

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers with dismal patient outcomes. The underlying core genetic drivers of disease have been identified in human tumor specimens and described in genetically engineered mouse models. These genetic drivers of PDAC include KRAS signaling, TP53 mutations, and genetic loss of the SMAD4 tumor suppressor protein. Beyond the known mutational landscape of PDAC genomes, alternative disrupted targets that extend beyond conventional genetic mutations have been elusive and understudied in the context of PDAC cell therapeutic resistance and survival. This last point is important because PDAC tumors have a unique and complex tumor microenvironment that includes hypoxic and nutrient-deprived niches that could select for cell populations that garner therapeutic resistance, explaining tumor heterogeneity in regards to response to different therapies. We and others have embarked in a line of investigation focused on the key molecular mechanism of posttranscriptional gene regulation that is altered in PDAC cells and supports this pro-survival phenotype intrinsic to PDAC cells. Specifically, the key regulator of this mechanism is a RNA-binding protein, HuR (ELAVL1), first described in cancer nearly two decades ago. Herein, we will provide a brief overview of the work demonstrating the importance of this RNA-binding protein in PDAC biology and then provide insight into ongoing work developing therapeutic strategies aimed at targeting this molecule in PDAC cells.
Keywords: ELAVL1, HuR, pancreatic cancer, pancreatic ductal adenocarcinoma, RNA binding proteins
1 |. INTRODUCTION
Polymerase chain reaction (PCR)-amplified based sequencing of isolated pancreatic ductal adenocarcinoma (PDAC) genomes followed by high throughput next generation sequencing (NGS) platforms have provided a genetic landscape of the progression model of PDAC (Knudsen, O’Reilly, Brody, & Witkiewicz, 2016). These later studies have subtyped PDAC tumors, which may have implications for prognostic clinical value and for therapeutic targeting (Bailey et al., 2016; Collisson et al., 2011). To highlight how far this work has come, NGS sequencing of PDAC tumors have now become routine in some institutions and numerous commercial platforms exist. In fact, a pioneer in this field and one of the leading advocacy groups in the country, the Pancreatic Cancer Action Network (PanCAN), has launched into a program attempting to democratize sequencing of PDAC tumors for the ultimate purpose of matching patients with the appropriate clinical trial (Pishvaian et al., 2016). Additionally, PanCAN is pushing the limits on personalized medicine for pancreatic cancer patients, by initiating the launch of the Precision Promise Program—a match your tumor-to-therapy initiative that brings together a multidisciplinary group of investigators, institutions, and platforms to accelerate the pace of molecular tailored trials and research. Certainly, these tour de force clinically driven initiatives are timely, logical, and cutting-edge, and the amount of data the research community will obtain will be invaluable. However, these personalized medicine initiatives will take time and will most likely not directly translate into clinical practice in the near future.
The long term future of precision medicine for pancreatic cancer seems more promising than the short term, in part, because the targeted therapies available for treating pancreatic cancer cells and their complex tumor microenvironment lack specificity and activity. For instance, a one size fits all approach of targeting KRAS mutations have proven to be challenging most likely due to the PDAC cell’s ability to compensate through different signaling pathways. Besides compensation, many genetic lesions that have been identified as critical for tumor progression may not be the most valuable targets at the time the tumor presents clinically. Therefore, a target that meets the following criteria should be considered in order to target PDAC cells efficiently: (a) an available target: one that is abundant in cancer cells compared to normal cells, (b) having a functional purpose that can be targeted in cancer cells, (c) activated by the unique tumor microenvironment in PDAC tumors (e.g., hypoxia and low glucose), and (d) provide an important survival advantage for PDAC cells. Herein, we will provide a brief account of the evidence describing the role of a target that meets these criteria, the RNA-binding protein HuR that is a master regulator of pancreatic tumorigenesis and cell survival.
2 |. AN ALTERNATIVE TO DRIVER GENETIC EVENTS: A CRITICAL POSTTRANSCRIPTIONAL GENE REGULATORY MECHANISM
Many laboratories, including our own work, have established a line of investigation that dates back over 10 years, demonstrating that gastrointestinal (GI) cancer cells and tumors (i.e., PDAC and colorectal cancer) survive under cancer-associated stress conditions (e.g., chemotherapy) through posttranscriptional gene regulation (Blanco et al., 2016; Costantino et al., 2009; Dixon et al., 2001; Lal et al., 2014; Lal et al., 2017; McAllister et al., 2014; Pineda et al., 2012; Richards et al., 2010; Romeo et al., 2016; Subbaramaiah, Marmo, Dixon, & Dannenberg, 2003; Williams et al., 2010; Young et al., 2009; Young, Moore, Sokol, Meisner-Kober, & Dixon, 2012). Primarily through the actions of RNA-binding proteins (RBPs) and micro-RNAs (miRNAs), posttranscriptional gene regulation can rapidly alter the proteome in response to cellular signals by directly impacting gene expression at the level of mRNA stability (Cheadle et al., 2005; Fan et al., 2002; Garneau, Wilusz, & Wilusz, 2007). While the majority of RBPs and miRNAs are noted for their effects on promoting mRNA decay and/or suppressing translation, the RBP Human antigen R (HuR, ELAVL1) is the most intriguing and prominent antagonist of cancer-associated mRNA degradation.
HuR is a member of the ELAV family of RBPs (for an extensive review of this family of proteins please see Hinman & Lou (2008)) and consists of two-tandem RNA recognition motif (RRM) domains, followed by a hinge region and a third RRM domain (Brennan & Steitz, 2001; Meisner & Filipowicz, 2010). HuR binds to adenylate uridylate (AU)-rich elements (AREs) typically located in the 3′-untranslated regions (3′UTRs) of specific target genes involved in cell survival and tumorigenesis (Abdelmohsen & Gorospe, 2010; Blanco, Jimbo, et al., 2016; Brody & Gonye, 2011; Costantino et al., 2009; Khabar, 2017; Lal et al., 2014). HuR is predominantly localized to the nucleus (>90%) and can shuttle between the nucleus and the cytoplasm. Nucleocytoplasmic trafficking is mediated through a basic 32-amino acid HuR Nucleocytoplasmic Shuttling (HNS) sequence contained in the hinge region and involves several transport machinery components including exportin-1 (XPO1, CRM1), transportins, and importins (Fan & Steitz, 1998a; Fan & Steitz, 1998b; Gallouzi & Steitz, 2001; Rebane, Aab, & Steitz, 2004; Wang et al., 2004). It is hypothesized that the ability of HuR to promote mRNA stabilization requires its translocation to the cytoplasm (Brennan & Steitz, 2001; Keene, 1999). In the context of cancer, HuR becomes functionally active as an mRNA stability factor in response to various cancer-associated stressors (e.g., DNA damage, low glucose), where it binds to selective mRNAs and translocates to the cytoplasm.
3 |. HUR IN PANCREATIC CANCER
The Gorospe and Dixon laboratories have initiated the discovery that HuR is both abundant in GI cancer cells and tumors, and is functionally active as a pro-survival network important for tumorigenesis (Dixon et al., 2001; Lopez de Silanes et al., 2003; Mazan-Mamczarz et al., 2003; Wang et al., 2000). HuR has been identified as a prognostic marker or relevant to at least a dozen tumor types (Abdelmohsen & Gorospe, 2010; Kotta-Loizou, Giaginis, & Theocharis, 2014; Wang et al., 2013; Zucal et al., 2015). Mechanistically, it was shown that HuR is upregulated and dysregulated in cancer cells, in part, through posttranscriptional gene regulation (Abdelmohsen et al., 2010; Abdelmohsen, Srikantan, Kuwano, & Gorospe, 2008; Pullmann Jr. et al., 2007).
The first study to demonstrate that HuR was abundant in PDAC cells was published in 2009 (Costantino et al., 2009). This study showed that overexpression of HuR in PDAC cell lines did not dramatically change the in vitro phenotype, but did specifically make PDAC cells highly sensitive to the commonly utilized chemotherapeutic agent, gemcitabine. These data were also supported by a retrospective study of patient data that showed if patient’s tumor specimens had a high cytoplasmic HuR score they were more likely to respond to gemcitabine adjuvant therapy. These findings were expanded to show that PDAC cells exposed to gemcitabine induced HuR’s translocation to the cytoplasm. This allowed for binding of the HuR target mRNA encoding deoxycytidine kinase (dCK) and its subsequent upregulation, leading to enhanced metabolism and efficacy of the prodrug (gemcitabine) in PDAC cells. These results provided a mechanistic explanation when PDAC cells are exposed to the stressor gemcitabine. This study laid the framework for future studies linking a pancreatic cancer-associated stress with a novel HuR target that conferred a pro-survival PDAC phenotype (see Table 1 and Figure 1).
TABLE 1.
Pancreatic cancer-associated stressors associated with HuR target mRNAs
| PDAC stress | HuR target identified |
|---|---|
| Gemcitabine | dCK (deoxycytidine kinase) |
| Hypoxia | PIM1 (Pim-1 proto-oncogene, serine/threonine kinase) |
| Apoptosis | DR4/5 (TNF receptor superfamily member 10a and 10b; death receptors 4 and 5) |
| DNA damage | WEE1 (WEE1 G2 checkpoint kinase) |
| Low glucose | IDH1 (isocitrate dehydrogenase (NADP(+)) 1, cytosolic) |
| PARP inhibitor | PARG (poly(ADP-ribose) glycohydrolase) |
FIGURE 1.
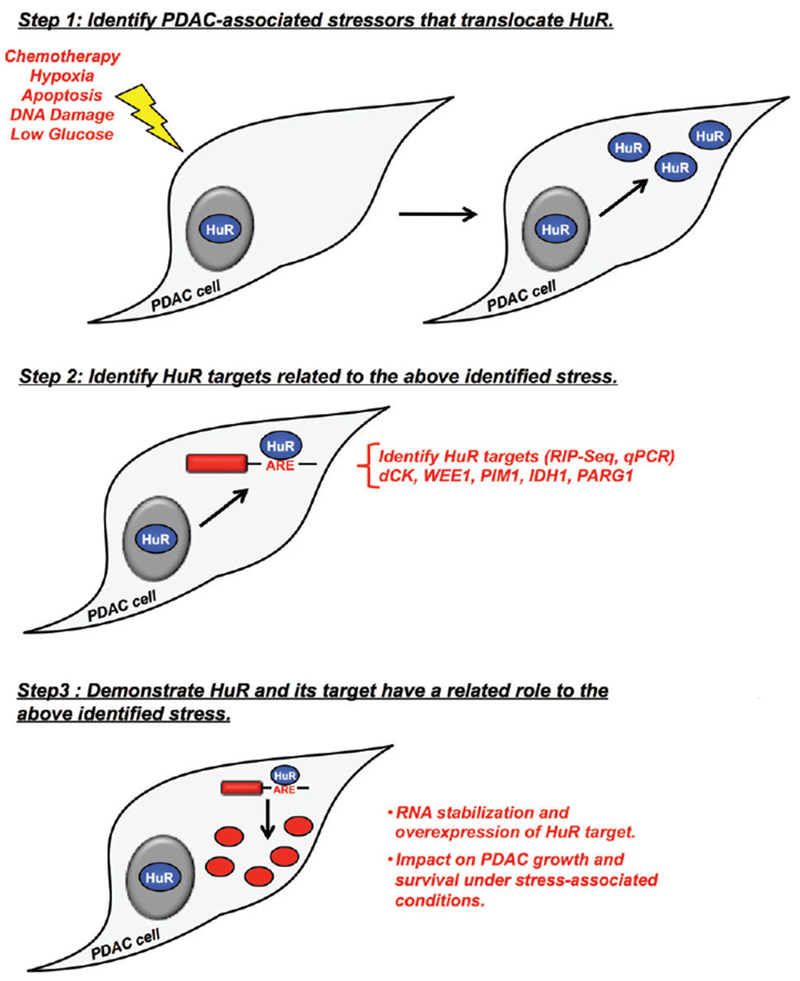
Framework for linking a pancreatic cancer-associated stress with a novel HuR target that confers a pro-survival pancreatic ductal adenocarcinoma (PDAC) phenotype
3.1 |. Pre-clinical evidence that HuR is a good target in PDAC cells
More recently, we generated and evaluated HuR-null cells in two different PDAC cell lines by CRISPR/Cas9 deletion of the ELAVL1 (HuR) gene (Lal, Cheung, et al., 2017). While HuR(−/−) cells displayed a mild growth phenotype in vitro, deletion of HuR generated a dramatic xenograft lethal phenotype in vivo, supporting the notion that HuR is required for tumor initiation and progression in vivo. Specifically, deletion of HuR in two PDAC cell lines were unable to engraft in mice, compared to isogenic proficient HuR PDAC lines. To demonstrate that this phenotype was directly due to the loss of HuR, addition of a HuR cDNA rescued the ability of the PDAC cells to establish tumors in nude mice (Lal, Cheung, et al., 2017). These data support our previous published work that inducible knockdown of HuR using short hairpin RNA-based silencing in PDAC cells in vivo can reduce the tumor size by roughly fourfold (Jimbo et al., 2015). Similarly, we recently demonstrated the efficacy of targeting HuR in GI cancer in vivo by using small molecule inhibitors of HuR function (Blanco et al., 2016; Lal et al., 2017).
4 |. HUR IS A POTENT MODULATOR OF PDAC DRUG RESISTANCE WITHIN THE ELEMENTS OF THE PDAC ENVIRONMENT
The selective pressure imposed by the harsh tumor microenvironment favors growth of the most aggressive and fit PDAC cells, which tend to be the most resistant to cytotoxic chemotherapeutic agents (Jones et al., 2008; Vineis, 2000; Von Hoff et al., 2013). Previously, it has been shown that PDAC tumors are embedded in a highly hypoxic and nutrient-deprived tumor microenvironment wherein clonal populations of PDACs with aggressive traits thrive and expand (Anderson, Mack, & Silverman, 2006; Koong et al., 2000; Vineis, 2000). In order to overcome the harsh stress imposed by chronic hypoxia (e.g., low oxygen pressure [pO2] and intratumoral perfusion) PDAC cells orchestrate a multifaceted response by activating hypoxia-inducible factors (HIFs) such as HuR and PIM1 (Bertout, Patel, & Simon, 2008; Burkhart et al., 2013). Additionally, we have recently demonstrated that HuR translocation from the nucleus to the cytoplasm occurs under nutrient deprivation (e.g., low glucose) (Zarei et al., 2017). Specifically, under these conditions HuR regulates the key metabolic enzyme, wild-type IDH1 (Table 1). These potent acute cellular reprogramming events regulated by HuR activates pathways responsible for regulating cell motility, intracellular pH, mitochondrial function, angiogenesis, cellular metabolism, DNA repair, and cell survival (Buchler et al., 2004; Chang, Jurisica, Do, & Hedley, 2011; Humar, Kiefer, Berns, Resink, & Battegay, 2002; Zarei et al., 2017). Indeed, we have shown that HuR plays critical roles in both hypoxia and low glucose-induced chemoresistance that represents a major barrier for the clinical efficacy of chemotherapeutic regimens in PDAC (Blanco, Jimbo, et al., 2016; Zarei et al., 2017; Figure 1 and Table 1). In future, more sophisticated studies will address the additive effect of hypoxia and nutrient deprivation on HuR’s subcellular localization and function.
4.1 |. Example 1 of a validated HuR target: The proto-oncogene PIM1
PIM1 (Proviral Integration site for Moloney murine leukemia virus 1), a serine-threonine kinase, has emerged as a prominent modulator of therapeutic resistance in head and neck squamous cell carcinoma, prostate carcinoma, and, most recently, PDAC (Blanco, Jimbo, et al., 2016). PIM1 drives chemoresistance by phosphorylating and inactivating key apoptotic and tumor suppressive proteins (Blanco, Jimbo, et al., 2016), thus rendering cells resistant to the stress imposed by DNA-damaging cytotoxic chemotherapy. Until recently (Blanco, Jimbo, et al., 2016), the mechanism behind PIM1 overexpression in PDAC was unknown, especially in the context of the hypoxic tumor microenvironment (1% oxygen). Histologic analysis of PIM1 expression in PDAC tumors revealed a strong correlation with tumor hypoxia markers (Blanco, Jimbo, et al., 2016). Since hypoxia-mediated PIM1 overexpression occurs in a HIF-1α-independent manner (Blanco, Jimbo, et al., 2016), and no known PIM1 mutations have been identified in PDAC, the contribution of posttranscriptional mechanisms was evaluated. We identified that a cis-acting AREs present in the 3′UTR of the PIM1 mRNA mediates interaction with HuR under conditions of hypoxic stress. This regulatory mechanism results in enhanced PIM1 mRNA stability and consequently PIM1 protein overexpression.
4.2 |. Example 2 of validated HuR target: The metabolic enzyme IDH1
As discussed, HuR protects PDACs not only from hypoxia but also nutrient-related stress (Blanco, Jimbo, et al., 2016; Burkhart et al., 2013). Since nutrient deprivation and chemotherapy induce a surge in reactive oxygen species (ROS) (Chio & Tuveson, 2017; Zarei et al., 2017), the adaptive mechanisms required by PDAC cells to survive oxidative stress in the PDAC microenvironment likely also contribute to chemotherapy resistance. For example, HuR regulates a critical anti-ROS defense system through the posttranscriptional stabilization of the nicotinamide adenine dinucleotide phosphate (NAPDH)-generating enzyme, isocitrate dehydrogenase 1 (IDH1) (Zarei et al., 2017). It was observed that diminished nutrient availability (e.g., low glucose levels) promotes chemotherapy resistance in PDAC cells and mouse xenografts, as well as in a retrospective cohort of patients who underwent pancreatic resection for PDAC. HuR silencing by RNAi resulted in impaired PDAC cell viability under nutrient (glucose) withdrawal, and also abrogated chemotherapy resistance related to nutrient withdrawal. Ribonucleoprotein immunoprecipitation studies of HuR confirmed the binding interaction of HuR to IDH1 mRNA. Importantly, expression of IDH1 RNA and protein was almost completely absent in two different HuR-null PDAC cell lines, and transient IDH1 overexpression restored chemotherapy resistance under low nutrient conditions. Furthermore, stable expression of IDH1 in HuR-null PDAC cells restored the ability of these cells to successfully implant and grow in nude mice at the same rate as HuR-proficient control cells.
4.3 |. Example 3 of a validated HuR target: The mitotic checkpoint inhibitor WEE1
Previously it has been shown that conventional DNA damaging agents such as arsenite (Bhattacharyya, Habermacher, Martine, Closs, & Filipowicz, 2006), actinomycin D (Rattenbacher & Bohjanen, 2012), and hydrogen peroxide (Martin-Garrido et al., 2011) can activate (translocate) HuR (Wang et al., 2000). We have shown that other DNA damaging agents used clinically promotes activation of HuR, as assessed by the rapid translocation of HuR from the nucleus to the cytoplasm (Lal et al., 2014). Additionally, mitomycin C (MMC) treatment within in hours induced γ-H2AX foci in both control and HuR-silenced cells, indicating the presence of DNA damage breaks (Lal et al., 2014). Remarkably, after MMC treatment the number of foci per nuclei increased significantly in HuR-silenced cells compared to the control cells, demonstrating that DNA damage persisted and DNA repair was considerably delayed in the absence of HuR. Mechanistically to prove that this was a HuR-dependent event, HuR depletion resulted in significant downregulation of WEE1 (a mitotic inhibitor) mRNA and protein, both in treated and untreated cells. Ribonucleoprotein immunoprecipitation analysis validated that WEE1 mRNA was bound to HuR, demonstrating WEE1 mRNA as a novel HuR target in the DNA damage repair pathway. These observations were further supported by findings that showed silencing HuR followed by stress resulted in enhanced phosphorylation of CDK1 (a key cell cycle regulator) that phosphorylates HuR and restricts its localization to the nucleus (Kim et al., 2008). Together, these data indicate that HuR regulates the DNA damage repair in part via WEE1, which may lead to the inhibitory phosphorylation of CDK1 and G2/M cell cycle arrest.
In summary, it was demonstrated that the harsh tumor microenvironment (i.e., low glucose, hypoxia, and DNA damage) of PDAC renders these cells resistant to chemotherapy, and that posttranscriptional regulation of IDH1, PIM1, and WEE1 by HuR, in part, drives an underlying adaptive survival mechanism. These data may explain why conventional chemotherapy shows limited effectiveness against PDAC, and highlights HuR as a compelling therapeutic target in the context of the harsh and oxidative PDAC tumor microenvironment. Further effort to understand the significance of reported posttranslational modifications on HuR’s subcellular localization status (i.e., specifically phosphorylation) (Abdelmohsen et al., 2007; Abdelmohsen & Gorospe, 2010; Chemnitz, Pieper, Gruttner, & Hauber, 2009; Doller et al., 2007; Doller et al., 2011; Doller, Schlepckow, Schwalbe, Pfeilschifter, & Eberhardt, 2017; Eberhardt, Doller, & Pfeilschifter, 2012; Grammatikakis, Abdelmohsen, & Gorospe, 2017; Kim et al., 2008; Liu et al., 2009; Scheiba, Aroca, & Diaz-Moreno, 2012; Wang et al., 2004; Yoon et al., 2014; Yu et al., 2011; Zou et al., 2008) is currently being investigated. Understanding the specific kinases and protein motifs affected under the above mentioned conditions may be important for targeting HuR in unique PDA tumor microenvironment niches. Based on this work, we hypothesize that this gene regulatory mechanism serves as a backbone of chemoresistance in PDAC that can be therapeutically disrupted using agents targeting HuR.
5 |. HUR AS A BIOMARKER IN PDAC AND OTHER CANCERS: “TO BE OR NOT TO BE A BIOMARKER”
HuR is abundant in most cancers and reduced in normal cells, and the cytoplasmic HuR status in tumor specimens have been shown to correlate with poor prognostic value in many tumor types (Abdelmohsen & Gorospe, 2010; Kotta-Loizou et al., 2014; Wang et al., 2013; Zucal et al., 2015). Specifically, we have demonstrated that HuR is overexpressed and functionally active (i.e., cytoplasmic status) in patient-derived cell lines and in clinical samples (Costantino et al., 2009; Dixon et al., 2001; Young et al., 2009). Importantly, we have demonstrated that cytoplasmic HuR correlates with an identified HuR target in patient samples (Blanco, Jimbo, et al., 2016; McAllister et al., 2014; Pineda et al., 2012). In line with previous work in other tumor systems, HuR cytoplasmic expression was shown to be a poor prognostic marker, as it has been shown to correlate with tumor T staging (Richards et al., 2010). In regards to cytoplasmic HuR as a predictive biomarker for gemcitabine-based adjuvant therapy, we have followed up our evaluation of this with multiple publications (Costantino et al., 2009; McAllister et al., 2014; Richards et al., 2010; Tatarian et al., 2018). Many possibilities could account for why HuR status correlates as a predictive marker for gemcitabine and/or 5-fluorouracil-based therapies in these settings: (a) We performed these studies primarily in the adjuvant setting (postoperatively) in stage I and II patients. Thus, these studies analyzed the primary tumors, which were resected, and adjuvant therapy would most likely be targeting the micrometastatic and metastatic tumor cells, which may have a different cytoplasmic HuR status depending on the microenvironment. (b) Different therapeutic regimens may include radiation that we know from previous work may disrupt HuR’s biology (Masuda et al., 2011). Ultimately, validating HuR as a predictive marker should be performed in a prospective fashion in a clinical trial focused in patients with advanced, metastatic disease.
6 |. ONGOING THERAPEUTIC STRATEGIES TO TARGET HUR
Revisiting our proposed criteria for the therapeutic value of a target in PDAC, HuR checks off many of these boxes: (a) it is an available target: HuR is overexpressed in PDAC cells compared to normal cells, (b) HuR has a functional purpose that can be targeted in PDAC cells as a pro-survival hub, (c) HuR is activated by the unique tumor microenvironment in PDAC tumors, and (d) HuR provides an important survival advantage for PDAC cells. Therefore, based on nearly a decade’s worth of published data, strategies to target HuR as a candidate molecular target are gaining significant attention (Zucal et al., 2015).
Our recent efforts have led the evaluation of lead small molecule HuR inhibitors (Blanco, Jimbo, et al., 2016; Blanco, Preet, et al., 2016; Lal, Cerofolini, et al., 2017; Romeo et al., 2016; Wu et al., 2015; Young et al., 2012). Currently, there are a number of HuR small molecule inhibitors that have been identified by high-throughput biochemical screens with the ability to inhibit HuR activity (Chae et al., 2009; D’Agostino, Adami, & Provenzani, 2013; Guo et al., 2016; Meisner et al., 2007; Wu et al., 2015; Zucal et al., 2015). MS-444 leads the small molecule inhibitors of HuR in citations and evaluation in different models and pathways. MS-444 is a small natural product produced by gram-positive bacteria Micromonospora and chrysanthone-like in structure that was originally identified as an anti-tumorigenic, anti-inflammatory and anti-HIV agent (Aotani & Saitoh, 1995; Nakanishi, Chiba, Yano, Kawamoto, & Matsuda, 1995). MS-444 and a set of chemically related polyketides were identified as high-affinity small molecule HuR inhibitors by our collaborators (Novartis, Basel, Switzerland) (Meisner et al., 2007). Mechanistically, MS-444 has been shown to inhibit HuR homodimerization which prevents the binding of AREs (Meisner et al., 2007). This prevents translocation of HuR (and its associated mRNA cargos) to the cytoplasm. In a number of tumor cells, HuR inhibition by MS-444 leads to a dose-dependent reduction in cell proliferation by promoting apoptosis. Moreover, MS-444 inhibits HuR-dependent tumorigenic potential of colon cancer cells and GI tumorigenesis (Blanco, Preet, et al., 2016; Lang et al., 2017). While these studies provide proof-of-principal in small molecule targeting of HuR, limitations with MS-444 exist such as high IC50 values, large-scale drug solubility, and in vivo drug modifications (our personal observations). Other prominent small molecules have been described as HuR inhibitors include: CMLD-2, which maintains a coumarin-derived core and binds HuR to disrupt RNA binding (Wu et al., 2015); DHTS (15,16-dihydrotanshinone-I) derived from Salvia miltiorrhiza, is in the family of diterpenic tanshinones and inhibits the HuR-RNA complex formation (D’Agostino et al., 2015; Lal, Cerofolini, et al., 2017); and pyrvinium pamoate, an FDA-approved anthelminthic drug that blocks HuR nucleocytoplasmic translocation (Guo et al., 2016) and has been shown effectively reduce in vivo tumor growth for several different cancers, including pancreatic cancer(Esumi, Lu, Kurashima, & Hanaoka, 2004; Guo et al., 2016; Li et al., 2014; Lim et al., 2014; Sugimoto et al., 2015; Xu et al., 2013). Ongoing in vivo studies and chemical refinements of these and other compounds will aid in identifying a best-in-class molecule to test in the clinic.
7 |. FUTURE TARGETING OF HUR
Working in collaboration with Genisphere LLC (Hatfield, PA), we are using the novel 3DNA dendrimer for siHuR delivery; it is unique both in structure and functional capabilities as compared to other nanomaterials (Huang et al., 2016). Its dendrimeric structure is built entirely from interconnected monomeric subunits of CpG-free DNA through a directed assembly process that yields a single molecular species. Derivatization of individual DNA arms with therapeutic payloads (e.g., siRNA, miRNA, DNA, or small molecules), targeting moieties (e.g., antibodies or ligands), and imaging tags allows for versatility in the design of nanocarriers having potential for a wide range of applications. The ability to easily modify the targeting moiety (e.g., targeting epidermal growth factor receptor [EGFR], transferrin) opens the possibility to move towards a realistic personalized approach. Current efforts have tested PDAC-specific targeting moieties for delivery of HuR-specific siRNAs and showed promising results both in vitro and in vivo indicating the feasibility of this novel targeting approach (Brody, Dixon laboratories unpublished).
8 |. CONCLUSION
As the clinical medicine field continues to advance the concept of targeting a nonmutated gene or pathway, HuR is emerging as an attractive target in PDAC and other cancers. For example, HuR has been demonstrated to either be a marker for malignancy or have an oncogenic role in numerous tumor systems including breast, ovarian, and colon (Heinonen et al., 2005; Huang et al., 2016; Young et al., 2009). As a means to halt tumorigenesis or treat cancer, HuR functions analogous to other gene regulatory mechanisms and proteins that govern these mechanisms, allowing it to be considered as a candidate target. However, the concept of targeting HuR is a new approach and many questions are still unanswered. For instance, what are the most critical HuR-target mRNAs in PDAC? Will targeting HuR be effective as a monotherapy or serve best in the context of combination therapy? How long lasting is the HuR pro-survival effect in a given tumor (e.g., minutes, hours, and days), and will inhibition of HuR be effective enough to compensate for this effect? Are the other elements that will compensate for HuR inhibition and render a PDAC cell resistant to this type of therapy? These and other questions are intriguing, but leave plenty for the field to explore and prioritize. As we and others continue to understand HuR’s role in tumorigenesis and cancer cell survival, we strongly believe enough evidence exists to initiate therapeutic strategies, some described herein, to target this molecule in pancreatic cancer.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01 CA212600 [J.R.B.], R01 CA134609 [D.A.D.]), American Cancer Society grant MRSG-14-019-01-CDD (J.R.B.), NIH/NCI Cancer Center support grants P30 CA056036 and P30 CA168524, Fund A Cure and the Michele Barnett Rudnick Fund (J.R.B.), and 2015 Pancreatic Cancer Action Network American Association for Cancer Research Acceleration Network grant (15-90-25-BROD). Of note, J.R.B. works with Pan-CAN on the KYT initiative mentioned within the project. We would like to acknowledge the art work of Jennifer Brumbaugh (TJU).
Funding information
2015 Pancreatic Cancer Action Network American Association for Cancer Research Acceleration Network, Grant/Award number: 15-90-25-BROD; Fund A Cure and the Michele Barnett Rudnick Fund; NIH/NCI Cancer Center, Grant/Award numbers: P30 CA168524, P30 CA056036; American Cancer Society, Grant/Award number: MRSG-14-019-01-CDD; National Institutes of Health, Grant/Award numbers: R01 CA134609, R01 CA212600
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
RELATED WIREs ARTICLE
This article is categorized under: RNA in Disease and Development > RNA in Disease
REFERENCES
- Abdelmohsen K Gorospe M Posttranscriptional regulation of cancer traits by HuR WIREs RNA 2010. 1 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K Kim MM Srikantan S Mercken EM Brennan SE Wilson GM Gorospe M miR-519 suppresses tumor growth by reducing HuR levels Cell Cycle 2010. 9 1354–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K Pullmann R Jr. Lal A Kim HH Galban S Yang X Gorospe M Phosphorylation of HuR by Chk2 regulates SIRT1 expression Molecular Cell 2007. 25 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K Srikantan S Kuwano Y Gorospe M miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels Proceedings of the National Academy of Sciences of the United States of America 2008. 105 20297–20302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KE Mack TM Silverman D Schottenfeld D JFJ F Cancer of the pancreas Cancer epidemiology and prevention 2006. 721–762 Oxford University Press; New York, NY: 3rd ed. [Google Scholar]
- Aotani Y Saitoh Y Structure determination of MS-444: a new myosin light chain kinase inhibitor Journal of Antibiotics (Tokyo) 1995. 48 952–953 [DOI] [PubMed] [Google Scholar]
- Bailey P Chang DK Nones K Johns AL Patch AM Gingras MC Grimmond SM Genomic analyses identify molecular subtypes of pancreatic cancer Nature 2016. 531 47–52 [DOI] [PubMed] [Google Scholar]
- Bertout JA Patel SA Simon MC The impact of O2 availability on human cancer Nature Reviews. Cancer 2008. 8 967–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN Habermacher R Martine U Closs EI Filipowicz W Relief of microRNA-mediated translational repression in human cells subjected to stress Cell 2006. 125 1111–1124 [DOI] [PubMed] [Google Scholar]
- Blanco FF Jimbo M Wulfkuhle J Gallagher I Deng J Enyenihi L Brody JR The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells Oncogene 2016. 35 2529–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FF Preet R Aguado A Vishwakarma V Stevens LE Vyas A Dixon DA Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis Oncotarget 2016. 7 74043–74058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CM Steitz JA HuR and mRNA stability Cellular and Molecular Life Sciences 2001. 58 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody JR Gonye GE HuR’s role in gemcitabine efficacy: An exception or opportunity? WIREs RNA 2011. 2 435–444 [DOI] [PubMed] [Google Scholar]
- Buchler P Reber HA Buchler MW Friess H Lavey RS Hines OJ Antiangiogenic activity of genistein in pancreatic carcinoma cells is mediated by the inhibition of hypoxia-inducible factor-1 and the down-regulation of VEGF gene expression Cancer 2004. 100 201–210 [DOI] [PubMed] [Google Scholar]
- Burkhart RA Pineda DM Chand SN Romeo C Londin ER Karoly ED Winter JM HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer RNA Biology 2013. 10 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae MJ Sung HY Kim EH Lee M Kwak H Chae CH Park WY Chemical inhibitors destabilize HuR binding to the AU-rich element of TNF-alpha mRNA Experimental & Molecular Medicine 2009. 41 824–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q Jurisica I Do T Hedley DW Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer Cancer Research 2011. 71 3110–3120 [DOI] [PubMed] [Google Scholar]
- Cheadle C, Fan J, Cho-Chung YS, Werner T, Ray J, Do L, Becker KG. Control of gene expression during T cell activation: Alternate regulation of mRNA transcription and mRNA stability. BMC Genomics. 2005;6:75. doi: 10.1186/1471-2164-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemnitz J Pieper D Gruttner C Hauber J Phosphorylation of the HuR ligand APRIL by casein kinase 2 regulates CD83 expression European Journal of Immunology 2009. 39 267–279 [DOI] [PubMed] [Google Scholar]
- Chio IIC Tuveson DA ROS in cancer: The burning question Trends in Molecular Medicine 2017. 23 411–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA Sadanandam A Olson P Gibb WJ Truitt M Gu S Gray JW Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy Nature Medicine 2011. 17 500–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino CL Witkiewicz AK Kuwano Y Cozzitorto JA Kennedy EP Dasgupta A Brody JR The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase Cancer Research 2009. 69 4567–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino VG Adami V Provenzani A A novel high throughput biochemical assay to evaluate the HuR protein-RNA complex formation PLoS One 2013. 8 e72426–e72434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino VG, Lal P, Mantelli B, Tiedje C, Zucal C, Thongon N, Provenzani A. Dihydrotanshinone-I interferes with the RNA-binding activity of HuR affecting its post-transcriptional function. Scientific Reports. 2015;5:16478. doi: 10.1038/srep16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DA Tolley ND King PH Nabors LB McIntyre TM Zimmerman GA Prescott SM Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells Journal of Clinical Investigation 2001. 108 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doller A Huwiler A Muller R Radeke HH Pfeilschifter J Eberhardt W Protein kinase C alpha-dependent phosphorylation of the mRNA-stabilizing factor HuR: Implications for posttranscriptional regulation of cyclooxygenase-2 Molecular Biology of the Cell 2007. 18 2137–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doller A Schlepckow K Schwalbe H Pfeilschifter J Eberhardt W Tandem phosphorylation of serines 221 and 318 by protein kinase C delta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR Molecular and Cellular Biology 2017. 30 1397–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doller A Winkler C Azrilian I Schulz S Hartmann S Pfeilschifter J Eberhardt W High-constitutive HuR phosphorylation at Ser 318 by PKC {delta} propagates tumor relevant functions in colon carcinoma cells Carcinogenesis 2011. 32 676–685 [DOI] [PubMed] [Google Scholar]
- Eberhardt W Doller A Pfeilschifter J Regulation of the mRNA-binding protein HuR by posttranslational modification: Spotlight on phosphorylation Current Protein & Peptide Science 2012. 13 380–390 [DOI] [PubMed] [Google Scholar]
- Esumi H Lu J Kurashima Y Hanaoka T Antitumor activity of pyrvinium pamoate, 6-(dimethylamino)-2-[2-(2,5-dimethyl-1-phenyl-1H-pyrrol-3-yl) ethenyl]-1-methyl-qu inolinium pamoate salt, showing preferential cytotoxicity during glucose starvation Cancer Science 2004. 95 685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J Yang X Wang W Wood WH 3rd Becker KG Gorospe M Global analysis of stress-regulated mRNA turnover by using cDNA arrays Proceedings of the National Academy of Sciences of the United States of America 2002. 99 10611–10616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XC Steitz JA HNS, a nuclear-cytoplasmic shuttling sequence in HuR Proceedings of the National Academy of Sciences of the United States of America 1998a. 95 15293–15298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XC Steitz JA Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs The EMBO Journal 1998b. 17 3448–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallouzi IE Steitz JA Delineation of mRNA export pathways by the use of cell-permeable peptides Science 2001. 294 1895–1901 [DOI] [PubMed] [Google Scholar]
- Garneau NL Wilusz J Wilusz CJ The highways and byways of mRNA decay Nature Reviews. Molecular Cell Biology 2007. 8 113–126 [DOI] [PubMed] [Google Scholar]
- Grammatikakis I, Abdelmohsen K, Gorospe M. Posttranslational control of HuR function. WIREs RNA. 2017;8(1):e1372. doi: 10.1002/wrna.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J Lv J Chang S Chen Z Lu W Xu C Pang X Inhibiting cytoplasmic accumulation of HuR synergizes genotoxic agents in urothelial carcinoma of the bladder Oncotarget 2016. 7 45249–45262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen M Bono P Narko K Chang SH Lundin J Joensuu H Ristimaki A Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma Cancer Research 2005. 65 2157–2161 [DOI] [PubMed] [Google Scholar]
- Hinman MN Lou H Diverse molecular functions of Hu proteins Cellular and Molecular Life Sciences 2008. 65 3168–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH Peng W Furuuchi N DuHadaway JB Jimbo M Pirritano A Sawicki JA Insights from HuR biology point to potential improvement for second-line ovarian cancer therapy Oncotarget 2016. 7 21812–21824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH Peng W Furuuchi N Gerhart J Rhodes K Mukherjee N Sawicki JA Delivery of therapeutics targeting the mRNA-binding protein HuR using 3DNA nanocarriers suppresses ovarian tumor growth Cancer Research 2016. 76 1549–1559 [DOI] [PubMed] [Google Scholar]
- Humar R Kiefer FN Berns H Resink TJ Battegay EJ Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling The FASEB Journal 2002. 16 771–780 [DOI] [PubMed] [Google Scholar]
- Jimbo M Blanco FF Huang YH Telonis AG Screnci BA Cosma GL Brody JR Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells Oncotarget 2015. 6 27312–27331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S Zhang X Parsons DW Lin JC Leary RJ Angenendt P Kinzler KW Core signaling pathways in human pancreatic cancers revealed by global genomic analyses Science 2008. 321 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene J Why is Hu where? Shuttling of early-response messenger RNA subsets Proceedings of the National Academy of Sciences of the United States of America 1999. 96 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabar KS. Hallmarks of cancer and AU-rich elements. WIREs RNA. 2017;8:e1368. doi: 10.1002/wrna.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH Abdelmohsen K Lal A Pullmann R Jr. Yang X Galban S Gorospe M Nuclear HuR accumulation through phosphorylation by Cdk1 Genes & Development 2008. 22 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES O’Reilly EM Brody JR Witkiewicz AK Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine Gastroenterology 2016. 150 48–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koong AC Mehta VK Le QT Fisher GA Terris DJ Brown JM Vierra M Pancreatic tumors show high levels of hypoxia International Journal of Radiation Oncology, Biology, Physics 2000. 48 919–922 [DOI] [PubMed] [Google Scholar]
- Kotta-Loizou I, Giaginis C, Theocharis S. Clinical significance of HuR expression in human malignancy. Medical Oncology. 2014;31:161. doi: 10.1007/s12032-014-0161-y. [DOI] [PubMed] [Google Scholar]
- Lal P Cerofolini L D’Agostino VG Zucal C Fuccio C Bonomo I Provenzani A Regulation of HuR structure and function by dihydrotanshinone-I Nucleic Acids Research 2017. 45 9514–9527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal S Burkhart RA Beeharry N Bhattacharjee V Londin ER Cozzitorto JA Brody JR HuR posttranscriptionally regulates WEE1: Implications for the DNA damage response in pancreatic cancer cells Cancer Research 2014. 74 1128–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal S Cheung EC Zarei M Preet R Mambelli-Lisboa NC Chand SN Brody JR CRISPR knockout of the HuR gene in pancreatic and colorectal cancer cells causes a xenograft lethal phenotype Molecular Cancer Research 2017. 15 696–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M Berry D Passecker K Mesteri I Bhuju S Ebner F Gasche C HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis Cancer Research 2017. 77 2424–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Flaveny CA, Giambelli C, Fei DL, Han L, Hang BI, Robbins DJ. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS One. 2014;9:e101969. doi: 10.1371/journal.pone.0101969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M Otto-Duessel M He M Su L Nguyen D Chin E Jones JO Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium ACS Chemical Biology 2014. 9 692–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L Rao JN Zou T Xiao L Wang PY Turner DJ Wang JY Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation Molecular Biology of the Cell 2009. 20 4885–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Silanes I Fan J Yang X Zonderman AB Potapova O Pizer ES Gorospe M Role of the RNA-binding protein HuR in colon carcinogenesis Oncogene 2003. 22 7146–7154 [DOI] [PubMed] [Google Scholar]
- Martin-Garrido A Gonzalez-Ramos M Griera M Guijarro B Cannata-Andia J Rodriguez-Puyol D Saura M H2O2 regulation of vascular function through sGC mRNA stabilization by HuR Arteriosclerosis, Thrombosis, and Vascular Biology 2011. 31 567–573 [DOI] [PubMed] [Google Scholar]
- Masuda K Abdelmohsen K Kim MM Srikantan S Lee EK Tominaga K Gorospe M Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation The EMBO Journal 2011. 30 1040–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K Galban S Lopez de Silanes I Martindale JL Atasoy U Keene JD Gorospe M RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation Proceedings of the National Academy of Sciences of the United States of America 2003. 100 8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister F Pineda DM Jimbo M Lal S Burkhart RA Moughan J Brody JR dCK expression correlates with 5-fluorouracil efficacy and HuR cytoplasmic expression in pancreatic cancer: A dual-institutional follow-up with the RTOG 9704 trial Cancer Biology & Therapy 2014. 15 688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner NC Filipowicz W Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression Advances in Experimental Medicine and Biology 2010. 700 106–123 [PubMed] [Google Scholar]
- Meisner NC Hintersteiner M Mueller K Bauer R Seifert JM Naegeli HU Auer M Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR Nature Chemical Biology 2007. 3 508–515 [DOI] [PubMed] [Google Scholar]
- Nakanishi S Chiba S Yano H Kawamoto I Matsuda Y MS-444, a new inhibitor of myosin light chain kinase from Micromonospora sp. KY7123 Journal of Antibiotics (Tokyo) 1995. 48 948–951 [DOI] [PubMed] [Google Scholar]
- Pineda DM Rittenhouse DW Valley CC Cozzitorto JA Burkhart R Leiby B Brody JR HuR’s post-transcriptional regulation of death receptor 5 in pancreatic cancer cells Cancer Biology & Therapy 2012. 13 946–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishvaian MJ Bender RJ Matrisian LM Rahib L Hendifar A Hoos WA Brody JR A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative Oncotarget 2016. 8 83446–83456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullmann R Jr. Kim HH Abdelmohsen K Lal A Martindale JL Yang X Gorospe M Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs Molecular and Cellular Biology 2007. 27 6265–6278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattenbacher B Bohjanen PR Evaluating posttranscriptional regulation of cytokine genes Methods in Molecular Biology 2012. 820 71–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebane A Aab A Steitz JA Transportins 1 and 2 are redundant nuclear import factors for hnRNP A1 and HuR RNA 2004. 10 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards NG Rittenhouse DW Freydin B Cozzitorto JA Grenda D Rui H Witkiewicz AK HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients Annals of Surgery 2010. 252 499–505 [DOI] [PubMed] [Google Scholar]
- Romeo C Weber MC Zarei M DeCicco D Chand SN Lobo AD Brody JR HuR contributes to TRAIL resistance by restricting death receptor 4 expression in pancreatic cancer cells Molecular Cancer Research 2016. 14 599–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiba RM Aroca A Diaz-Moreno I HuR thermal stability is dependent on domain binding and upon phosphorylation European Biophysics Journal 2012. 41 597–605 [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K Marmo TP Dixon DA Dannenberg AJ Regulation of cyclooxgenase-2 mRNA stability by taxanes: Evidence for involvement of p38, MAPKAPK-2, and HuR Journal of Biological Chemistry 2003. 278 37637–37647 [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Hayakawa F, Shimada S, Morishita T, Shimada K, Katakai T, Naoe T. Discovery of a drug targeting microenvironmental support for lymphoma cells by screening using patient-derived xenograft cells. Scientific Reports. 2015;5:13054. doi: 10.1038/srep13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatarian T Jiang W Leiby BE Grigoli A Jimbo M Dabbish N Brody JR Cytoplasmic HuR status predicts disease-free survival in resected pancreatic cancer: A post-hoc analysis from the international phase III ESPAC-3 clinical trial Annals of Surgery 2018. 267 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineis P Exposures, mutations and the history of causality Journal of Epidemiology and Community Health 2000. 54 652–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD Ervin T Arena FP Chiorean EG Infante J Moore M Renschler MF Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine The New England Journal of Medicine 2013. 369 1691–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J Guo Y Chu H Guan Y Bi J Wang B Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis International Journal of Molecular Sciences 2013. 14 10015–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W Furneaux H Cheng H Caldwell MC Hutter D Liu Y Gorospe M HuR regulates p21 mRNA stabilization by UV light Molecular and Cellular Biology 2000. 20 760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W Yang X Kawai T Lopez de Silanes I Mazan-Mamczarz K Chen P Gorospe M AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: Involvement in the nuclear import of RNA-binding protein HuR Journal of Biological Chemistry 2004. 279 48376–48388 [DOI] [PubMed] [Google Scholar]
- Williams TK, Costantino CL, Bildzukewicz NA, Richards NG, Rittenhouse DW, Einstein L, Brody JR. pp32 (ANP32A) expression inhibits pancreatic cancer cell growth and induces gemcitabine resistance by disrupting HuR binding to mRNAs. PLoS One. 2010;5:e15455. doi: 10.1371/journal.pone.0015455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X Lan L Wilson DM Marquez RT Tsao WC Gao P Xu L Identification and validation of novel small molecule disruptors of HuR-mRNA interaction ACS Chemical Biology 2015. 10 1476–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lacerda L, Debeb BG, Atkinson RL, Solley TN, Li L, Woodward WA. The antihelmintic drug pyrvinium pamoate targets aggressive breast cancer. PLoS One. 2013;8:e71508. doi: 10.1371/journal.pone.0071508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH Abdelmohsen K Srikantan S Guo R Yang X Martindale JL Gorospe M Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs Nucleic Acids Research 2014. 42 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LE Moore AE Sokol L Meisner-Kober N Dixon DA The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2 Molecular Cancer Research 2012. 10 167–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LE Sanduja S Bemis-Standoli K Pena EA Price RL Dixon DA The mRNA binding proteins HuR and tristetraprolin regulate cyclooxygenase 2 expression during colon carcinogenesis Gastroenterology 2009. 136 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TX Wang PY Rao JN Zou T Liu L Xiao L Wang JY Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function Nucleic Acids Research 2011. 39 8472–8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei M Lal S Parker SJ Nevler A Vaziri-Gohar A Dukleska K Winter JM Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells Cancer Research 2017. 77 4460–4471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou T Liu L Rao JN Marasa BS Chen J Xiao L Wang JY Polyamines modulate the subcellular localization of RNA-binding protein HuR through AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1 The Biochemical Journal 2008. 409 389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucal C D’Agostino V Loffredo R Mantelli B Natthakan T Lal P Provenzani A Targeting the multifaceted HuR protein, benefits and caveats Current Drug Targets 2015. 16 499–515 [DOI] [PubMed] [Google Scholar]