

Abstract
Thiamin thiazole biosynthesis in eukaryotes is still not completely understood. In this report, a late intermediate, tightly bound to the active site of the Saccharomyces cerevisiae thiazole synthase, was identified as an adenylated thiazole tautomer. The reactivity of this unusual compound was evaluated. Its identification provides an additional molecular snapshot of the complex reaction sequence catalyzed by the eukaryotic thiazole synthase and identifies the final step of the thiamin-thiazole biosynthesis.
Introduction
Thiamin pyrophosphate is an essential cofactor in all living systems where it plays a key role in amino acid and carbohydrate metabolism.1 Prokaryotes and some eukaryotes (plants and fungi) can biosynthesize this cofactor, whereas other higher eukaryotes (mammals) are unable to do so and require it in their diet as thiamin (vitamin B1). Thiamin consists of a thiazole linked to a pyrimidine. These heterocycles are biosynthesized separately and then coupled.2 The mechanism of thiazole biosynthesis in prokaryotes is now relatively well understood and requires five proteins to catalyze the assembly of the thiazole by a complex oxidative condensation.3,4 In eukaryotes, a different pathway leads to the thiamin-thiazole. The mechanistic details of this pathway are only beginning to emerge (Figure 1a).5–7
Figure 1.
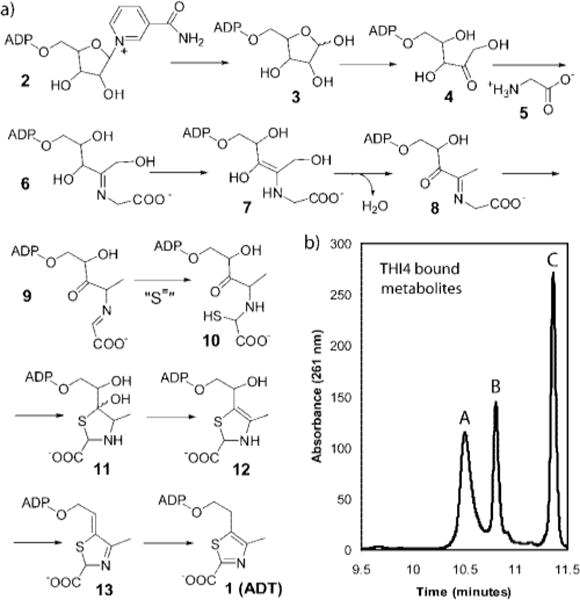
(a) A mechanistic proposal for the THI4p catalyzed biosynthesis of ADT 1. The sulfur source, indicated by ”S=” has not yet been identified. (b) HPLC analysis of the three major metabolites that copurify with THI4p.
Genetic studies in Saccharomyces cereVisiae have demonstrated that THI4 is the only gene required for thiamin thiazole formation.8 The THI4p protein, overexpressed in Escherichia coli, copurified with three adenylated metabolites (peaks A, B and C in Figure 1b). One of these (peak C) was isolated and identified as the adenylated thiazole 1 (ADT).7 The unanticipated structure of this molecule suggested that the thiamin thiazole is biosynthesized from NAD 2. This hypothesis could not be tested initially because overexpressed THI4p was inactive. However, two mutants of THI4p (H200N and C204A) showed partial activity and catalyzed the conversion of NAD and glycine to the Peak A compound via the intermediacy of ADP-ribose 3 and ADP-ribulose 4.5 The peak A compound was identified as 8, following its conversion to a stable quinoxaline derivative, by trapping with ortho-phenylenediamine.5 A mechanistic hypothesis for the THI4p-catalyzed reaction, consistent with these observations, is outlined in Figure 1a. While these results clarified the early steps in the thiamin-thiazole assembly catalyzed by THI4p, the later steps in the process still lacked experimental validation. Here we report the characterization of a labile THI4p bound metabolite that represents a late inter-mediate (13 in Figure 1) in the biosynthetic pathway and corresponds to the earlier reported “peak B” (Figure 1b). The unexpected reactivity of this molecule clarifies the final step in the biosynthesis of the thiamin-thiazole.
Results and Discussion
Peak B Compound Is a Late Intermediate
As reported earlier, the THI4p bound metabolite corresponding to the peak B is chemically unstable and its production varied widely between protein preparations. A systematic investigation of different growth conditions enabled us to develop a protocol that yielded the peak B compound as the major bound metabolite (Figure 2a). This protocol involved induction of protein over-expression at a lower cell density (OD600 of 0.2–0.3), storage of the cell pellets at a very low temperature (−80 °C) and a rapid protein purification at low temperature (4 °C). Incubation of the THI4p-peak B complex at room temperature resulted in the slow conversion of peak B to ADT 1 (peak C, Figure 2a). An identical reaction, using heat denatured THI4p, did not show this transformation. These observations suggested that the peak B compound is an intermediate in the THI4p-catalyzed reaction sequence.
Figure 2.
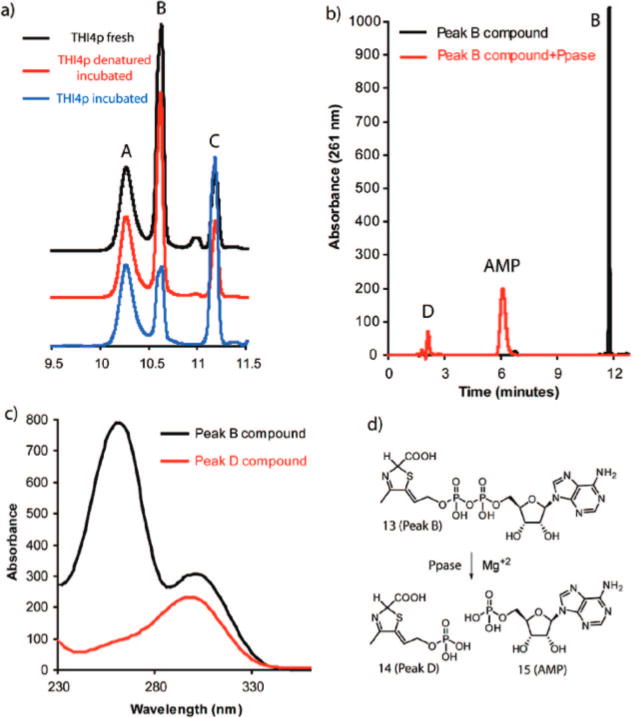
Characterization of the Peak B compound. (a) Room temperature conversion of protein bound peak B to ADT 1 (peak C). All traces represent the absorption at 261 nm, detected by a diode-array detector. Black trace represents a fresh THI4p sample whereas the blue trace represents the same after 6 h of incubation at room temperature. The red trace represents a sample identical to that of the blue trace except, in this case THI4p was heat denatured prior to incubation. (b) Pyrophosphatase catalyzed cleavage of the peak B compound to produce AMP 15 and peak D. Black trace: peak B compound, Red trace: peak B compound after ppase treatment. (c) UV absorption spectra of peak B (black) and peak D (red). (d) Schematic representation of the pyrophosphatase catalyzed reaction of the peak B compound. For the structural determination of 13, vide infra.
Isolation and Characterization of the Peak B Compound
The peak B compound was purified by HPLC. Its UV absorption spectrum showed maxima at 261 and 300 nm (Figure 2c). Treatment of purified peak B compound with nucleotide pyrophosphatase resulted in the formation of AMP 15, suggesting that the peak B compound is adenylated, and a second product with a short retention time of 2 min (peak D, Figure 2b). Negative mode ESI-MS analysis revealed a molecular mass of 596 Da (monoanionic m/z = 595), identical to that of ADT 1 (Figure 3a). However, the fragmentation pattern, producing mainly ADP, m/z = 426, was different from the fragmentation pattern of ADT, which showed mainly the decarboxylation product at m/z = 551. Together, the THI4p catalyzed isomerization and the MS data suggest that the peak B compound is an isomer of ADT 1.
Figure 3.
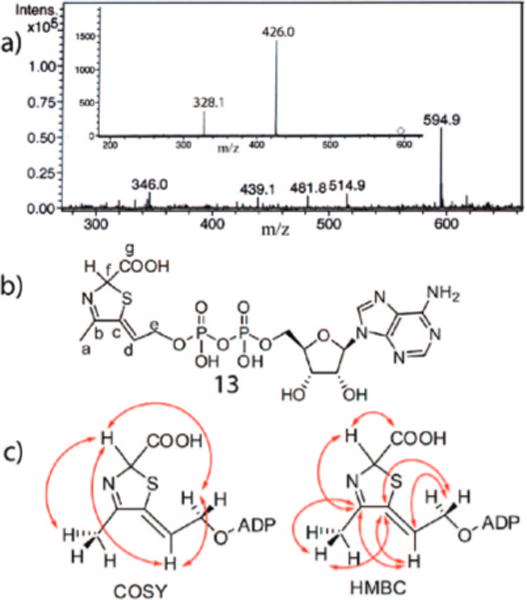
Spectroscopic analysis of the Peak B compound. (a) Negative mode ESI-MS analysis of the peak B compound. The inset shows the fragmentation pattern of the major species with m/z = 594.9. (b) Structure of the peak B compound deduced from NMR, MS and biochemical analyses. (c) Key COSY and HMBC correlations used to determine the structure the thiazole-tautomer.
Extensive spectroscopic analyses of the peak B compound (1H, COSY, HMBC, HMQC, ROESY, UV-visible and MS) confirm that it has the structure 13 shown in Figure 3b. The 5′-phosphorylated adenosine unit was readily identified from the NMR spectroscopic experiments. Additionally, COSY analysis revealed a CH{d}-CH2{e}-OP spin system (CH{d} at 5.95 ppm, t; CH2{e} at 4.3–4.4 ppm, m), a CH3{a} group (2.1 ppm, d) and a CH{f} proton (5.78 ppm, broad s). The CH{f} proton shows a weak COSY correlation to the CH3{a} protons and very weak COSY correlations to the CH{d} and the CH2{e} protons, indicating small long-range couplings between these protons. The HMBC showed strong correlations of the CH3{a} protons and the CH{d} proton to two quaternary olefininc or aromatic carbons (C{b}: 169.12 ppm and C{c}: 139.23 ppm). Additional HMBC correlations of the CH2{e} protons with one of the two quaternary olefinic carbons suggested a linear carbon skeleton with the CH3{a} and CH{d}-CH2{e}-OP groups flanking the two quarternary carbons (Figure 3). The CH{f} proton also shows correlations to the quaternary carbon {b} at 169.12 ppm and a carboxylate carbon {g} at 173.06 ppm. The ROESY analysis helped to determine the Z geometry of the presumed exocyclic double bond by demonstrating an NOE between the CH3{a} and the CH{d} protons, and the absence of an NOE between the CH3{a} and the CH2{e} protons. The resulting structure 13 is consistent with the mechanistic proposal outlined in Figure 1a.
While these spectroscopic analyses did not reveal the stereochemistry at C2 of the thiazole, the structure of the THI4p/ ADT complex (Figure 4a) shows a conserved arginine (Arg301) residue of THI4p interacting with the carboxylate functionality of ADT in the active site. A conserved histidine residue (His237) is positioned close to the C2 of the thiazole unit (4.1 Å). These residues are likely to function in carboxylate binding and the deprotonation at C2, required for the aromatization of 13 (Figure 4b), suggesting that the configuration at C2 of the thiazole-tautomer in 13 is R.
Figure 4.
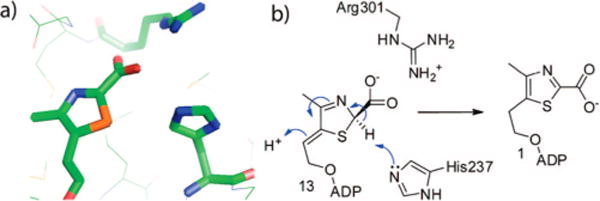
Determination of the absolute stereochemistry at C2 of the thiazole tautomer. (a) Crystal structure of ADT bound to the active site of THI4p (2GJC).6 The Arg301 residue of THI4p interacts with the carboxylate functionality of ADT. (b) Proposed model for 13 at the active site of THI4p, showing Arg301 interacting with the carboxylate functionality. In the proposed R configuration of 13, His237 and the beta phosphate of the ADP moiety are suitably positioned to catalyze the deprotonation/protonation reactions required for the aromatization of 13 leading to ADT 1.
Chemical Reactivity of 13
The intrinsic chemical reactivity of 13 was also evaluated. This compound slowly decomposes to produce ADP and a second compound with a retention time of 20 min (peak E, Figure 5). This second product, with a molecular mass of 169 Da, has a UV absorption maximum at 312 nm (Figure 5, insert). This is considerably shifted to a longer wavelength relative to 13 (300 nm, Figure 2c) suggesting a more delocalized chromophore. Analysis of a double quantum filtered COSY spectrum acquired for a partially degraded sample of 13 (Figure 6) revealed a spin system corresponding to a vinyl group, suggesting that the structure of the second degradation product was 16 (Figure 7c). When the degradation of 13 was monitored by 1H NMR spectroscopy at room temperature in D2O, further conversion of 16 to a new species, containing a similar vinyl unit could be observed (Figure 7a). Comigration with an authentic standard by HPLC identified this new species as 17, the decarboxylation product of 16 in D2O. The half-life of 13 was found to be 9.3 days (at room temperature in water). The surprisingly slow aromatization rate of 13 most likely reflects the reduced resonance energy of the thiazole relative to benzene due to the presence of the two heteroatoms in the ring.9,10 Interestingly, the nonenzymatic degradation of 13 did not produce any ADT 1. This suggests that the conversion of 13 to 1 has an absolute requirement for enzyme catalysis.
Figure 5.
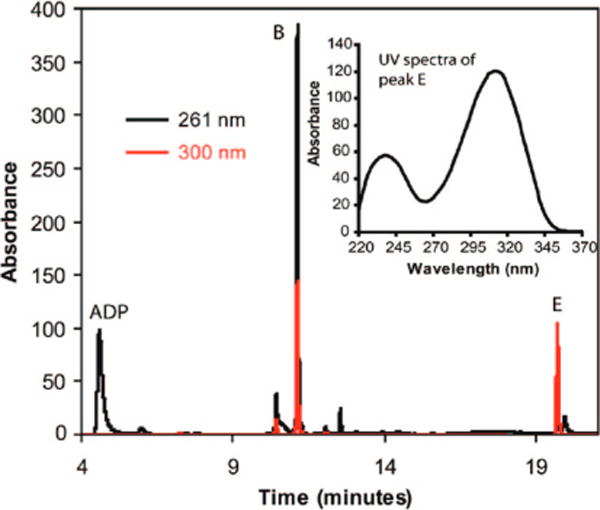
HPLC analysis of the degradation of the peak B compound to ADP and the peak E compound after three days. Black and red traces represent the same sample analyzed at 261 and 300 nm respectively. (Inset) UV absorption spectrum of the peak E compound.
Figure 6.
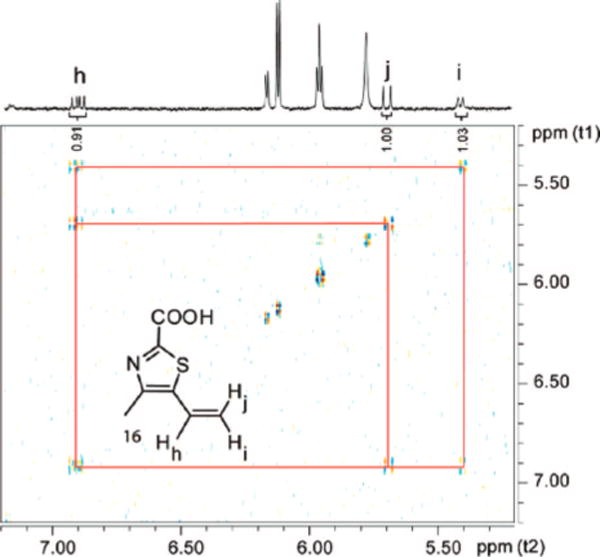
COSY analysis of a partially degraded sample of 13 reveals the presence of the vinyl unit of 16.
Figure 7.
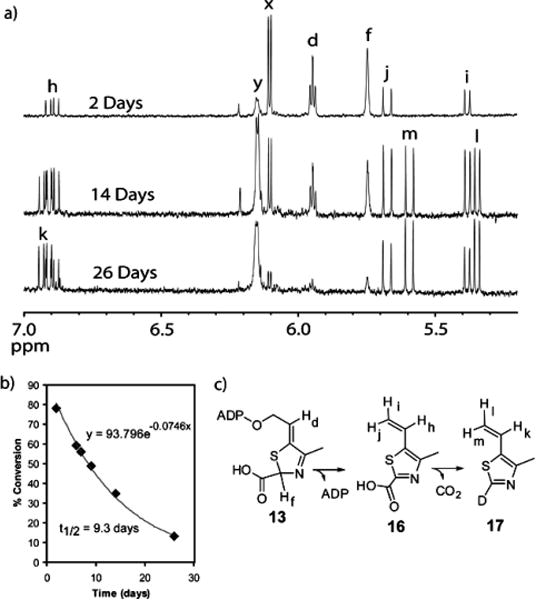
Analysis of the degradation of 13. (a) 1H NMR spectroscopic analysis of the degradation of 13 in D2O. Resonances x and y represent the anomeric protons on the ribose of 13 and ADP respectively. (b) Rate of the degradation of 13 fitted to a single exponential decay function, yielding a half-life of 9.3 days. (c) Degradation reaction of 13.
The identification of thiazole tautomer 13 at the active site of purified THI4p provides an additional informative molecular snapshot of the complex reaction catalyzed by the eukaryotic thiazole synthase (Figure 1) and further demonstrates that the characterization of enzyme-bound metabolites is a powerful strategy for studying enzyme function and mechanism.5–7
Experimental Methods
All of the chemicals and snake venom nucleotide pyrophosphatase were purchased from Sigma-Aldrich Corporation (USA) unless otherwise mentioned. LB medium was obtained from EMD Biosciences. Kanamycin and IPTG were purchased from LabScientific Inc. Analytical HPLC (Agilent 1100 instrument) was carried out using a Supelco LC-18-T (150 × 4.6 mm, 3 μm ID) reverse phase column and HPLC purifications were carried out using a semiprep Supelco LC-18-T (250 × 10 mm, 5 μm ID) column. HPLC grade solvents were obtained from Fisher Scientific.
Overexpression and Purification of THI4p
E. coli BL21(DE3) containing the overexpression plasmid (THI4 in the pET28 vector) was grown in LB medium containing kanamycin (40 μg/mL) with shaking at 37 °C until the OD600 reached 0.2–0.3. At this point, protein overexpression was induced with isopropyl-β-D-thiogalac-topyranoside (IPTG) (final concentration = 2 mM) and cell growth was continued at 15 °C for 16 h. The early induction was important for achieving higher relative ratio of the compound 13. The cells were harvested by centrifugation and the resulting cell pellets were stored at −80 °C. To purify the protein, the cell pellets from 1L of culture were resuspended in 25 mL lysis buffer (10 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8) and lysed by sonication (Heat systems Ultrasonics model W-385 sonicator, 2 s cycle, 50% duty). The resulting cell lysate was clarified by centrifugation and the THI4p protein was purified on Ni-NTA resin following the manufacturer’s (Qiagen) instructions. After elution, the protein was desalted using a 10-DG column (BioRad) pre-equilibrated with 50 mM potassium phosphate buffer, pH 7.8.
Metabolite Analysis
Analysis for bound metabolites was performed as previously described.5
Purification of 13
All the THI4p protein obtained from 4 L of culture (∼200 mg, 10 mL) was divided into twenty 500 μL aliquots and heat denatured (100 °C, 2 min). The precipitated protein was removed by centrifugation and the supernatants were combined and filtered through a 10 kDa MW cut off filter (Amicon Ultra-4, Millipore). The peak B compound was purified by HPLC using the following linear gradient at a flow rate of 3 mL/min: solvent A is water, solvent B is 100 mM KPi, pH 6.6, solvent C is methanol. 0 min: 100% B; 3 min: 10% A, 90%B; 17 min: 34% A, 60% B, 6% C; 21 min: 35% A, 25% B, 40% C; 23 min: 100%B. The fractions containing the peak B compound were collected and stored at −80 °C. Attempts at lyophilization of 13 from the high concentration phosphate buffer led to its degradation. A second HPLC purification, using a low concentration of volatile ammonium acetate buffer, was therefore performed on the pooled fractions from the first purification step using the following linear gradient at a flow rate of 3 mL/min: solvent A is ammonium acetate buffer (20 mM, pH 7), solvent B is methanol; 0 min: 100% A; 2 min: 100% A; 10 min: 50% A, 50% B; 12 min: 100% A. The pooled fractions containing the peak B compound from the second purification step were successfully lyophilized to yield 13.
Cleavage of 13 by Nucleotide Pyrophosphatase
To a 100 μL solution of purified 13 (∼200 μM) was added 1 U of lyophilized snake venom nucleotide pyrophosphatase (EC 3.6.1.9, Sigma) and MgCl2 to a final concentration of 1 mM (20 mM Tris-HCl, pH 8.0). A control reaction was set up with only MgCl2 and no enzyme. Both the samples were incubated at 37 °C for two hours, filtered (10 kDa MW cut off filter) and the filtrates were analyzed by HPLC.
UV, NMR, and MS Analyses
The UV–vis spectra were obtained using an inline diode array detector on the HPLC instrument.
The lyophilized product was dissolved in 0.5 mL of D2O and was relyophilized. The residue thus obtained was dissolved in about 0.3 mL of D2O and the solution was placed in a Shigemi NMR tube (susceptibility-matched for D2O). All NMR experiments were acquired on a Varian INOVA 600 MHz instrument equipped with a 5 mm triple-gradient inverse-detection HCN probe.
Degradation of 13 was monitored by 1H NMR by incubating a sample (produced as described above) in a sealed Shigemi NMR tube at room temperature (25 °C), and collecting its spectra at different time intervals. The half-life of 13 was determined by plotting its % conversion (to 16 and 17) over time and fitting to a single exponential decay function using the program Grafit.
For ESI-MS analysis, purified 13 (isolated as described above) was dissolved in 100 μL of water after lyophilization. 100 μL methanol containing 0.2% triethyl amine was added to this solution immediately before analysis by ESI-MS. The sample was analyzed in the negative ion mode on a Bruker Esquire Ion-Trap instrument, and selected peaks were isolated and fragmented.
Characterization of the Degradation Reaction of 13
A sample of 13 containing a trace of 16 and ADP was incubated at room temperature over a period of weeks. HPLC analysis of an aliquot removed from it after 3 days revealed a substantial enhancement in the concentrations of ADP and 16. From the 1H NMR of the reaction mixture, vinyl and methyl protons of 16 could be characterized. (D2O, 600 MHz) δ 6.89 (dd, 1H, 11, 17.3 Hz), 5.67 (d, 1H, 17.3 Hz), 5.38 (d, 1H, 11 Hz), 2.41 (s, 3H).
From a 500 μL solution of partially degraded 13 (~200 μM initial concentration), 16 was isolated by HPLC and lyophilized. The residue was redissolved in 100 μL of methanol with 0.1% triethylamine and was subjected to negative mode ESI-MS analysis to reveal a molecular mass of 169 Da.
HPLC analysis of another aliquot of the same degradation mixture after 40 days revealed the presence of 17 (identified by the comigration experiment with a standard sample of 17). Because of its hydrophobicity a different HPLC method was used with a linear gradient, at 1 mL/min, for its analysis: Solvent A is water, solvent B is methanol. 0 min: 100% A; 2 min: 100% A; 8 min: 100% B; 10 min: 100% A.
From the 1H NMR of the reaction mixture, vinyl and methyl protons of 17 could be identified. (D2O, 600 MHz) δ 6.92 (dd, 1H, 11, 17.3 Hz), 5.59 (d, 1H, 17.3 Hz), 5.35 (d, 1H, 11 Hz), 2.42 (s, 3H).
Supplementary Material
Acknowledgments
This research was funded by NIH grant DK44083 (to T.P.B.) and DK67081 (to S.E.E.).
Footnotes
Supporting Information Available
2D NMR data, summary of chemical shifts and NMR correlations for compound 13, further NMR analysis for the degradation of 13, and identification of 17. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jordan F. Nat Prod Rep. 2003;20(2):184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 2.Begley TP. Nat Prod Rep. 2006;23(1):15–25. doi: 10.1039/b207131m. [DOI] [PubMed] [Google Scholar]
- 3.Settembre E, Begley TP, Ealick SE. Curr Opin Struct Biol. 2003;13(6):739–747. doi: 10.1016/j.sbi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Begley TP, Downs DM, Ealick SE, McLafferty FW, Van Loon APGM, Taylor S, Campobasso N, Chiu H-J, Kinsland C, Reddick JJ, Xi J. Arch Microbiol. 1999;171(5):293–300. doi: 10.1007/s002030050713. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. J Am Chem Soc. 2007;129(10):2914–2922. doi: 10.1021/ja067606t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jurgenson CT, Chatterjee A, Begley TP, Ealick SE. Biochemistry. 2006;45(37):11061–11070. doi: 10.1021/bi061025z. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. J Am Chem Soc. 2006;128(22):7158–7159. doi: 10.1021/ja061413o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Praekelt UM, Byrne KL, Meacock PA. Yeast. 1994;10(4):481–90. doi: 10.1002/yea.320100407. [DOI] [PubMed] [Google Scholar]
- 9.Mansson M, Sunner S. Acta Chem Scand. 1966;20(3):845–848. [Google Scholar]
- 10.Bird CW. Tetrahedron. 1985;41(7):1409–1414. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.