

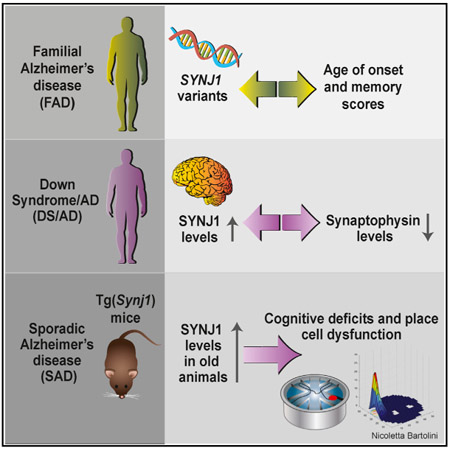
SUMMARY
The phosphoinositide phosphatase synaptojanin 1 (SYNJ1) is a key regulator of synaptic function. We first tested whether SYNJ1 contributes to phenotypic variations in familial Alzheimer’s disease (FAD) and show that SYNJ1 polymorphisms are associated with age of onset in both early- and late-onset human FAD cohorts. We then interrogated whether SYNJ1 levels could directly affect memory. We show that increased SYNJ1 levels in autopsy brains from adults with Down syndrome (DS/AD) are inversely correlated with synaptophysin levels, a direct readout of synaptic integrity. We further report age-dependent cognitive decline in a mouse model overexpressing murine Synj1 to the levels observed in human sporadic AD, triggered through hippocampal hyperexcitability and defects in the spatial reproducibility of place fields. Taken together, our findings suggest that SYNJ1 contributes to memory deficits in the aging hippocampus in all forms of AD.
Graphical Abstract
INTRODUCTION
Synaptic function is under the rigorous control of phosphoinositide turnover, and phosphatidylinositol-4,5-bisphosphate (PtdIns[4,5]P2) is particularly important in this process (Di Paolo et al., 2004). The PtdIns(4,5)P2 phosphatase synaptojanin 1 (Synj1) is a key regulator of synaptic vesicle endocytosis and reavailability on the pre-synaptic side (Cremona et al., 1999; Kim et al., 2002; Mani et al., 2007; McPherson et al., 1996; Verstreken et al., 2003), while on the post-synaptic side, it controls the endocytosis of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors (Gong and De Camilli, 2008).
A body of literature supports the importance of SYNJ1 in neurodegenerative disorders, including Alzheimer’s disease (AD). Clinically, AD is presented with memory loss and spatial disorientation. Neuropathology hallmarks of this disorder include amyloid plaques, composed primarily of Aᵦ peptides that result from the sequential cleavage of the amyloid precursor protein (APP), and neurofibrillary tangles of hyperphosphorylated Tau (Querfurth and LaFerla, 2010). The three forms of AD—familial AD (FAD), Down syndrome-related AD (DS/AD), and sporadic AD (SAD)—share common clinical and neuropathology signatures. Although early-onset FAD is caused by mutations in the APP, PSEN1, or PSEN2 gene (Reitz et al., 2011) and DS/AD is due to triplication of human chromosome 21 (Hsa21) (Antonarakis, 2017; Wiseman et al., 2015), the most potent genetic risk factor for SAD is the ε4 allele of the APOE gene (APOE4) (Lambert et al., 2013; Strittmatter et al., 1993).
SYNJ1 was reported to be crucial for the enlargement of early endosomes (Cossec et al., 2012), one of the earliest cellular phenotypes associated with AD, observed before amyloid accumulation and cognitive decline (Cataldo et al., 2000). Enlarged endosomes are present in neurons of APOE4 carriers (Cataldo et al., 2000) as well as in fibroblasts and lymphocytes from individuals with DS and SAD (Corlier et al., 2015; Cossec et al., 2012). C99, the C-terminal APP fragment resulting from the activity of ᵦ-secretase, has been reported to be required for early endosomal enlargement (Jiang et al., 2010). Importantly though, overexpressing APP alone is not sufficient to alter endosomal size (Cataldo et al., 2003), and endosomal size is unaffected in APP microduplications (Cossec et al., 2012). However, Synj1 overexpression alone is sufficient to produce enlarged endosomes in the brain of transgenic mice (Cossec et al., 2012), and SYNJ1 trisomy results in increased endosomal size in cell lines derived from individuals with partial or full trisomy of Hsa21 (Cossec et al., 2012). In addition, SYNJ1 has also been linked to amyloid toxicity. Oligomers of Aᵦ peptides disrupt PtdIns(4,5)P2 metabolism in cultured primary cortical neurons, and genetically decreasing Synj1 levels protects from the inhibitory effect of Aᵦ oligomers on hippocampal long-term potentiation in brain slices (Berman et al., 2008).
A recent study reported that APOE4 carriers show increased levels of SYNJ1 compared with non-carriers (Zhu et al., 2015). SYNJ1 is encoded by SYNJ1, mapping to Hsa21 (Cremona et al., 2000), and is increased in individuals with DS and DS/AD (Arai et al., 2002; Martin et al., 2014). SYNJ1 levels are thus elevated in individuals at high risk for developing SAD and DS/AD, but very little is known on a potential role for synaptojanin 1 in FAD. To our knowledge, only indirect evidence in model systems has currently been published. Specifically, it has been reported that PtdIns(4,5)P2 metabolism is disrupted in cells expressing FAD-mutant forms of presenilin 1 (Landman et al., 2006), and earlier studies support that genetically decreasing Synj1 levels rescues cognitive deficits in murine models of FAD (McIntire et al., 2012; Zhu et al., 2013), although the mechanisms involved are still controversial.
The present study addresses whether SYNJ1 is associated with human FAD. It also explores whether elevated levels of SYNJ1 directly affect cognition in an age-dependent manner. Our work, combining human genetics, human autopsy brain samples, and behavior and in vivo electrophysiology studies in a transgenic mouse model overexpressing murine Synj1, Tg(Synj1) (Voronov et al., 2008), strongly supports that SYNJ1 plays a role in the function of place cells in the aging hippocampus, with critical implications for memory deficits in all three forms of AD and their possible treatment.
RESULTS
Variants of SYNJ1 Are Associated with Memory Performance in FAD
Individuals with DS/AD (Martin et al., 2014) and APOE4 carriers (Zhu et al., 2015) show increased levels of SYNJ1. We thus targeted SYNJ1 as a candidate gene that may contribute to phenotypic variations in FAD. Specifically, we examined whether SYNJ1 was associated with memory performance and age of onset in early-onset FAD by testing a cohort of Caribbean Hispanic families with the PSEN1-G206A mutation (Table S1) (Athan et al., 2001; Lee et al., 2015). Intriguingly, we observed a genewise association of SYNJ1 with age of onset of AD (p = 0.0195) and long-term recall performance (p = 0.0443) in this cohort (Table S2). Our subsequent SNP analysis within SYNJ1, based on whole-genome sequencing (WGS) data, yielded four SNPs that were significantly associated with age of onset (p < 0.01; Table 1). Furthermore, we observed three additional SNPs associated with long-term recall scores and global memory scores (p < 0.05; Table 1).
Table 1.
Association of SNP within SYNJ1 Gene with Age at Onset and Memory Scores in a Cohort of Caribbean Hispanic Families with the PSEN1-G206A Mutation
| Age at Onseta |
Global Memoryb |
Long-Term Recallb |
Delayed Recallb |
||||||
|---|---|---|---|---|---|---|---|---|---|
| SNP | Location (bp) | Beta | p Value | Beta | p Value | Beta | p Value | Beta | p Value |
| 21:34004976 | 34,004,976 | −0.77 | 0.6602 | 5.90 | 0.0397c | 6.81 | 0.0083c | 1.04 | 0.0504 |
| 21:34006054:G:T | 34,006,054 | 14.91 | 0.0094c | 9.88 | 0.3092 | 5.71 | 0.5150 | 1.45 | 0.4203 |
| 21:34012999 | 34,012,999 | 0.22 | 0.8909 | 6.46 | 0.0137c | 7.14 | 0.0024c | 1.10 | 0.0230c |
| 21:34019201 | 34,019,201 | −16.14 | 0.0061c | 7.06 | 0.5553 | 6.84 | 0.5278 | 3.00 | 0.1862 |
| 21:34041167 | 34,041,167 | −10.23 | 0.0010c | 2.94 | 0.6074 | 1.23 | 0.8125 | −0.21 | 0.8402 |
| rs200644223:34057206: ACGGCCGGG:A |
34,057,206–34,057,214 | −0.98 | 0.7395 | −15.13 | 0.0004c | −11.55 | 0.0029c | −2.14 | 0.0066c |
| 21:34078985 | 34,078,985 | 22.46 | 0.0050c | −0.09 | 0.9939 | −4.05 | 0.7115 | 2.39 | 0.2821 |
See also Figure S2 and Tables S1 and S2 for additional information.
Covariates included AD, sex, PSEN1-G206A, education, APOE4, and principal components (PC1, PC2, and PC3). Age at onset is defined as age at onset for affected individuals and age at last examination for unaffected individuals (see text for details).
Covariates included age at onset, sex, education, APOE4, and principal components (PC1, PC2, and PC3).
p < 0.05.
We then determined whether the observed effect was present in late-onset FAD, a form of the disease defined by having multiple family members affected with late-onset AD (Romas et al., 2002; Zhao et al., 2013), by extending our study to the EFIGA cohort (Lee et al., 2011; Romas et al., 2002) (Table S1). Because the EFIGA dataset lacked WGS data, we analyzed the candidate SYNJ1 region (bp 34,004,976–34,078,985) identified from our early-onset FAD results, using seven tagSNPs that were found to be significant from the genome-wide association study (GWAS) dataset available for the EFIGA cohort (Figure S1 and Table S3). Our subsequent sliding-window haplotype analysis indicated that window 5 in a 3-mer analysis (bp 34,020,786–34,027,774) and window 4 in a 4-mer approach (bp 34,020,653–34,027,774) were the primary candidates for harboring the variant(s) that contribute to age of onset of AD (Table 2). Furthermore, we observed that carriers of the minor haplotype (AGA or AAGA) were protected against AD, as their age of onset was delayed by 8–10 years on average (Table 2). The effect of APOE4 on age at onset was not significant. Our findings in human cohorts thus support an association of SYNJ1 with both early-onset and late-onset FAD.
Table 2.
Sliding-Window Haplotype Analysis of SYNJ1 Gene in Late-Onset FAD (EFIGA)
| rs11702774 |
rs2833930 |
rs2833931 |
rs10470165 |
rs17694546 |
rs2833934 |
rs2833935 |
Model 1 | Model 2 | H1/H1 |
H1/− |
−/− |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A/G(a) | A/G | A/G | G/A | A/C | A/G | G/A | Haplotype | p Valueb | p Valuec | Mean | SD | Mean | SD | Mean | SD |
| Window1 | GAA | 0.1145 | 0.1019 | 67.9 | 10.5 | 70.7 | 9.8 | 71.2 | 9.8 | ||||||
| Window2 | AAA | 0.2072 | 0.1828 | 69.8 | 10.4 | 71.1 | 9.8 | 71.2 | 9.7 | ||||||
| Window3 | AAA | 0.0709 | 0.0580 | 80.0 | 1.4 | 72.4 | 10.7 | 71.0 | 9.8 | ||||||
| Window4 | AAG | 0.0709 | 0.0580 | 80.0 | 1.4 | 72.4 | 10.7 | 71.0 | 9.8 | ||||||
| Window5 | AGA | 0.0418d | 0.0342d | 80.0 | 1.4 | 72.5 | 10.7 | 70.7 | 9.8 | ||||||
| Window1 | GAAA | 0.1184 | 0.1059 | 67.9 | 10.5 | 70.7 | 9.8 | 71.2 | 9.8 | ||||||
| Window2 | AAAC | 0.3410 | 0.2979 | 70.4 | 10.3 | 71.0 | 9.8 | 71.2 | 9.7 | ||||||
| Window3 | AAAG | 0.0714 | 0.0584 | 80.0 | 1.4 | 72.4 | 10.7 | 71.0 | 9.8 | ||||||
| Window4 | AAGA | 0.0323d | 0.0256d | 80.0 | 1.4 | 72.4 | 10.7 | 70.7 | 9.8 | ||||||
See also Figure S1 and Tables S1 and S3 for additional information.
A minor allele is presented first.
Covariates for model 1: Alzheimer’s disease, sex, education, APOE4, principal components (PCs), and genetic relationship matrix (GRM).
Covariates for model 2: similar to model 1 but excludes APOE4.
p < 0.05.
To determine whether the candidate SNPs we identified in early-onset FAD may affect SYNJ1 expression in the brain, we examined the expression quantitative trait loci (eQTL) data in the GTEx Portal, restricting our analysis to the tissues in the frontal cortex. In the frontal cortex, we found that, on the basis of 129 tissue samples, rs2833943 located at 34,041,650 bp and rs66528773 located at 34,080,468 bp were differentially expressed (p = 0.00788771 for each). Their m values, representing the likelihood of functional relevance, were 0.978 and 0.995, respectively. Interestingly, the set of AD associated SNPs that were identified from early-onset FAD (Table 1) was in linkage disequilibrium with the eQTL identified in the frontal cortex tissues in the GTEx dataset (Figure S2), suggesting that the identified SNPs (or adjacent SNPs) are likely to influence SYNJ1 expression.
Elevated SYNJ1 Levels Are Associated with Synaptic Deficits in DS/AD
In light of earlier reports that SYNJ1 levels are elevated in individuals at high risk for developing SAD (Zhu et al., 2015) and DS/AD (Martin et al., 2014), our results in human FAD cohorts strongly suggested that SYNJ1 may play a role in all three forms of AD. This motivated us to investigate whether elevated SYNJ1 affects cognition in an age-dependent manner, an AD hallmark. We hypothesized that a large increase in SYNJ1 levels, such as the one observed in populations at high risk for developing DS/AD (+155% compared with age-matched disomic controls) (Martin et al., 2014), could influence synaptic integrity. To test this hypothesis, we used data previously collected on post-mortem brain samples of individuals with DS at different ages (Martin et al., 2014). We focused on the age range of 40–52 years, when most individuals with DS develop AD. For each individual, we plotted the levels of SYNJ1 against the levels of synaptophysin, a pre-synaptic protein that we used as a direct measure of synaptic integrity (Figure 1). Indeed, synaptophysin levels have been extensively and consistently found to be decreased in individuals with AD and DS/AD (e.g., (Downes et al., 2008; Masliah et al., 1989, 1991; Reddy et al., 2005; Terry et al., 1991). We found that levels of SYNJ1 were inversely correlated (p = 0.0151, R2 = 0.2862) with levels of synaptophysin; that is, the higher the levels of SYNJ1, the more synaptic integrity was compromised. In contrast, no such correlation (p = 0.1784, R2 = 0.2790) was observed in younger individuals with DS (age range 1–39 years), whose SYNJ1 levels are only mildly higher (+36%) than those of disomic controls (Figure S3). Our results thus strongly suggest that elevated levels of SYNJ1 observed in populations at high risk for developing AD could directly affect synaptic structure, function, or both.
Figure 1. Elevated SYNJ1 Levels Are Associated with Synaptic Deficits in DS/AD.

Western blot analysis of SYNJ1 and synaptophysin in human post-mortem brain samples from the mid-frontal cortex (BA46) of individuals with DS, aged 40–52 years (Martin et al., 2014) (n = 20). The line represents the linear regression (R2 = 0.29, p = 0.015). See also Figure S3 for additional information.
Elevated Synj1 Levels Drive Age-Dependent Hippocampal Cognitive Deficits
To dissect the effect of elevated levels of synaptojanin 1 on cognition, we used a transgenic mouse model overexpressing murine Synj1, Tg(Synj1) (Voronov et al., 2008). We observed 76% more Synj1 in the brain of transgenic mice than in littermate controls (wild-type [WT]) (Figure 2A). This closely recapitulates the overexpression levels (+73%) described in APOE4 carriers with early AD (clinical dementia rating [CDR] 0.5–1) (Zhu et al., 2015) but is milder than the overexpression levels in individuals with DS/AD (+155% compared with age-matched disomic controls) (Martin et al., 2014). Indeed, we found that levels of pre-synaptic (synaptophysin) and post-synaptic (PSD 95)proteins were not altered in the hippocampi of 19-month-old transgenic versus WT animals (Figure S4A), suggesting no gross synaptic loss, even in older animals. Tg(Synj1) mice thus appeared as a good model system to dissect the effect of elevated levels of Synj1 on synaptic dysfunction and cognitive deficits.
Figure 2. Overexpression of Synj1 Drives Hippocampal-Dependent Cognitive Deficits in an Age-Dependent Manner.

(A) Western blot analysis of Synj1 in 19-month-old WT and Tg(Synj1) mice (n = 4). Tubulin was used as an equal loading marker. Synj1 protein levels were 76% higher in Tg(Synj1) (1.76 ± 0.11) than in WT (1.00 ± 0.05) mice. ***p < 0.001 in unpaired Student’s t test.
(B) Performance of WT and Tg(Synj1) mice at 9 (n = 8 WT and 7 Tg[Synj1] mice) and 19 (n = 9 mice for both genotypes) months in the radial arm water maze (RAWM). Mice were administered 30 trials over a 2-day period, and the number of errors was averaged over three trials. Two-way ANOVA revealed an interaction between genotype and trial block at 19 months but not at 9 months. ns, p > 0.05, and **p < 0.01 for the overall effect of genotype in two-way ANOVA. In trial 6, ***p < 0.001 for the effect of genotype in two-way ANOVA with Bonferroni post-test.
(C) Age-dependent modification of RAWM performance of WT and Tg(Synj1) mice. Age-dependent cognitive deficits were more severe in Tg(Synj1) mice (188 ± 25%, n = 9) than in WT mice (100 ± 20%, n = 9). *p < 0.05 in unpaired Student’s t test.
(D and E) Freezing response in the contextual and cued FC paradigm in (D) 9-month-old WT (n = 10) and Tg(Synj1) (n = 9) mice and in (E) 19-month-old WT (n = 11) and Tg(Synj1) (n = 7) mice. *p < 0.05 in unpaired Student’s t test.
Data are represented as mean ± SEM. See also Figure S4 for additional information.
We focused on two hippocampus-dependent behavior tasks, as this region is critically affected in AD (Stoub et al., 2006), and investigated whether the performance of transgenic Tg(Synj1) mice in these tasks declined with age. We first used the radial arm water maze (RAWM) paradigm to probe working memory. At 9 months, Tg(Synj1) and WT mice performed similarly in the RAWM test (Figure 2B). In contrast, at 19 months, Tg(Synj1) mice showed a significantly higher number of errors in the RAWM compared with WT mice (Figure 2B). Although WT mice experienced cognitive decline with age, age-dependent cognitive deficits were significantly more pronounced in Tg(Synj1) than in WT littermates (188% of normal aging; Figure 2C). Although 19-month-old Tg(Synj1) swam slightly slower than age-matched WT mice, their ability to reach a visible platform remained unchanged (Figure S4B), ruling out any visual or motivational impairment.
To assess whether other forms of learning were impaired in transgenic mice, we used a fear conditioning (FC) paradigm. Whereas contextual fear learning depends on both hippocampus and amygdala, cued testing only depends on the amygdala. No difference in contextual or cued freezing behavior was observed between Tg(Synj1) and WT mice at 9 months (Figure 2D). However, at 19 months, Tg(Synj1) mice showed a specific decrease in freezing in contextual but not in cued conditioning compared with WT mice, suggesting hippocampal but not amygdala impairment (Figure 2E). Taken together, our behavior results strongly suggest that increased levels of Synj1 drive age-dependent cognitive deficits in the hippocampus.
Elevated Synj1 Levels Trigger Hippocampal Place Cell Dysfunction
We then pursued the functional basis underlying hippocampal cognitive deficits in older Tg(Synj1) animals. Specifically, we investigated whether increased Synj1 levels could alter hippocampal synaptic function using in vivo electrophysiological recordings in the hippocampus (Figure S5A). Although the firing of hippocampal inhibitory neurons was not affected (Figure S5B), the average and peak firing rates of hippocampal excitatory neurons were significantly increased in Tg(Synj1) mice (+57% and +67% increases compared with WT, respectively; Figure 3A). Because these recorded excitatory neurons were place cells (Hussaini et al., 2011) (Figure 3B), we asked whether place field properties were altered in transgenic animals. The average and peak field firing rates were increased in Tg(Synj1) mice compared with WT (Figure 3C). The mean place field size was comparable between transgenic animals and controls (Figure 3D). However, place field size distribution was broader in Tg(Synj1) mice, highlighting a higher size variability in transgenic animals (Figure 3D). Information content and spatial coherence were comparable between transgenic and control mice (Figures 3Eand 3F). More important, the stability of place fields after 18–24hr was decreased by more than 3-fold in Tg(Synj1) mice (Figure 3G), suggesting a memory retention deficit.
Figure 3. Overexpression of Synj1 Results in Hippocampal Hyperexcitability and Decreased Place Field Stability.

(A) Left: average firing rate of hippocampal excitatory (pyramidal) neurons in 24-month-old Tg(Synj1) mice (3.2 ± 0.2 Hz, n = 72 neurons from six animals) and controls (2.0 ± 0.2 Hz, n = 98 neurons from five animals). Right: peak firing rate of pyramidal neurons in Tg(Synj1) mice (9.3 ± 0.7 Hz) and controls (5.6 ± 0.4 Hz). ***p < 0.001 in Mann Whitney test.
(B) Representative examples of firing rate maps showing place fields obtained after WT and Tg(Synj1) mice explored a 50-cm-diameter cylindrical arena for 20 min. The firing rate is represented by a heatmap ranging from blue (no firing) to red (peak firing). White spaces indicate locations not visited by the animal.
(C) Left: average field firing rate in Tg(Synj1) mice (3.8 ± 0.3 Hz, n = 72 neurons) and controls (2.2 ± 0.2 Hz, n = 98 neurons). Right: peak field firing rate in Tg(Synj1) mice (9.2 ± 0.7 Hz) and controls (5.5 ± 0.4 Hz). ***p < 0.001 in Mann-Whitney test.
(D) Size of place fields in Tg(Synj1) and WT mice. The average size (left) of place fields was comparable (p > 0.05 in Mann-Whitney test) between Tg(Synj1) (1,066 ± 58 cm2, n = 72 neurons) and WT (1,043 ± 24 cm2, n = 98 neurons) mice, although the distribution (right) of place field sizes was significantly different (***p < 0.001 in chi-square test).
(E) Comparable (p > 0.05, Mann-Whitney test) information content between Tg(Synj1) (0.29 ± 0.05 bits/spike, n = 72) and WT (0.19 ± 0.03 bits/spike, n = 98) neurons.
(F) Similar spatial coherence (p > 0.05, Mann-Whitney test) between Tg(Synj1) (2.25 ± 0.03, n = 72) and WT (2.21 ± 0.02, n = 98) neurons.
(G) Place field stability at 18–24 hr was significantly decreased (*p < 0.05 in unpaired Student’s t test) in Tg(Synj1) mice (0.05 ± 0.04, n = 20 neurons) compared with controls (0.17 ± 0.03, n = 24 neurons).
Overall, our findings indicate that elevated levels of Synj1 trigger acute hyperexcitability as well as dramatic defects in the spatial reproducibility of place fields in the hippocampus of older Tg(Synj1) animals. Our data from mouse model systems strongly argue that the elevated levels of SYNJ1 observed in DS/AD and SAD could be the cause of the age-dependent long-term memory defects observed in AD patients.
DISCUSSION
The present study addressed whether the elevated levels of SYNJ1 observed in populations at high risk to develop AD could be a common feature underlying age-dependent cognitive deficits. This study is supported by earlier reports in AD mouse models that described an important role for synaptojanin 1 in mechanisms of neuronal toxicity (Berman et al., 2008; Cossec et al., 2012) and human data that showed elevated levels of SYNJ1 in APOE4 carriers (Zhu et al., 2015) and in individuals with DS/AD (Martin et al., 2014) and answers two previously unaddressed questions. Specifically, this work addresses whether SYNJ1 is associated with human FAD as well as whether elevated levels of Synj1 directly affect cognition in an age-dependent manner.
Our targeted gene approach extended the relevance of alterations in SYNJ1 to FAD by showing that variants in SYNJ1 are associated with age of onset and long-term memory deficits in an early-onset FAD cohort of Caribbean Hispanic families with the PSEN1-G206A founder mutation. We further showed that variants in SYNJ1 are also associated with age of onset in the EFIGA cohort of late-onset FAD. Our findings highlight the relevance of studying the impact of SYNJ1 alterations on memory performance for AD. We observed that in DS/AD brain samples, higher SYNJ1 levels correlated with compromised synaptic integrity. We then mimicked milder SYNJ1 overexpression levels, as observed in SAD, in a previously described transgenic mouse model (Voronov et al., 2008). Three- to 4-month-old mice with a mixed FVB/C57BL/6 background showed no anxiety-related behavior (Voronov et al., 2008). They also did not exhibit deficits in the Morris water maze paradigm but performed slightly worse than control animals in the reverse platform test variation of this paradigm (Voronov et al., 2008). For this study, the BAC was backcrossed on the C57BL/6 background for eight generations, and we focused on older, and thus more AD-relevant, animals. Our RAWM and FC behavior studies showed that increased levels of Synj1 triggered age-dependent cognitive deficits in the hippocampus of transgenic Tg(Synj1) mice. Our findings support that increased levels of Synj1 did not impair learning per se in older animals, as evident from the non-null slope in trials 1–5 and trials 6–10 in the RAWM, but caused a specific defect in long-term memory retention. This is particularly well illustrated by the very large number of errors in the first trial of day 2 (trial 6; Figure 2B) in the RAWM as well as by the reduced freezing behavior after 24 hr in contextual conditioning (Figure 2E). Using in vivo recordings, we showed that this defect is due to hippocampal hyperexcitability and, more specifically, to a dramatic alteration in the spatial reproducibility of hippocampal place fields. Taken together, our data strongly argue that the elevated levels of SYNJ1 observed in populations at high risk to develop AD could be sufficient to trigger age-dependent long-term memory retention impairment, a signature trait of the cognitive deficits observed in AD patients.
A key finding of our study is that levels of synaptojanin 1 can regulate the properties, specifically the spatial reproducibility, of hippocampal place fields. Indeed, although the unique role of Synj1 in synaptic function has been very well described (Cremona et al., 1999; Gong and De Camilli, 2008; Kim et al., 2002; Mani et al., 2007; Verstreken et al., 2003), how it translates to hippocampal spatial memory encoding remained unknown. This is particularly important in light of the emerging role of SYNJ1 as a crucial regulator in neurodegenerative diseases, including AD and Parkinson’s disease. Remarkably, mutations in SYNJ1 have recently been reported to be associated with early-onset progressive parkinsonism (Kirola et al., 2016; Krebs et al., 2013; Olgiati et al., 2014; Quadri et al., 2013). These mutations affect the role of synaptojanin 1 at the synapse and result in defects in clathrin uncoating (Cao et al., 2017), in autophagosome maturation (Vanhauwaert et al., 2017), or both.
Another central finding highlights the importance of SYNJ1 in AD. Our results establish synaptojanin 1 as a key regulator of age-related cognitive decline and support that modifications of SYNJ1 levels could be a unifying feature of memory deficits observed in the three types of AD (familial, sporadic, and DS/AD). To our knowledge, the only other protein described to be involved in all three forms of AD is APP. Indeed, APP can be mutated in FAD, it maps to Hsa21, and variants in the APP gene promoter region are a risk factor for SAD (Guyant-Maréchal et al., 2007; Lv et al., 2008).
Our findings thus strongly argue that developing specific SYNJ1 inhibitors is an attractive therapeutic strategy for AD. This is supported by earlier reports showing that genetically decreasing Synj1 levels rescues cognitive deficits in murine models of FAD (APP and PSEN1 mutations) (McIntire et al., 2012; Zhu et al., 2013) and SAD (ApoE4 knockins) (Zhu et al., 2015). If successful, this therapy would be protective against both toxic effects of oligomeric Aᵦ (Berman et al., 2008) and cognitive decline linked to age and could be extended to all three forms of AD. Importantly, SYNJ1 can serve as an ideal drug target, as it is a brain-specific phosphatase. That is, its activity can be targeted by small molecules without affecting its structural roles, with limited secondary effects on peripheral organs. Our findings also provide disease-relevant functional readouts, e.g., accuracy of hippocampal spatial encoding, to evaluate the efficacy of these future drugs.
EXPERIMENTAL PROCEDURES
Genetics and Population
Early-Onset FAD
For the genetic study of early-onset FAD, all participating subjects were at least 35 years of age. FAD patients, with the age at onset <65 years, met the research criteria of the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the AD and Related Disorders Association (ADRDA) for probable or possible AD (McKhann et al., 1984). We studied 305 family members from 45 Caribbean Hispanic families that had at least one G206A founder mutation in the PSEN1 gene (PSEN1-G206A), in which approximately 50% of the family members were carriers of the PSEN1-G206A (Athan et al., 2001; Lee et al., 2015).
To obtain WGS data on all family members while minimizing sequencing costs, we first selected two to four highly informative family members from each branch of the pedigree using the GIGI algorithm (Cheung et al., 2013) and performed WGS on the Illumina HiSeq 2500 platform. In addition, we alsoperformed GWAS on all clinically evaluated family members in the pedigree. To generate WGS data for all family members, we applied SHAPIT2 (Delaneau et al., 2013) and IMPUTE2 (Howie et al., 2009) to impute sequence data into GWAS in family members who were not sequenced. To ensure high-quality imputation in this admixed cohort, we used in-house WGS data generated from 608 Caribbean Hispanics from Puerto Rico and the Dominican Republic, plus the 1000 Genomes data (n = 2,504) as a reference panel. Standard quality control (QC) procedures were performed (Howie et al., 2012). To determine whether genetic variants in SYNJ1 were associated with variation in age at onset of AD and memory traits, specifically global memory, long-term recall and delayed recall, we first performed a gene-wise analysis while taking into account AD status, sex, PSEN1-G206A, level of education, APOE4, and principal components 1–3, as implemented in FamSKAT (Chen et al., 2013) for each trait. For the purpose of analysis, we followed the convention of survival analysis and defined age at onset of AD as follows: if affected, age at onset was used as the age at onset; if unaffected, age at last examination was used. To determine whether certain variants within SYNJ1 were significantly associated with the traits, we performed a SNP-wise analysis for the variants in SYNJ1 while controlling for the same set of covariates as well as kinship coefficient to take into account non-independence among family members. Linear mixed modeling was performed using R (http://cran.r-project.org/web/packages/kinship2/kinship2.pdf).
Replication in Late-Onset AD
To determine whether the observed genetic association between SYNJ1 and early-onset FAD was present in late-onset AD as well, we examined the role of SYNJ1 by evaluating the EFIGA cohort, which comprises both Caribbean Hispanic families with late-onset AD (Lee et al., 2011; Romas et al., 2002) and unrelated Caribbean Hispanics with SAD (Tosto et al., 2015) (see Table S1 for their characteristics). Genotyping data were obtained using multiple batches of SNP microarray platforms (see Table S3). We performed a sliding-window haplotype analysis using the GMMAT algorithm (Chen et al., 2016), taking three or four tagSNPs at a time. We then compared the mean age at onset associated with the risk haplotype.
Recruitment, informed consent, and study procedures for the above two studies were approved by the institutional review boards of the Columbia University Medical Center (AAA R5816 for the Genetic modifier study and AAA PO477 for EFIGA).
Human Subjects, Autopsy Brain Tissue, and Western Blot
Characteristics of autopsy cases, as well as brain tissue preparation protocol and western blotting procedures, were previously described in full detail (Martin et al., 2014).
Mouse Models
Two different mouse models were tested: (1) Tg(Synj1) mice and (2) their C57BL/6 control littermates (WT). The Tg(Synj1) line was a kind gift from the Antonarakis lab. It was generated on the FVB background using mouse BAC RPCI-23 402J16, as described previously (Voronov et al., 2008). This BAC also contains two additional complete genes, the mouse orthologs of C21orf59 and C21orf66 (Voronov et al., 2008). The expression of C21orf59 is enriched in the cerebellum (https://gtexportal.org/home/gene/C21ORF59). The C21orf59 protein controls primary cilia motility and polarization (Austin-Tse et al., 2013; Jaffe et al., 2016). The expression of C21orf66, also called PAXBP1, is enriched in the cerebellum too (https://gtexportal.org/home/gene/PAXBP1). PAXBP1 is an adaptor protein linking the transcription factors PAX3 and PAX7 to the histone methylation machinery in muscle precursor cells (Diao et al., 2012). A variant of PAXBP1 was recently linked to myopathic hypotonia (Alharby et al., 2017). Contributing effects from these genes cannot be excluded. For this study, the BAC was backcrossed on the C57BL/6 background for eight generations. Genotypes were assessed using PCR. All animals were hemizygous for the BAC transgene. All procedures were performed following NIH guidelines in accordance with Institutional Animal Care and Use Committee (IACUC) protocols. Tests were performed on 4–14 mice for each genotype group. All experiments were performed blind with respect to the genotype. Behavior experiments were performed on two age groups: 9 months (range 8–11 months) and 19 months (range 18–22 months). Because of technical considerations, in vivo electrophysiology recordings were performed on a larger age range (19–31 months), with an average age of 24 months at recording. All experiments were performed on age-matched mice for each genotype group. Separate tests were performed for males and females. Because no sex-specific differences were found, results from both genders were pooled.
Statistical Analysis
Statistical calculations were performed using GraphPad Prism version 5.02. All data are expressed as mean ± SEM. In most cases, when comparing two samples, two-tailed Student’s t test was performed. When variances were not comparable, Welch’s correction was applied. When the distribution could not be assumed to be Gaussian, we used a non-parametric Mann-Whitney test. When more samples were compared and Bartlett’s test showed that variances could be compared, we used one-way ANOVA with Dunnett’s post-test or two-way ANOVA with the Bonferroni post-test. If variances could not be compared (p value in Bartlett’s test < 0.05), we used t tests. When more samples were compared and when the distribution could not be assumed to be Gaussian, we used the Kruskal-Wallis test. The chi-square test was used to compare distributions. Outliers, defined as values that were superior to (mean + 3 SDs) or inferior to (mean − 3 SDs) were excluded.
DATA AND SOFTWARE AVAILABILITY
The authors declare that all the data supporting the findings of this study are available within the article and its Supplemental Information files or are available from the corresponding author on request. The accession number for the WGS data obtained from members of families with early onset PSEN1-G206A mutation carriers reported in this paper is National Institute on Aging Genetics of AD Data Storage Site (NIAGADS): NG00064.
Supplementary Material
In Brief.
Miranda et al. combine human genetics, human brain samples, and behavior and electrophysiology in a transgenic mouse model to show that synaptojanin 1 levels regulate the function of place cells in the aging hippocampus. The results have implications for memory deficits in all types of Alzheimer’s disease.
Highlights.
• SYNJ1 variants associate with age of onset in familial Alzheimer’s disease
• SYNJ1 and synaptophysin are inversely correlated in adults with Down syndrome
• Aged mice overexpressing Synj1 exhibit cognitive decline and place field defects
ACKNOWLEDGMENTS
We would like to thank Agnieszka Staniszewski and Dr. Ottavio Arancio for their expert help with behavior studies. We would like to thank Eric Doran and Dr. Sarah B. Martin for their help with DS studies. We want to acknowledge Valerie Savage, Alejandro J. Mercado-Capote, Adithi Jayaraman, and Masayuki Yanagiba for help with microdrive construction and spike sorting. We also want to thank Nicoletta Barolini for her expert help with illustrations and graphics. This work was supported by grants from Fundação para a Cieêcia e Tecnologia (PD/BD/105915/2014 to A.M.M.), the Philippe Chatrier Foundation (C.M.), the Lejeune Foundation (1149 to G.D.P.), the Alzheimer’s Association (2015-NIRG-341570 to S.A.H.), ANR Investissements d’Avenir (ANR-10-IAIHU-06 to M.-C.P.), and the NIH (R01AG050425 to S.A.H.; RO1HD064993 to E.H. and F.A.S.; and RO1HD065160, P50AG16573, and U01AG051412 to I.T.L.). Data collection on Caribbean Hispanic families with at least one G206A founder mutation in the PSEN1 gene was supported by the BrightFocus Foundation (A2015633S) and NIH/National Institute on Aging (NIA) (R56 AG051876-01A1) to J.H.L. Data collection for the EFIGA project was supported by the Genetic Studies of Alzheimer’s Disease in Caribbean Hispanics (EFIGA), funded by the NIH/NIA (5R37AG015473, RF1AG015473, and R56AG051876). We acknowledge the EFIGA study participants and the EFIGA research and support staff for their contributions to this study.
15Present address: Denali Therapeutics, South San Francisco, CA 94080, USA
17Lead Contact
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.011.
AUTHOR CONTRIBUTIONS
C.M. conceived most of the research. G.D.P. conceived a subset of behavioral experiments. A.M.M., M.H., E.N., E.M., G.F., and C.M. performed experiments. A.M.M., G.B., S.A.H., and C.M. analyzed experiments. R.C. analyzed genetic data from human cohorts. E.H., F.A.S., and I.T.L. contributed key data on individuals with DS. I.Z.J.-V. played a key role in the recruitment of mutation carriers. S.E.A. contributed a key animal model. M.-C.P., J.H.L., S.A.H., and C.M. supervised the work. C.M., S.A.H., and J.H.L. wrote the manuscript, and all authors critically discussed the data and edited the manuscript.
DECLARATION OF INTERESTS
G.D.P. is a full-time employee of Denali Therapeutics, Inc. All other authors declare no competing interests.
Data are represented as mean ± SEM. See also Figure S5 for additional information.
REFERENCES
- Alharby E, Albalawi AM, Nasir A, Alhijji SA, Mahmood A, Ramzan K, Abdusamad F, Aljohani A, Abdelsalam O, Eldardear A, et al. (2017). A homozygous potentially pathogenic variant in the PAXBP1 gene in a large family with global developmental delay and myopathic hypotonia. Clin. Genet 92, 579–586. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet 18, 147–163. [DOI] [PubMed] [Google Scholar]
- Arai Y, Ijuin T, Takenawa T, Becker LE, and Takashima S (2002). Excessive expression of synaptojanin in brains with Down syndrome. Brain Dev 24, 67–72. [DOI] [PubMed] [Google Scholar]
- Athan ES, Williamson J, Ciappa A, Santana V, Romas SN, Lee JH, Rondon H, Lantigua RA, Medrano M, Torres M, et al. (2001). A founder mutation in presenilin 1 causing early-onset Alzheimer disease in unrelated Caribbean Hispanic families. JAMA 286, 2257–2263. [DOI] [PubMed] [Google Scholar]
- Austin-Tse C, Halbritter J, Zariwala MA, Gilberti RM, Gee HY, Hellman N, Pathak N, Liu Y, Panizzi JR, Patel-King RS, et al. (2013). Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet 93, 672–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DE, Dall’Armi C, Voronov SV, McIntire LBJ, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, and Di Paolo G (2008). Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci 11, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Wu Y, Ashrafi G, McCartney AJ, Wheeler H, Bushong EA, Boassa D, Ellisman MH, Ryan TA, and De Camilli P (2017). Parkinson Sac domain mutation in synaptojanin 1 impairs clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron 93, 882–896.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, and Nixon RA (2000). Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol 157, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Petanceska S, Peterhoff CM, Terio NB, Epstein CJ, Villar A, Carlson EJ, Staufenbiel M, and Nixon RA (2003). App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci 23, 6788–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Meigs JB, and Dupuis J (2013). Sequence kernel association test for quantitative traits in family samples. Genet. Epidemiol 37, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wang C, Conomos MP, Stilp AM, Li Z, Sofer T, Szpiro AA, Chen W, Brehm JM, Celedón JC, et al. (2016). Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am. J. Hum. Genet 98, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung CYK, Thompson EA, and Wijsman EM (2013). GIGI: an approach to effective imputation of dense genotypes on large pedigrees. Am. J. Hum. Genet 92, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlier F, Rivals I, Lagarde J, Hamelin L, Corne H, Dauphinot L, Ando K, Cossec JC, Fontaine G, Dorothée G, et al. ; Clinical ImaBio3 Team (2015). Modifications of the endosomal compartment in peripheral blood mononuclear cells and fibroblasts from Alzheimer’s disease patients. Transl. Psychiatry 5, e595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossec JC, Lavaur J, Berman DE, Rivals I, Hoischen A, Stora S, Ripoll C, Mircher C, Grattau Y, Olivomarin JC, et al. (2012). Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet 21, 3156–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Lüthi A., Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, et al. (1999). Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188. [DOI] [PubMed] [Google Scholar]
- Cremona O, Nimmakayalu M, Haffner C, Bray-Ward P, Ward DC, and De Camilli P (2000). Assignment of SYNJ1 to human chromosome 21q22.2 and Synj12 to the murine homologous region on chromosome 16C3-4 by in situ hybridization. Cytogenet. Cell Genet 88, 89–90. [DOI] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, and Marchini J (2013). Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, and De Camilli P (2004). Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422. [DOI] [PubMed] [Google Scholar]
- Diao Y, Guo X, Li Y, Sun K, Lu L, Jiang L, Fu X, Zhu H, Sun H, Wang H, and Wu Z (2012). Pax3/7BP is a Pax7- and Pax3-binding protein that regulates the proliferation of muscle precursor cells by an epigenetic mechanism. Cell Stem Cell 11, 231–241. [DOI] [PubMed] [Google Scholar]
- Downes EC, Robson J, Grailly E, Abdel-All Z, Xuereb J, Brayne C, Holland A, Honer WG, and Mukaetova-Ladinska EB (2008). Loss of synaptophysin and synaptosomal-associated protein 25-kDa (SNAP-25) in elderly Down syndrome individuals. Neuropathol. Appl. Neurobiol 34, 12–22. [DOI] [PubMed] [Google Scholar]
- Gong LW, and De Camilli P (2008). Regulation of postsynaptic AMPA responses by synaptojanin 1. Proc. Natl. Acad. Sci. U S A 105, 17561–17566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyant-Maréchal L, Rovelet-Lecrux A, Goumidi L, Cousin E, Hannequin D, Raux G, Penet C, Ricard S, Macé S, Amouyel P, et al. (2007). Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology 68, 684–687. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, and Marchini J (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, and Abecasis GR (2012). Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet 44, 955–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussaini SA, Kempadoo KA, Thuault SJ, Siegelbaum SA, and Kandel ER (2011). Increased size and stability of CA1 and CA3 place fields in HCN1 knockout mice. Neuron 72, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe KM, Grimes DT, Schottenfeld-Roames J, Werner ME, Ku TSJ, Kim SK, Pelliccia JL, Morante NFC, Mitchell BJ, and Burdine RD (2016). c21orf59/kurly Controls Both Cilia Motility and Polarization. Cell Rep 14, 1841–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM, and Nixon RA (2010). Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U S A 107, 1630–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WT, Chang S, Daniell L, Cremona O, Di Paolo G, and De Camilli P (2002). Delayed reentry of recycling vesicles into the fusion-competent synaptic vesicle pool in synaptojanin 1 knockout mice. Proc. Natl. Acad. Sci. U S A 99, 17143–17148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirola L, Behari M, Shishir C, and Thelma BK (2016). Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Parkinsonism Relat. Disord 31, 124–128. [DOI] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, et al. (2013). The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat 34, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. ; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (2013). Metaanalysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, Kang MS, Park MK, Di Paolo G, Chung S, and Kim TW (2006). Presenilin mutations linked to familial Alzheimer’s disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U S A 103, 19524–19529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cheng R, Barral S, Reitz C, Medrano M, Lantigua R, Jiménez-Velazquez IZ, Rogaeva E, St George-Hyslop PH, and Mayeux R (2011). Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol 68, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cheng R, Vardarajan B, Lantigua R, Reyes-Dumeyer D, Ortmann W, Graham RR, Bhangale T, Behrens TW, Medrano M, et al. (2015). Genetic modifiers of age at onset in carriers of the G206A mutation in PSEN1 with familial Alzheimer disease among Caribbean Hispanics. JAMA Neurol 72, 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, Jia L, and Jia J (2008). Promoter polymorphisms which modulate APP expression may increase susceptibility to Alzheimer’s disease. Neurobiol. Aging 29, 194–202. [DOI] [PubMed] [Google Scholar]
- Mani M, Lee SY, Lucast L, Cremona O, Di Paolo G, De Camilli P, and Ryan TA (2007). The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron 56, 1004–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SB, Dowling ALS, Lianekhammy J, Lott IT, Doran E, Murphy MP, Beckett TL, Schmitt FA, and Head E (2014). Synaptophysin and synaptojanin-1 in Down syndrome are differentially affected by Alzheimer’s disease. J. Alzheimers Dis 42, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Terry RD, DeTeresa RM, and Hansen LA (1989). Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett 103, 234–239. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Alford M, DeTeresa R, and Hansen LA (1991). Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol 138, 235–246. [PMC free article] [PubMed] [Google Scholar]
- McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, and Kim TW (2012). Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J. Neurosci 32, 15271–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, and Stadlan EM (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Garcia EP, Slepnev VI, David C, Zhang X, Grabs D, Sossin WS, Bauerfeind R, Nemoto Y, and De Camilli P (1996). A presynaptic inositol-5-phosphatase. Nature 379, 353–357. [DOI] [PubMed] [Google Scholar]
- Olgiati S, De Rosa A, Quadri M, Criscuolo C, Breedveld GJ, Picillo M, Pappatà S, Quarantelli M, Barone P, De Michele G, and Bonifati V (2014). PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 15, 183–188. [DOI] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, et al. ; International Parkinsonism Genetics Network (2013). Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum. Mutat 34, 1208–1215. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, and LaFerla FM (2010). Alzheimer’s disease. N. Engl. J. Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W Jr., Kaye J, and Manczak M (2005). Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J. Alzheimers Dis 7, 103–117, discussion 173–180. [DOI] [PubMed] [Google Scholar]
- Reitz C, Brayne C, and Mayeux R (2011). Epidemiology of Alzheimer disease. Nat. Rev. Neurol 7, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romas SN, Santana V, Williamson J, Ciappa A, Lee JH, Rondon HZ, Estevez P, Lantigua R, Medrano M, Torres M, et al. (2002). Familial Alzheimer disease among Caribbean Hispanics: a reexamination of its association with APOE. Arch. Neurol 59, 87–91. [DOI] [PubMed] [Google Scholar]
- Stoub TR, deToledo-Morrell L, Stebbins GT, Leurgans S, Bennett DA, and Shah RC (2006). Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 10041–10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, and Roses AD (1993). Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, and Katzman R (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol 30, 572–580. [DOI] [PubMed] [Google Scholar]
- Tosto G, Fu H, Vardarajan BN, Lee JH, Cheng R, Reyes-Dumeyer D, Lantigua R, Medrano M, Jimenez-Velazquez IZ, Elkind MSV, et al. (2015). F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol 2, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 36, 1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, and Bellen HJ (2003). Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40, 733–748. [DOI] [PubMed] [Google Scholar]
- Voronov SV, Frere SG, Giovedi S, Pollina EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, et al. (2008). Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down’s syndrome. Proc. Natl. Acad. Sci. U S A 105, 9415–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, and Strydom A (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci 16, 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Marchani EE, Cheung CYK, Steinbart EJ, Schellenberg GD, Bird TD, and Wijsman EM (2013). Genome scan in familial late-onset Alzheimer’s disease: a locus on chromosome 6 contributes to age-at-onset. Am. J. Med. Genet. B. Neuropsychiatr. Genet 162B, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zhong M, Zhao J, Rhee H, Caesar I, Knight EM, Volpicelli-Daley L, Bustos V, Netzer W, Liu L, et al. (2013). Reduction of synaptojanin 1 accelerates Aᵦ clearance and attenuates cognitive deterioration in an Alzheimer mouse model. J. Biol. Chem 288, 32050–32063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zhong M, Elder GA, Sano M, Holtzman DM, Gandy S, Cardozo C, Haroutunian V, Robakis NK, and Cai D (2015). Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. U S A 112, 11965–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.