
| BSc5192 |

|
0.78 | 22 | n.d. | BSc5204 |

|
0.41 | 12 | n.d. |
| BSc5193 |

|
>10 | 54 | n.d. | BSc5205 |

|
0.61 | 18 | 0.23 |
| BSc5194 |

|
0.64 | 19 | n.d. | CAPF |

|
~50 | n.d. | n.d. |
The IC50 of CAPF for GlpG is a value from the literature.47
Figure 3.

(A) Evaluation of saccharin inhibitors in the GlpG in vitro activity assay. The E. coli rhomboid GlpG was recombinantly expressed and preincubated with each saccharin inhibitor at concentrations ranging from 0.1 to 250 μM for 30 min at 37 °C prior to the addition of the Gurken substrate. The SDS–PAGE gel shows the reaction products at the end of a 90 min incubation period. The arrows indicate the full-length Gurken substrate (S, ~66 kDa) and the N-terminal Gurken cleavage product (P, ~55 kDa). Signal intensities of the N-terminal Gurken product were normalized to the DMSO control condition and plotted against log(inhibitor concentration). A nonlinear regression curve fit of the log(inhibitor concentration) vs percent N-terminal Gurken product was used to determine IC50 values. The SDS–PAGE gel was stained with Coomassie Brilliant Blue and quantified with ImageJ, and the values were plotted in GraphPad Prism. Two independent IC50 determinations were performed for each compound, and one representative experiment is shown. (B) In vivo inhibition of GlpG in live E. coli cells. Wild-type strain NR698 with endogenous GlpG activity53 or GlpG-deficient strain NR698 ΔglpG57 was transformed with the MBP-FLAG-LacYTM2-Trx substrate.39,57 Increasing concentrations of the indicated saccharin inhibitors from 5 nM to 100 μM were added to the culture media; substrate expression was induced with 1 mM L-rhamnose, and cells were grown in the presence of the inhibitors for 4 h. Cleavage fragments of the substrate (P, ~46 kDa) were quantified by SDS–PAGE and near-infrared Western blotting of cell lysates, and IC50 values were determined as described previously.57 Note that in the GlpG-deficient cells the substrate (S) was not processed and no cleavage fragment was produced (lane KO). Two independent IC50 determinations were performed for each compound, and one representative experiment is shown.
Effectiveness of Saccharins against Rhomboid Embedded in the Lipid Membrane
Until now, we have evaluated the activity of the saccharin-based inhibitors only in the GlpG in vitro cleavage assay with purified enzyme and substrate solubilized in detergent micelles. In this assay, the enzyme active site is principally more accessible to incoming compounds than in the native environment of rhomboids, which is the lipid membrane. We therefore chose to evaluate four potent compounds in an in vivo system of E. coli.57 The LacYTM2-based chimeric substrate was expressed in E. coli in the presence of increasing concentrations of rhomboid inhibitors, and substrate cleavage was assessed by SDS–PAGE and quantitative Western blotting. Compounds BSc5195, BSc5196, BSc5198, and BSc5205 inhibited the processing of LacYTM2 by GlpG in living cells with IC50 values comparable to those obtained in the in vitro cleavage assay with purified enzyme and substrate (Figure 3B and Table 1).
Mechanism of Saccharin-Based Inhibitors
Our saccharin-based inhibitors were supposed to react with the nucleophilic side chain of the catalytic serine of a serine protease (Ser201 in the case of the rhomboid protease GlpG).24 The resulting electronic rearrangements should then lead to the release of the acidic leaving group. Compound BSc5193 is an N-methylated saccharin derivative with the fluorescent anthranilic acid (o-aminobenzoic acid) as a LG. Indeed, incubation of BSc5193 with the rhomboid protease GlpG caused an increase in fluorescence emission at 400 nm, while there was no fluorescence increase with compound BSc5195, which was used as a negative control (Figure 4A). This supported the idea that the reaction mechanism of N-methylated saccharin-based acid derivatives involved the release of the acidic leaving group.
On the basis of the reaction mechanism of saccharins with classical soluble serine proteases,24 we assumed that after release of the LG the generated Michael acceptor would further react with a nucleophilic residue in its immediate vicinity to form a double-bonded covalent end product (Figure 1B). In the direct vicinity of the catalytic Ser201, two histidine residues could possibly be modified by the saccharin inhibitors: His150 and His254. Alternatively, the Michael acceptor might further react with water, leaving only the catalytic serine modified by the saccharin. To differentiate between these possibilities, we reacted purified wild-type GlpG in vitro with five different saccharin inhibitors and studied the difference in mass between free GlpG and the inhibitor–GlpG adduct by electrospray mass spectrometry. All compounds (BSc5156, BSc5195, BSc5196, BSc5198, and BSc5205) generated identical mass shifts of 195 Da, which matched well the theoretical mass difference of 196 Da that would be expected from the reaction of a saccharin with a classical serine protease (Figure 4B). Next, we expressed and purified mutants of GlpG, in which either Ser201 was changed to threonine (S201T) or His150 and His254 were changed to alanine (H150A and H254A, respectively). Again, these GlpG mutants were examined alone or after incubation with saccharin inhibitor BSc5196 by mass spectrometry. BSc5196 was able to bind wild-type GlpG and the H150A mutant, but not the S201T, H254A, or H150A/H254A mutants (Figure 4C).
Overall, the mass spectrometry data were consistent with a reaction mechanism in which the saccharins would modify both the catalytic Ser201 and the catalytic His254, resulting in an end product that forms a covalent cross-link in the enzyme. Nevertheless, we did not consider these data fully conclusive for two reasons. First, in the catalytic Ser201-His254 dyad, the histidine is indispensable for proper activation of the serine.54 Consequently, it is highly likely that in the H254A mutant Ser201 is not nucleophilic enough to react with the carbonyl of the saccharin, providing an alternative explanation for why the saccharin failed to bind this mutant. Second, Ser201 and His254 are located on different transmembrane domains, and earlier work had demonstrated that cross-linking the active-site dyad with the isocoumarin inhibitor JLK-6 changed the mobility of the GlpG–inhibitor complex on a SDS–PAGE gel in comparison to that of unmodified GlpG.23,30 However, while we could easily reproduce the mobility shift with JLK-6, we did not observe a change in the apparent molecular weight of the rhomboid on SDS–PAGE gels after in vitro reaction with any of our saccharin inhibitors (data not shown). One possible explanation could be that the Michael acceptor did not react with His254 but with another nucleophilic residue closer to Ser201 on the same transmembrane domain, facilitating a crosslink that did not result in a conformational change and a corresponding mobility shift. Modeling the spatial proximity of the Michael acceptor to neighboring nucleophilic residues indicated that, aside from His254 (4.9 Å from the Michael acceptor carbon) and His150 (6.4 Å), Tyr205 (3.8 Å) was close in distance, whereas Trp157 and Trp236 were more distant. Other explanations for the lack of a gel shift could be that the saccharin-based inhibitors modified only the catalytically active serine and did not result in an intramolecular cross-link, or that in the double-bonded (cross-linked) end product the bond to the catalytic serine was deacylated, reverting to a single-bonded species (via the Michael acceptor). However, for the latter explanation, mass spectrometry analysis did not provide direct evidence, as it should have resulted in a mass shift of 214 kDa. We explored the possible hydrolysis of the Ser201–saccharin ester bond further by investigating the reversibility of covalent binding over time using the rapid dilution method and a previously described fluorogenic peptidic rhomboid sub-strate. 40,55 The inhibitors were preincubated with GlpG for 1 h at 10 times their IC50 concentrations. The reaction mixtures were then rapidly diluted 100-fold, and the substrate peptide was added. Processing of this substrate by the rhomboid releases a quencher peptide and activates a red fluorophore, which indicates recovery of enzyme activity. When the assay was performed with the irreversible inhibitor JLK-6, which forms a double-bonded end product with the rhomboid,23,30 no recovery of enzyme activity was observed (Figure 4D). Conversely, with the β-lactam L29, a known reversible inhibitor of rhomboids,27 substantial recovery of activity was evident over a time period of 100 min. With the saccharin inhibitor BSc5195, less but noticeable recovery of GlpG activity was observed, demonstrating partial reversibility of the inhibition mechanism (Figure 4D). Taken together, these data do not easily conform to the reaction mechanism of saccharins with classical soluble serine proteases. Instead, they point to a heterogeneous mechanism in which the saccharin-based inhibitors might form a double-bonded end product by cross-linking Ser201 with another spatially close nucleophilic residue, or might modify only Ser201 because of the low reactivity of the Michael acceptor. This may be followed by slow hydrolysis of the formed ester bond, explaining the partial reversibility of the reaction at low inhibitor concentrations.
Selectivity for Rhomboid Proteases
The saccharin inhibitors were designed to fit into the active-site cleft of the E. coli rhomboid protease GlpG. However, we were further interested in knowing whether the compounds would inhibit other rhomboid proteases. To address this issue, we expressed and purified the rhomboid protease from A. aeolicus (AaROM) and performed an in vitro cleavage assay with AaROM and the Gurken substrate in the presence of the three most potent saccharin derivatives (BSc5188, BSc5195, and BSc5204). All three compounds were able to inhibit the AaROM activity with IC50 values between 60 and 90 μM, indicating an approximately 100-fold lower potency toward AaROM in comparison to that of GlpG (Supplementary Table 5). Next, to investigate whether the saccharin-based compounds might also inhibit the enzymatic activity of a more distant member of the rhomboid protease family, a previously established cell-based assay for murine rhomboid RHBDL2 was employed.27,56 Human HEK293T cells were transiently co-transfected with a HA-tagged RHBDL2 construct and a FLAG-tagged Gurken substrate, and three saccharins with IC50 values of <1 μM (BSc5156, BSc5205, and BSc5192) were added to the cell culture media at concentrations of ≤250 μM. Forty-eight hours after the transfection, cell lysates were prepared and Gurken cleavage fragments were detected by Western blotting. The saccharin-based compounds displayed no inhibitory effect on the processing of Gurken by RHBDL2 (Supplementary Figure 4).
To further investigate the selectivity of the saccharin-based inhibitors against different rhomboids, we took advantage of a recently described activity-based probe (ABP) competition assay.23,36 In this assay, small molecule ABPs were used to specifically label the active site of recombinantly expressed and proteolytically active rhomboids. One such ABP is the commercially available TAMRA-FP serine hydrolase probe, which allows the visualization of the labeled enzyme by SDS–PAGE and fluorescence scanning. This fluorophosphonatederived compound binds to the active site of rhomboid proteases and covalently modifies the catalytic serine. However, if after preincubation of the rhomboids with a saccharin-based inhibitor the catalytic site of the enzyme is already modified, the fluorescent ABP would not be able to bind and the fluorescent signal would be reduced. In total, nine different rhomboids from all three phylogenetic trees (archaea, bacteria, and eukaryota) were selected and screened against three potent saccharin-based inhibitors (BSc5195, BSc5196, and BSc5205) and the nonspecific pan-rhomboid inhibitor DCI as a positive control. All compounds were used at a final concentration of 50 μM and were preincubated with the rhomboids before being labeled with the ABP. The results are shown in a heat map to visualize the selectivity of the saccharin-based inhibitors. As expected, the isocoumarin DCI inhibited around 80–100% of the activity of all nine rhomboids (Figure 5A). Interestingly, the saccharin-based inhibitors were active against only four bacterial rhomboids (E. coli GlpG, Haemophilus inf luenzae HiGlpG, Vibrio cholerae VcROM, and Providencia stuartii PsAarA) and one archaeal rhomboid (Methanocaldococcus jannaschii MjROM). Two other bacterial rhomboids (A. aeolicus AaROM and Thermotoga maritima TmROM) were only partially inhibited, whereas two eukaryotic rhomboids (Drosophila melanogaster DmRho1 and Mus musculus RHBDL3) were completely spared (Figure 5A). These results are fully consistent with the low potency of the saccharin inhibitors in the AaROM in vitro activity assay (Supplementary Table 5) and the lack of an effect in the cell-based assay for eukaryotic murine rhomboid RHBDL2 (Supplementary Figure 4). Taken together, these data indicated that the saccharin-based inhibitors, in contrast to DCI, did not indiscriminately inhibit all rhomboids but were more specific and might have a preference for the evolutionarily older bacterial and archaeal rhomboid proteases.
Figure 5.

Selectivity of saccharin-based inhibitors. (A) Heat map representation of the activity-based probe (ABP) competition assay with three potent saccharin inhibitors against nine different rhomboid proteases. Recombinantly expressed rhomboids were preincubated with saccharins (50 μM), labeled with the ABP TAMRA-FP, and the labeled enzymes were visualized by SDS–PAGE and fluorescence scanning. The isocoumarin DCI was used as a positive control and inhibited around 80–100% of the activity of all nine rhomboids. Two independent experiments were performed, and average values for each experimental condition are shown. (B) A high-throughput activity-based probe competition assay was used to profile saccharins against human soluble serine hydrolases. Isocoumarins S006 and S016 inhibited multiple human serine hydrolases. In contrast, at concentrations sufficient to inhibit the rhomboid GlpG in the in vitro activity assay, saccharin-based compounds BSc5195, BSc5196, and BSc5205 showed far less activity against the panel of human serine hydrolases (no inhibition, white; weak inhibition, gray to light blue; strong inhibition, dark blue). Two independent experiments were performed, and one representative experiment is shown.
Selectivity over Classical Serine Proteases
A common problem of inhibitors against broadly distributed enzymes such as proteases is off-target activity. In addition to finding more potent rhomboid inhibitors, we sought to identify more specific inhibitors with a clear preference for rhomboids over soluble serine proteases. Therefore, we first investigated the potency of 18 aryl acid-substituted saccharins against the well-characterized soluble serine protease α-chymotrypsin in an in vitro assay.41 α-Chymotrypsin was preincubated with saccharins for 30 min at 25 °C prior to the addition of the substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide. This substrate is processed by α-chymotrypsin under alkaline conditions to yield p-nitroaniline, which can be detected spectroscopically at 410 nm. The positive control DCI inhibited α-chymotrypsin with an IC50 value of 3.5 μM, which is close to the value reported in the literature (2.8 μM).27 The IC50 values of saccharins against α-chymotrypsin were 20–370-fold higher than the IC50 values for inhibition of GlpG in the in vitro activity assay (Table 1). This indicated that the saccharin-based inhibitors offered an at least 20-fold window of selectivity for the inhibition of the rhomboid GlpG over the soluble serine protease α-chymotrypsin. However, it is important to note that the IC50 values for α-chymotrypsin and GlpG were determined in different assays and are not directly comparable. In addition, a more exhaustive specificity profiling against 71 serine hydrolases using a high-throughput activity-based probe competition assay37 showed that, while two isocoumarins S006 and S016 inhibited more than a dozen other human serine hydrolases, saccharins BSc5195, BSc5196, and BSc5205 displayed appreciable inhibition of only a few enzymes [e.g., neutrophil elastase, ELA2 (see also Supplementary Figure 5)] in the same concentration range, in which they inhibited the rhomboid GlpG (Figure 5B). Overall, these experiments led us to conclude that N-methylene saccharin-derived compounds are potent inhibitors of a subset of rhomboids with selectivity far better than that of isocoumarins.
CONCLUSIONS
In this study, we have described the computer-aided discovery and biological characterization of a novel and improved class of partially reversible inhibitors of rhomboid proteases. In comparison to previously known rhomboid inhibitors, the N-methylene saccharins presented here have increased potency and selectivity, and a certain preference for archaeal and bacterial rhomboids. Further structural studies will be needed to determine how saccharins might be able to distinguish between different rhomboids. Importantly, N-methylene saccharin-based rhomboid protease inhibitors are easily amenable to derivatization and have a clear potential for further development.
Supplementary Material
Acknowledgments
Funding
P.G. was supported by a scholarship of the iGRASPseed graduate school of the Heinrich-Heine-University Duesseldorf. K.S. was a recipient of Purkyne Fellowship of the Academy of Sciences of the Czech Republic and also acknowledges support from EMBO (Installation Grant 2329), Ministry of Education, Youth and Sports of the Czech Republic (Projects LK11206 and LO1302), a Marie Curie Career Integration Grant (Project 304154), and the National Subvention for Development of Research Organisations (RVO, 61388963) to the Institute of Organic Chemistry and Biochemistry. D.A.B. was supported by the Josie Robertson Foundation and MSKCC Core Grant P30 CA008748 and D.C.J. by National Institutes of Health Grant T32 GM115327-Tan. M.T.N.N. and S.V. were supported by the Deutsche Forschungsgemeinschaft, the Ministerium für Innovation, Wissenschaft and Forschung des Landes Nordrhein-Westfalen, the Senatsverwaltung für Wirtschaft, Technologie and Forschung des Landes Berlin, and the Bundesministerium für Bildung and Forschung.
The authors thank Professors Matthew Freeman (University of Oxford), Sinisa Urban (Johns Hopkins University), and Ya Ha (Yale School of Medicine) for various expression constructs.
Footnotes
Author Contributions
P.G. and T.J. contributed equally to this work.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem. 7b01066.
Five supplementary figures showing chemical synthesis schemes, docking data, a cell-based activity assay for murine rhomboid RHBDL2, and an in vitro activity assay for human neutrophil elastase; five tables displaying chemical structures and summarizing IC50 values of saccharin inhibitors in various biological assays; and detailed synthesis and analytical information about saccharin inhibitors (PDF)
References
- 1.Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 2.Rawlings ND, Barrett AJ. Evolutionary families of peptidases. Biochem J. 1993;290(1):205–218. doi: 10.1042/bj2900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 4.Krane SM. Elucidation of the potential roles of matrix metalloproteinases in skeletal biology. Arthritis Res Ther. 2003;5:2–4. doi: 10.1186/ar600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lichtenthaler SF, Haass C, Steiner H. Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J Neurochem. 2011;117:779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- 6.Luttun A, Dewerchin M, Collen D, Carmeliet P. The role of proteinases in angiogenesis, heart development, restenosis, atherosclerosis, myocardial ischemia, and stroke: insights from genetic studies. Curr Atheroscler Rep. 2000;2:407–416. doi: 10.1007/s11883-000-0079-z. [DOI] [PubMed] [Google Scholar]
- 7.Mittl PR, Grutter MG. Opportunities for structure-based design of protease-directed drugs. Curr Opin Struct Biol. 2006;16:769–775. doi: 10.1016/j.sbi.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 8.Siefert SA, Sarkar R. Matrix metalloproteinases in vascular physiology and disease. Vascular. 2012;20:210–216. doi: 10.1258/vasc.2011.201202. [DOI] [PubMed] [Google Scholar]
- 9.Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discovery. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 10.Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, Makarova KS, Rogozin IB, Davidovic L, Letellier MC, Pellegrini L. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4:R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urban S. Making the cut: central roles of intra-membrane proteolysis in pathogenic microorganisms. Nat Rev Microbiol. 2009;7:411–423. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Storer AC, Menard R. Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol. 1994;244:486–500. doi: 10.1016/0076-6879(94)44035-2. [DOI] [PubMed] [Google Scholar]
- 14.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 15.Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005;24:464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 17.Freeman M. Rhomboids: 7 years of a new protease family. Semin Cell Dev Biol. 2009;20:231–239. doi: 10.1016/j.semcdb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Freeman M. The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu Rev Cell Dev Biol. 2014;30:235–254. doi: 10.1146/annurev-cellbio-100913-012944. [DOI] [PubMed] [Google Scholar]
- 19.Strisovsky K. Structural and mechanistic principles of intramembrane proteolysis–lessons from rhomboids. FEBS J. 2013;280:1579–1603. doi: 10.1111/febs.12199. [DOI] [PubMed] [Google Scholar]
- 20.Strisovsky K. Rhomboid protease inhibitors: Emerging tools and future therapeutics. Semin Cell Dev Biol. 2016;60:52–62. doi: 10.1016/j.semcdb.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci U S A. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 2010;29:3797–3809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vosyka O, Vinothkumar KR, Wolf EV, Brouwer AJ, Liskamp RM, Verhelst SH. Activity-based probes for rhomboid proteases discovered in a mass spectrometry-based assay. Proc Natl Acad Sci U S A. 2013;110:2472–2477. doi: 10.1073/pnas.1215076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 25.Powers JC, Kam CM, Narasimhan L, Oleksyszyn J, Hernandez MA, Ueda T. Mechanism-based isocoumarin inhibitors for serine proteases: use of active site structure and substrate specificity in inhibitor design. J Cell Biochem. 1989;39:33–46. doi: 10.1002/jcb.240390105. [DOI] [PubMed] [Google Scholar]
- 26.Wolf EV, Zeissler A, Verhelst SH. Inhibitor Fingerprinting of Rhomboid Proteases by Activity-Based Protein Profiling Reveals Inhibitor Selectivity and Rhomboid Autoprocessing. ACS Chem Biol. 2015;10:2325–2333. doi: 10.1021/acschembio.5b00514. [DOI] [PubMed] [Google Scholar]
- 27.Pierrat OA, Strisovsky K, Christova Y, Large J, Ansell K, Bouloc N, Smiljanic E, Freeman M. Monocyclic beta-lactams are selective, mechanism-based inhibitors of rhomboid intramembrane proteases. ACS Chem Biol. 2011;6:325–335. doi: 10.1021/cb100314y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue Y, Ha Y. Catalytic mechanism of rhomboid protease GlpG probed by 3,4-dichloroisocoumarin and diisopropyl fluorophosphonate. J Biol Chem. 2012;287:3099–3107. doi: 10.1074/jbc.M111.310482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf EV, Zeissler A, Vosyka O, Zeiler E, Sieber S, Verhelst SH. A new class of rhomboid protease inhibitors discovered by activity-based fluorescence polarization. PLoS One. 2013;8:e72307. doi: 10.1371/journal.pone.0072307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoll S, Stanchev S, Began J, Skerle J, Lepsik M, Peclinovska L, Majer P, Strisovsky K. Substrate binding and specificity of rhomboid intramembrane protease revealed by substrate-peptide complex structures. EMBO J. 2014;33:2408–2421. doi: 10.15252/embj.201489367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho S, Dickey SW, Urban S. Crystal Structures and Inhibition Kinetics Reveal a Two-Stage Catalytic Mechanism with Drug Design Implications for Rhomboid Proteolysis. Mol Cell. 2016;61:329–340. doi: 10.1016/j.molcel.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hlasta DJ, Ackerman JH, Court JJ, Farrell RP, Johnson JA, Kofron JL, Robinson DT, Talomie TG, Dunlap RP, Franke CA. A novel class of cyclic beta-dicarbonyl leaving groups and their use in the design of benzisothiazolone human leukocyte elastase inhibitors. J Med Chem. 1995;38:4687–4692. doi: 10.1021/jm00023a008. [DOI] [PubMed] [Google Scholar]
- 33.Walker B, Lynas JF. Strategies for the inhibition of serine proteases. Cell Mol Life Sci. 2001;58:596–624. doi: 10.1007/PL00000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong J, Groutas WC. Recent developments in the design of mechanism-based and alternate substrate inhibitors of serine proteases. Curr Top Med Chem. 2004;4:1203–1216. doi: 10.2174/1568026043387971. [DOI] [PubMed] [Google Scholar]
- 35.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discovery Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf EV, Seybold M, Hadravova R, Strisovsky K, Verhelst SH. Activity-Based Protein Profiling of Rhomboid Proteases in Liposomes. ChemBioChem. 2015;16:1616–1621. doi: 10.1002/cbic.201500213. [DOI] [PubMed] [Google Scholar]
- 37.Bachovchin DA, Koblan LW, Wu W, Liu Y, Li Y, Zhao P, Woznica I, Shu Y, Lai JH, Poplawski SE, Kiritsy CP, Healey SE, DiMare M, Sanford DG, Munford RS, Bachovchin WW, Golub TR. A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat Chem Biol. 2014;10:656–663. doi: 10.1038/nchembio.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molecular Operating Environment. 2013 http://github.com/Yelp/MOE.
- 39.Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36:1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ticha A, Stanchev S, Skerle J, Began J, Ingr M, Svehlova K, Polovinkin L, Ruzicka M, Bednarova L, Hadravova R, Polachova E, Rampirova P, Brezinova J, Kasicka V, Majer P, Strisovsky K. Sensitive Versatile Fluorogenic Trans-membrane Peptide Substrates for Rhomboid Intramembrane Proteases. J Biol Chem. 2017;292:2703–2713. doi: 10.1074/jbc.M116.762849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DelMar EG, Largman C, Brodrick JW, Geokas MC. A sensitive new substrate for chymotrypsin. Anal Biochem. 1979;99:316–320. doi: 10.1016/s0003-2697(79)80013-5. [DOI] [PubMed] [Google Scholar]
- 42.Zimmerman M, Morman H, Mulvey D, Jones H, Frankshun R, Ashe BM. Inhibition of elastase and other serine proteases by heterocyclic acylating agents. J Biol Chem. 1980;255:9848–9851. [PubMed] [Google Scholar]
- 43.Groutas WC, Brubaker MJ, Venkataraman R, Epp JB, Stanga MA, McClenahan JJ. Inhibitors of human neutrophil cathepsin G: structural and biochemical studies. Arch Biochem Biophys. 1992;294:144–146. doi: 10.1016/0003-9861(92)90148-p. [DOI] [PubMed] [Google Scholar]
- 44.Groutas WC, Chong LS, Venkataraman R, Kuang R, Epp JB, Houser-Archield N, Huang H, Hoidal JR. Amino acid-derived phthalimide and saccharin derivatives as inhibitors of human leukocyte elastase, cathepsin G, and proteinase 3. Arch Biochem Biophys. 1996;332:335–340. doi: 10.1006/abbi.1996.0350. [DOI] [PubMed] [Google Scholar]
- 45.Groutas WC, Epp JB, Venkataraman R, Kuang R, My Truong T, McClenahan JJ, Prakash O. Design, synthesis, and in vitro inhibitory activity toward human leukocyte elastase, cathepsin G, and proteinase 3 of saccharin-derived sulfones and congeners. Bioorg Med Chem. 1996;4:1393–1400. doi: 10.1016/0968-0896(96)00133-2. [DOI] [PubMed] [Google Scholar]
- 46.Groutas WC, Kuang R, Ruan S, Epp JB, Venkataraman R, Truong TM. Potent and specific inhibition of human leukocyte elastase, cathepsin G and proteinase 3 by sulfone derivatives employing the 1,2,5-thiadiazolidin-3-one 1,1 dioxide scaffold. Bioorg Med Chem. 1998;6:661–671. doi: 10.1016/s0968-0896(98)00006-6. [DOI] [PubMed] [Google Scholar]
- 47.Xue Y, Chowdhury S, Liu X, Akiyama Y, Ellman J, Ha Y. Conformational change in rhomboid protease GlpG induced by inhibitor binding to its S′ subsites. Biochemistry. 2012;51:3723–3731. doi: 10.1021/bi300368b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker RP, Urban S. Cytosolic extensions directly regulate a rhomboid protease by modulating substrate gating. Nature. 2015;523:101–105. doi: 10.1038/nature14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickey SW, Baker RP, Cho S, Urban S. Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity. Cell. 2013;155:1270–1281. doi: 10.1016/j.cell.2013.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vinothkumar KR, Pierrat OA, Large JM, Freeman M. Structure of rhomboid protease in complex with beta-lactam inhibitors defines the S2′ cavity. Structure. 2013;21:1051–1058. doi: 10.1016/j.str.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- 53.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell. 2005;121:307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 54.Ha Y, Akiyama Y, Xue Y. Structure and mechanism of rhomboid protease. J Biol Chem. 2013;288:15430–15436. doi: 10.1074/jbc.R112.422378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harper JW, Hemmi K, Powers JC. Reaction of serine proteases with substituted isocoumarins: discovery of 3,4-dichloroisocoumarin, a new general mechanism based serine protease inhibitor. Biochemistry. 1985;24:1831–1841. doi: 10.1021/bi00329a005. [DOI] [PubMed] [Google Scholar]
- 56.Adrain C, Strisovsky K, Zettl M, Hu L, Lemberg MK, Freeman M. Mammalian EGF receptor activation by the rhomboid protease RHBDL2. EMBO Rep. 2011;12:421–427. doi: 10.1038/embor.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ticha A, Stanchev S, Vinothkumar KR, Mikles DC, Pachl P, Began J, Skerle J, Svehlova K, Nguyen MTN, Verhelst SHL, et al. General and modular strategy for designing potent, selective and pharmacologically compliant inhibitors of rhomboid proteases. Cell Chem Biol. 2017;24:1–14. doi: 10.1016/j.chembiol.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.