

Abstract
OBJECTIVES
As most hepatocellular carcinoma (HCC) patients have cirrhosis, the association between diabetes and HCC may be confounded by the fact that diabetes is common in patients with cirrhosis. The aim of this study is to investigate whether diabetes increases the risk of HCC in patients with cirrhosis and whether the etiology of liver disease modifies the association between diabetes and HCC.
METHODS
All liver cirrhosis patients who had repeated radiographic evaluation of the liver (that is, ultrasound, computed tomography, or magnetic resonance image) at Mayo Clinic Rochester between January 2006 and December 2011 were included. The Cox proportional hazard regression analysis was used to investigate the effect of diabetes on the risk of HCC.
RESULTS
A total of 739 patients met the eligibility criteria, of whom 253 (34%) had diabetes. After a median follow-up of 38 months, 69 (9%) patients developed HCC. In patients without hepatitis C virus (HCV) infection, diabetes was significantly associated with the risk of developing HCC (hazard ratio (HR)=2.1, 95% confidence interval (CI)=1.1–4.1), whereas in patients with HCV, there was no association (HR=0.8, 95% CI=0.4–1.8). When adjusted for covariates, the interaction between HCV and diabetes remained significant (HR for non-HCV=1.9, 95% CI=0.9–3.7; HR for HCV=0.6, 95% CI=0.2–1.3). Lack of association between diabetes and HCC was externally validated in 410 patients with HCV cirrhosis enrolled in the HALT-C trial.
CONCLUSIONS
Diabetes increases the risk of HCC in patients with non-HCV cirrhosis. In HCV cirrhosis patients who already have very high risk, diabetes may not increase the risk any further.
INTRODUCTION
The incidence of hepatocellular carcinoma (HCC) in the US has tripled over the past three decades, from 1.5 to 4.9 cases per 100,000 per year (1). The main driver of this rise in HCC incidence is commonly attributed to increasing disease burden of hepatitis C virus (HCV) infection (2). Recently, non-alcoholic fatty liver disease (NAFLD) has been reported to be the third leading cause of HCC in the population-based/referral center based cohort studies (3,4). Of known risk factors for HCC, diabetes is reported to have the greatest population-attributable fraction in the US (5).
Most epidemiologic studies have reported a two-to three-fold increase in the risk of HCC in patients with diabetes, regardless of the study design (case-control studies, cohort studies and meta-analyses) (6,7). Some of the association may be attributable to HCC occurring in patients with NAFLD many of whom have diabetes. Moreover, even in patients with cirrhosis from etiology other than NAFLD, progression of liver dysfunction is accompanied by insulin resistance and higher prevalence of diabetes (8,9). In contrast, there are studies, oft en from hepatitis B virus (HBV) endemic areas, in which no association between diabetes and HCC is found (10–12). The association between diabetes and HCC could be further obscured given that HCV infection increases the risk of diabetes (13). These conflicting data may be in part due to the complex relationship between diabetes and cirrhosis from different etiologies.
It is clear that in studying the causal effect of diabetes on the risk of HCC, the etiology and severity of the underlying liver disease must be taken into account. Thus, in this work, we investigate (1) the degree to which diabetes is an independent risk factor for HCC in patients with cirrhosis and (2) the potential interaction between liver disease etiology and diabetes as a risk factor for HCC.
METHODS
Patients
The primary analysis in this study was based on all patients with the diagnosis of cirrhosis of the liver seen at Mayo Clinic Rochester between January 2006 and December 2011. By querying an institutional database, we identified all patients with cirrhosis, as defined by liver histology, features of portal hypertension (splenomegaly, esophageal varices, thrombocytopenia (platelet <150 K), ascites, or hepatic encephalopathy), or radiographical evidence in the setting of chronic liver disease. Our study population consisted of patients who underwent ultrasound, computed tomography, or magnetic resonance image more than once at least 6 months apart. Thus, patients who did not have more than 6 months of radiographical follow-up, including those who underwent liver transplantation (LT) or died within 6 months of initial assessment were excluded (N =746). In addition, patients with previous history of HCC or HCC diagnosed at the initial evaluation or within 6 months thereafter were excluded (N =132). Finally, patients who initially presented with cholangiocarcinoma in the setting of liver cirrhosis (N =3) were excluded. The study was approved by the institutional review board of Mayo Foundation.
We validated the results of our analysis using data from the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial (14). In brief, patients with chronic hepatitis C with compensated liver function, who failed to achieve sustained virologic response after previous interferon treatment and had histological evidence of advanced hepatic fibrosis or cirrhosis were enrolled in a randomized trial evaluating the potential benefit of 3.5 years of peginterferon alfa-2a treatment in reducing the progression of liver disease or development of HCC (ref. 15). Among 428 patients with cirrhosis at baseline, 18 patients were excluded (17- <6 month follow-up; 1-HCC outcome was not able to be determined per HALT-C investigators) (15). At the time of enrollment, patients were required to undergo ultrasound, computed tomography or magnetic resonance image to exclude possible HCC. During the follow-up, abdominal ultrasound was performed at the time of randomization, 6 months after randomization, and every 6–12 months thereafter.
HCC ascertainment
HCC was defined by histologic confirmation (n =30) or clinical diagnostic criteria (n =39) following the American Association For the Study of the Liver Disease (AASLD) guideline updated in 2011 (refs 16–18). The latter criteria included a new liver mass of at least 1 cm in diameter with characteristic features of HCC including both arterial enhancement and delayed washout on dynamic magnetic resonance image or four phasic computed tomography scan. In addition, we included patients who had lesions with compatible cross-sectional and angiographic imaging characteristics and underwent HCC-specific locoregional treatment such as transarterial chemoembolization or radioembolization (n =10).
Ascertainment of HCC in the HALT-C study was published previously–either by histology or following clinical diagnosis criteria (15). The latter criteria consisted of (1) a new mass lesion on imaging and (2) serum alpha fetoprotein levels increasing to ≥1,000 ng/ml. Clinically suspicious lesions with the following criteria were characterized as presumed HCC: (1) two or more imaging studies showing a mass lesion in the liver with arterial enhancement with/without washout, (2) progressively enlarging lesions on ultrasound leading to death of the patient, or (3) a mass lesion with arterial enhancement with/without washout that increased in size or was accompanied by rising serum levels of alpha fetoprotein. All cases of HCC were individually adjudicated by a panel of investigators.
Clinical information
In Mayo patients, clinical information at the time of initial evaluation was collected by medical record review. It included demographic data such as age, sex, and race and clinical characteristics including the etiology of underlying liver disease, severity of hepatic decompensation, as measured by the Child Turcotte Pugh (CTP) and Model for End-stage Liver Disease (MELD) scores, and laboratory results. Diabetes was defined any of the following criteria: (i) documented history of diabetes, (ii) administration of a diabetes medication, or (iii) fasting glucose ≥126 mg/dl or HgbA1C ≥6.5 on two separate occasions. HCV infection was defined by detectable HCV RNA or positive Anti-HCV antibody with a documented history of chronic liver disease. HBV infection was diagnosed by positive serum hepatitis B virus surface antigen. Alcoholic liver disease was designated by history of alcohol abuse or dependence, or documented alcohol consumption of more than 20 gm daily for men and 10 gm daily for women. NAFLD consisted of radiographical diagnosis of steatosis in the absence of alcoholic liver disease, other chronic liver disease (viral, autoimmune or inherited metabolic, or biliary liver disease), or crypto-genic cirrhosis with metabolic syndrome (19).
From the HALT-C data set, baseline demographics, severity of liver disease, diabetes, and laboratory data including HOMA2-IR at the time of enrollment were extracted. Diabetes was defined by self-reported medical history or fasting serum glucose concentrations ≥126 mg/dl on two separate occasions.
Statistical analysis
Clinical characteristics of the study population were compared using the Student t-test for continuous variables and the χ2 test for categorical variables. The incidence of HCC was described by the Kaplan–Meier method and was compared by the log-rank test. The Cox proportional hazard regression analysis was used to investigate the effect of diabetes on the risk of HCC. Patients without HCC were censored at the time of the last radiographical assessment of the liver or at the time of LT or death. Patients were followed until 15th April 2015. JMP 10 (SAS Institute, Cary, NC, USA) was used for the statistical analysis.
RESULTS
Patient characteristics
The primary analysis of this study included 739 patients at Mayo Clinic, of whom 253 (34%) had diabetes (Table 1). The mean age was 57 years and 59% were male. Most patients (90%) were Caucasian. Alcohol (32%), NAFLD (23%), and HCV (21%) were the main etiology of cirrhosis. Other etiology (22%) included cirrhosis from autoimmune hepatitis (n =34), primary biliary cirrhosis (n =33), primary sclerosing cholangitis (n =30), crypto-genic (n =20), cardiac (n =13), alpha 1-antitrypsin (n =10), hemochromatosis (n =8), sarcoidosis (n =4), Wilson’s disease (n =2), drug (n =2), and other rare causes (n =4).
Table 1.
Clinical characteristics
| Diabetes (+) (N =253; %) | Diabetes (−) (N=486; %) | P-value | |
|---|---|---|---|
| Age | 61±10 | 56±13 | <0.01 |
| Gender (male) | 150 (59) | 283 (58) | 0.78 |
| Race (Caucasian) | 221 (87) | 441 (91) | 0.16 |
| Etiology | <0.01 | ||
| HCV | 44 (17) | 110 (23) | |
| HBV | 4 (2) | 11 (2) | |
| Alcohol | 64 (25) | 173 (36) | |
| NAFLD | 118 (47) | 55 (11) | |
| Other | 23 (9) | 137 (28) | |
| BMI | 34±9 | 29±7 | <0.01 |
| Ascites | 0.05 | ||
| None | 143 (57) | 252 (52) | |
| Grades 1–2 | 79 (31) | 131 (27) | |
| Grades 3–4 | 31 (12) | 103 (21) | |
| Hepatic encephalopathy | 0.30 | ||
| None | 204 (81) | 407 (84) | |
| Grades 1–2 | 44 (17) | 69 (14) | |
| Grades 3–4 | 5 (2) | 10 (2) | |
| International normalized ratio | 1.3±0.5 | 1.3±0.5 | 0.34 |
| Albumin | 3.5±0.7 | 3.4±0.7 | 0.64 |
| Creatinine | 1.1±1.0 | 1.0±0.7 | 0.05 |
| Bilirubin | 1.9±3.2 | 2.9±4.6 | <0.01 |
| CTP score | 7.8±1.4 | 8.1±1.5 | 0.01 |
| Model for End-stage Liver Disease score | 11.6±5.1 | 12.4±5.7 | 0.04 |
NAFLD was the leading cause of cirrhosis in patients with diabetes while alcohol was most common among patients without diabetes (P<0.01). As expected, body mass index (BMI) was higher in patients with diabetes compared with patients without diabetes. Patients had mostly compensated or mildly decompensated liver disease: slightly more than half had no evidence of ascites and most patients did not have hepatic encephalopathy. The severity of hepatic decompensation, as measured by the mean CTP and MELD scores were lower in patients with diabetes compared with those without diabetes. In contrast, serum creatinine was higher in diabetic patients.
A total of 102 (40%) patients with diabetes and 139 (29%) without died during the follow-up. Patients with diabetes had a shorter survival (Supplementary Figure S1 online) with a univariate hazard ratio (HR) of 1.5 (95% CI: 1.2–1.9, P <0.01). In contrast, 21 (8%) patients with diabetes and 62 (13%) without underwent LT. There was a trend for patients with diabetes to be less likely to receive LT compared with those without diabetes (Supplementary Figure S2) with a univariate HR of 0.7 (95% CI: 0.4–1.1, P =0.08).
Association between diabetes and risk of HCC
Aft er a median follow-up of 38 months, 69 (9%) patients developed HCC including 27 with diabetes and 42 without. Figure 1 displays the effect of diabetes on HCC incidence in the five categories of liver disease. Although some of the categories only included small number of patients (e.g., HBV), patients with diabetes experienced a higher incidence of HCC except those with HCV. Univariate hazard ratios for diabetes were 0.8 (95% confidence interval (CI)=0.4–1.8) for HCV, compared with 2.4 (95% CI=0.6–15.8) for NAFLD, 4.7 (95% CI=0.2–119.9) for HBV, 1.7 (95% CI=0.6–4.6) for alcoholic liver disease, and 4.5 (95% CI=0.6–23.1) for other etiology. When all of the non-HCV groups were combined together, the association between diabetes and HCC became clearer (Figure 1f). The univariate hazard ratio for diabetes for non-HCV patients was 2.1 (95% CI=1.1–4.1, P =0.02).
Figure 1.
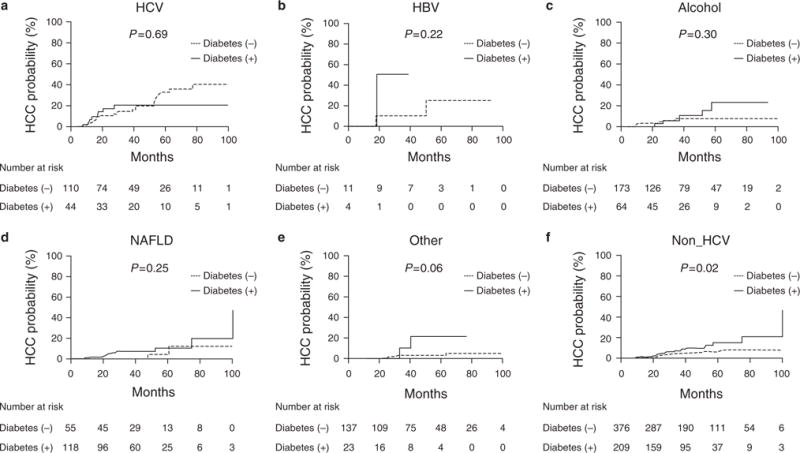
Diabetes and risk of hepatocellular carcinoma (HCC) in different HCC etiology.
Table 2 summarizes a series of proportional hazard regression analyses, considering baseline variables shown in Table 1 as potential predictors of HCC. In univariate analyses, male gender, cirrhosis etiology, and low serum albumin level were significant predictors of HCC development. With ‘other’ etiology as the reference group, HCV, HBV, alcoholic liver disease, and NAFLD were associated with higher hazards of HCC. However, no significant association with HCC was found for age, BMI, CTP, or MELD. Although diabetes was associated with higher BMI, BMI was not associated with the risk of HCC in the subgroup of HCV (HR: 1.0, 95% CI: 0.9–1.0, P =0.44), HBV (HR: 1.2, 95% CI: 1.0–1.6, P =0.06), Alcohol (HR: 1.0, 95% CI: 1.0–1.1, P =0.50), NAFLD (HR: 1.0, 95% CI: 0.9–1.0, P =0.20) and others (HR: 1.1, 95% CI: 1.0–1.2, P =0.19). Finally, the hazard ratio associated with diabetes in the entire data set was 1.4 (P =0.23).
Table 2.
Factors associated with the risk of HCC
| Univariate | Multivariate-1 | Multivariate-2 a | ||||
|---|---|---|---|---|---|---|
| Age (10 years) | 1.1 (0.9–1.4) | 0.21 | 1.4 (1.1–1.8) | 0.02 | 1.4 (1.1–1.9) | 0.02 |
| Male | 1.7 (1.1–3.0) | 0.03 | 1.2 (0.7–2.1) | 0.59 | 1.1 (0.7–2.0) | 0.67 |
| Non Caucasian Race | 1.9 (1.0–3.4) | 0.06 | 1.0 (0.4–2.0) | 0.93 | 1.1 (0.5–2.2) | 0.88 |
| Etiology | ||||||
| Other (Reference) | – | – | – | – | – | – |
| HCV | 6.4 (2.9–17.1) | <0.01 | 6.1 (2.5–17.0) | <0.01 | 8.5 (3.4–25.0) | <0.01 |
| HBV | 6.2 (1.3–23.5) | 0.03 | 5.0 (0.7–26.5) | 0.11 | 4.1 (0.5–21.8) | 0.16 |
| Alcohol | 2.0 (0.8–5.6) | 0.12 | 1.6 (0.6–4.5) | 0.33 | 1.5 (0.6–4.3) | 0.37 |
| NAFLD | 1.9 (0.8–5.6) | 0.17 | 1.4 (0.5–4.3) | 0.49 | 1.1 (0.4–3.3) | 0.91 |
| BMI | 1.0 (1.0–1.0) | 0.74 | ||||
| CTP | 1.0 (0.8–1.2) | 0.99 | ||||
| MELD (per 5 unit) | 1.1 (0.8–1.3) | 0.60 | ||||
| Diabetes | 1.4 (0.8–2.2) | 0.23 | 1.1 (0.6–1.9) | 0.68 | 2.0 (0.9–4.2) | 0.08 |
| Albumin | 0.5 (0.4–0.8) | <0.01 | 0.5 (0.4–0.8) | <0.01 | 0.5 (0.4–0.7) | <0.01 |
| International normalized ratio | 1.1 (0.7–1.6) | 0.54 | ||||
| Creatinine | 0.9 (0.5–1.2) | 0.51 | ||||
| Bilirubin | 1.0 (0.9–1.0) | 0.54 | ||||
| Diabetes a HCV | 0.3 (0.1–0.9) | 0.03 | ||||
BMI, body mass index; CTP, Child Turcotte Pugh; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; MELD, Model for End-stage Liver Disease.
Multivariate model 2 included an interaction term of HCV and diabetes.
Based on these univariate analyses, a multivariable model was constructed, in which age, serum albumin, and HCV etiology were significantly associated with HCC. In pursuing the dichotomous relationship between diabetes and HCC for HCV and non-HCV patients, we tested for interaction between HCV and diabetes on the risk of HCC. In a multivariable model that adjusted for age, sex, race, and albumin, a statistically significant interaction was found between diabetes and HCV. Table 3 explores this relationship further. In patients with HCV infection, diabetes was not associated with development of HCC (HR=0.6) in multivariable analysis in patients with HCV. In contrast, among non-HCV patients, diabetes was associated with nearly twofold increase in the risk of HCC (HR=1.9), although it did not reach statistical significance.
Table 3.
Diabetes and risk of HCC stratified by HCV status
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR | P-value | HR | P-value | |
| HCV | ||||
| Diabetes | 0.8 (0.4–1.8) | 0.68 | 0.6 (0.2–1.3) | 0.20 |
| Albumin | 0.4 (0.2–0.8) | <0.01 | 0.4 (0.2–0.8) | <0.01 |
| Age (10 years) | 1.3 (0.7–2.1) | 0.41 | 1.3 (0.7–2.2) | 0.35 |
| Non-HCV | ||||
| Diabetes | 2.1 (1.1–4.1) | 0.02 | 1.9 (0.9–3.7) | 0.07 |
| Albumin | 0.6 (0.4–0.9) | 0.02 | 0.6 (0.4–0.9) | 0.01 |
| Age (10 years) | 1.4 (1.0–1.8) | 0.02 | 1.4 (1.0–1.9) | 0.04 |
Association between diabetes and HCC in the HALT-C cohort
In the HALT-C data set, there were 410 patients with HCV cirrhosis with sufficient data to allow assessment of the effect of diabetes on the risk of HCC. Relevant characteristics of the study subjects are summarized in Table 4. In definition, all patients had compensated cirrhosis and their CTP and MELD scores were lower compared with the Mayo patients. Eighty (20%) patients had diabetes at baseline. As expected, diabetic patients were older and had higher BMI, and HOMA2-IR2 than non-diabetic patients. Overall, the CTP and MELD scores were comparable between diabetes and non-diabetes subjects, whereas the mean serum albumin was higher and serum bilirubin was lower in diabetic patients.
Table 4.
Clinical characteristics in HALT-C cohort
| Diabetes (+) (N =80; %) | Diabetes (−) (N =330; %) | P-value | |
|---|---|---|---|
| Age | 52±7 | 50±7 | 0.01 |
| Gender (male) | 53 (66) | 245 (74) | 0.16 |
| Race (Caucasian) | 50 (63) | 244 (74) | 0.05 |
| BMI | 32±5 | 30±5 | <0.01 |
| Ascites | 0 (0) | 0 (0) | 1.00 |
| Hepatic encephalopathy | 0 (0) | 0 (0) | 1.00 |
| International normalized ratio | 1.1±0.1 | 1.1±0.1 | 0.05 |
| HOMA2-IR2 | 8.3±4.9 | 5.5±3.7 | <0.01 |
| Albumin | 3.7±0.4 | 3.8±0.4 | 0.01 |
| Creatinine | 1.1±1.0 | 1.0±0.7 | 0.05 |
| Bilirubin | 1.9±3.2 | 2.9±4.6 | <0.01 |
| CTP score | 5.4±0.5 | 5.3±0.5 | 0.05 |
| Model for End-stage Liver Disease score | 7.3±1.4 | 7.5±1.5 | 0.12 |
After a median follow-up of 72 months, 21 diabetic patients (26%) and 63 non-diabetic patients (19%) died, while nine patients with diabetes (11%) and 45 patients without (14%) underwent LT. Diabetes did not affect either outcome. The hazard ratio for diabetes was 1.5 for death (95% CI: 0.9–2.3, P =0.15) and 1.2 for LT (95% CI=0.6–2.4, P =0.61). With regard to the HCC outcome, there were 46 (11%) patients who developed HCC during the follow-up, including 7 (8%) with diabetes and 39 (12%) without. As depicted in Figure 2, there was no difference in the incidence of HCC between diabetic and non-diabetic patients. Table 5 shows univariate and multivariable analyses replicating the analysis shown in Table 2. Diabetes was not associated with HCC in univariate and multivariable analyses. Moreover, the hazard ratios associated with diabetes, albumin, and age in the latter model were essentially identical to those derived in Mayo patients with HCV infection. When HOMA2-IR2 was considered instead of diabetes, there was no association with HCC (Supplementary Table; Supplementary Figure S3).
Figure 2.
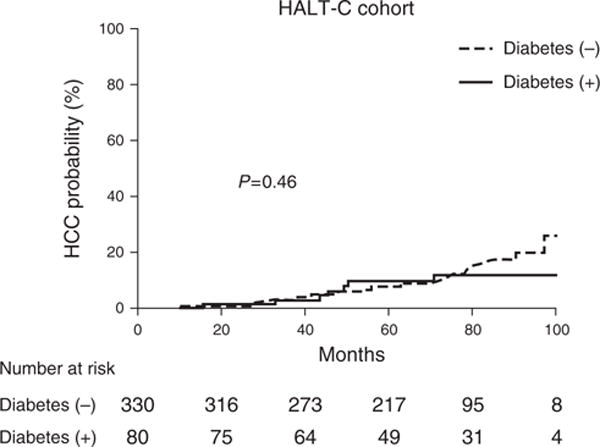
Diabetes and risk of hepatocellular carcinoma (HCC) in hepatitis C antiviral long-term treatment against cirrhosis cohort.
Table 5.
Diabetes and risk of HCC in HALT-C cohort
| Univariate | Multivariable | |||
|---|---|---|---|---|
| HR | P-value | HR | P-value | |
| Diabetes | 0.7 (0.3–1.6) | 0.45 | 0.7 (0.3–1.4) | 0.27 |
| Albumin | 0.4 (0.2–0.8) | 0.01 | 0.4 (0.2–0.8) | 0.01 |
| Age (10 years) | 1.3 (0.9–1.8) | 0.23 | 1.2 (0.8–1.8) | 0.28 |
DISCUSSION
The aim of the current study was to investigate the effect of diabetes on the risk of HCC in patients with cirrhosis from different etiologies. We demonstrate that the association was etiology-specific. In patients with non-HCV cirrhosis, diabetes was associated with approximately twofold increase (univariate HR=2.1 and multivariable HR=1.9) in HCC. In contrast, there was no association between diabetes and HCC in patients with HCV cirrhosis. Th is observation was consistent between the Mayo and HALT-C data.
Diabetes has been associated with several human cancers (20). Biologically plausible postulates for carcinogenic effects of diabetes on the liver have been proposed. Insulin resistance and subsequent production of reactive oxygen species triggering inflammatory cascades may play a role in hepatocarcinogenesis (21). Oxidative stress and release of proinflammatory cytokines such as TNF, IL-1, and IL-6 promote inflammation of the liver. Proinflammatory cytokines are also known to stimulate growth of cancer cells and enhance their survival, as well as to promote angiogenesis and subversion from immunity (21). In addition, insulin downregulates insulin-like growth factor binding protein1, which enhances bioavailability of insulin-like growth factor-1, promoting cellular proliferation and inhibiting apoptosis in the liver (22). Finally, free fatty acids and inflammatory cytokines in hyperinsulinemia are potent activators of c-Jun N-terminal kinase 1 (23). c-Jun N-terminal kinase 1 mediates cell death and compensatory proliferation of hepatocytes and plays important roles in hepatocarcinogenesis (24–26).
Despite these theories, epidemiological data linking diabetes to HCC have been conflicting, particularly for HCV-induced HCC. A multi-center study from Europe and Canada included 541 HCV patients with advanced fibrosis or cirrhosis (Ishak fibrosis score ≥4) who received interferon-based treatment with a median follow-up of 4 years (9). There was no significant association between diabetes and HCC in the entire study cohort, while in a subgroup of patients with Ishak score of 6 (N =303), diabetes was associated with a threefold increase in HCC aft er adjusting for covariates. A Japanese cohort study with 4302 HCV patients reported that diabetes was associated with 1.7-fold increase in HCC risk in patients with HCV who received interferon-based treatment (27). This association, however, was most pronounced in patients who had achieved sustained virologic response, in whom the risk increased 2.5-fold. In contrast, in a large population-based cohort study from Taiwan (n =54,979), no association was found between diabetes and HCC (HR=0.62, P =0.37) among patients with HCV, although the study did not address patients with cirrhosis separately. Finally, in a study from Japan based on 161 HCV patients (39% with cirrhosis) with 6.4 years of follow-up, there was no association between diabetes and HCC after adjusting for other risk factors including cirrhosis (28).
We believe that the results of this study may help understand these conflicting data by specifically studying patients with cirrhosis. As cirrhosis advances, the prevalence and severity of insulin resistance and diabetes increase (8,9,29). In cirrhosis, impaired insulin secretion by the pancreas and insulin resistance and impaired insulin clearance in the liver have been described (30–33). Conversely, diabetes may accelerate progression of chronic liver disease (29,34). Insulin resistance creates a proinflammatory milieu, which promotes hepatocyte injury, inflammation, and progression of liver disease (35). Insulin also stimulates proliferation and collagen production of hepatic stellate cells (36). Moreover, insulin and/or high glucose concentrations upregulates connective tissue factor, further contributing to liver fibrosis (37,38). Finally and importantly, cirrhosis remains by far the strongest risk factor for HCC and severity of fibrosis and hepatic decompensation parallel the risk of HCC (39,40). Thus, this complex relation among diabetes, cirrhosis, and HCC has likely created varying degrees of confounding in different directions in prior studies.
Our study has several limitations. First, the retrospective nature of the study prevents systematic definition of variables based on objective biomarkers. For example, cirrhosis and advanced fibrosis may be best defined by liver histology or, more recently, by liver stiffness measurement. Similarly, we may have under-diagnosed diabetes—however, we minimized misclassification by incorporating use of anti-diabetic medications and plasma levels of fasting glucose and hemoglobin A1C in the definition of diabetes, in addition to the patient’s report of diabetes. Second, as a single-center study, our patients were derived from a large referral practice at Mayo Clinic and the results might not be generalizable to cirrhotic patients at large. We demonstrate that our results are replicated almost exactly in the HALT-C data, a prospective, multi-center study. Third, while we started with a large number of patients with cirrhosis followed for a reasonable length of time, there were only a modest number of patients who developed HCC, especially when they were divided into several categories of liver disease, which limited the statistical power for multivariable analyses and led to relatively wide CIs. For example, for non-HCV patients in Table 3, the hazard ratio for diabetes did not change materially between the univariate and multivariable analyses, whereas the P value for the latter was outside statistical significance. For the HCV subgroup, lack of association between diabetes and HCC may constitute a type two error—although we believe that it is not very likely, because the point estimates are considerably <1 and consistent with each other (0.6 and 0.7) between the Mayo and the HALT-C data. Finally, the HALT-C study was conducted before modern non-invasive diagnostic criteria of HCC were established. Although there may be some cases that may not have met the current AASLD criteria, a sensitivity analysis performed while excluding patients with ‘presumed’ HCC (n =12) yielded a similar result (HR: 1.0, 95% CI: 0.4–2.3, P =0.92).
In summary, in this study of patients with cirrhosis, diabetes was not associated with increased risk of HCC in patients with HCV, whereas in non-HCV patients, the hazard of HCC was two times higher in patients with diabetes compared with those without. These results may be interpreted to indicate that in HCV patients who already have a very high risk of HCC, diabetes may not increase the risk any further. These data highlight the need for care with which data linking diabetes to HCC are to be interpreted in patients with and without cirrhosis from diverse etiologies. They also support further studies, particularly among non-HCV patients, to understand the impact of diabetes on HCC with higher specificity according to the underlying etiology. In light of the emerging trends of increasing number of patients with NAFLD developing HCC, there is an urgent need to strategize stratifying their risk according to the metabolic profile.
Supplementary Material
Study Highlights.
WHAT IS CURRENT KNOWLEDGE
✓ Diabetes increases the risk for hepatocellular carcinoma (HCC) in the general population.
✓ Effect of diabetes on the risk of HCC in patients with cirrhosis remained to be determined.
✓ Whether etiology of liver disease modifies the association between diabetes and HCC is not well known.
WHAT IS NEW HERE
✓ Hepatitis C virus (HCV) etiology modifies the association between diabetes and HCC risk in cirrhotic patients.
✓ Diabetes is not associated with increased risk of HCC in patients with HCV cirrhosis.
✓ Diabetes is associated with 1.9-fold increased risk of HCC in patients with non-HCV cirrhosis.
Acknowledgments
Financial support: This study was supported by the National Institutes of Diabetes and Digestive and Kidney Diseases (DK-34238, DK-92336; to W.R.K.) and T32 DK07198 (to J.D.Y.).
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/ajg
CONFLICT OF INTEREST
Guarantor of the article: W. Ray Kim, MD.
Specific author contributions: Planning, conducting the study, collecting, interpreting data, and drafting the manuscript:
Ju Dong Yang; collecting and interpreting data, and critical revision of the manuscript: Hager Amed Mohamed and Jessica L. Cvinar; planning and/or conducting the study, interpreting data and critical revision of the manuscript: Gregory J. Gores and Lewis R. Roberts; planning and conducting the study, interpreting data and critical revision, and final approval of the manuscript: W. Ray Kim; All authors (Ju Dong Yang; Hager Amed Mohamed; Jessica L. Cvinar; Gregory J. Gores; Lewis R. Roberts; W. Ray Kim) approved the final draft of the manuscript.
Potential competing interests: None.
References
- 1.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–91. doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7:448–58. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang JD, Harmsen WS, Slettedahl SW, et al. Factors that affect risk for hepatocellular carcinoma and effects of surveillance. Clin Gastroenterol Hepatol. 2011;9:617–23 e1. doi: 10.1016/j.cgh.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 4.Yang JD, Kim B, Sanderson SO, et al. Hepatocellular carcinoma in Olmsted County, Minnesota, 1976-2008. Mayo Clin Proc. 2012;87:9–16. doi: 10.1016/j.mayocp.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welzel TM, Graubard BI, Quraishi S, et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am J Gastroenterol. 2013;108:1314–21. doi: 10.1038/ajg.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4:369–80. doi: 10.1016/j.cgh.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Kang D, Cao W, et al. Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev. 2012;28:109–22. doi: 10.1002/dmrr.1291. [DOI] [PubMed] [Google Scholar]
- 8.Gentile S, Loguercio C, Marmo R, et al. Incidence of altered glucose tolerance in liver cirrhosis. Diabetes Res Clin Pract. 1993;22:37–44. doi: 10.1016/0168-8227(93)90130-w. [DOI] [PubMed] [Google Scholar]
- 9.Veldt BJ, Chen W, Heathcote EJ, et al. Increased risk of hepatocellular carcinoma among patients with hepatitis C cirrhosis and diabetes mellitus. Hepatology. 2008;47:1856–62. doi: 10.1002/hep.22251. [DOI] [PubMed] [Google Scholar]
- 10.Chen CT, Chen JY, Wang JH, et al. Diabetes mellitus, metabolic syndrome and obesity are not significant risk factors for hepatocellular carcinoma in an HBV)and HCV-endemic area of Southern Taiwan. Kaohsiung J Med Sci. 2013;29:451–9. doi: 10.1016/j.kjms.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ko WH, Chiu SY, Yang KC, et al. Diabetes, hepatitis virus infection and hepatocellular carcinoma: a case-control study in hepatitis endemic area. Hepatol Res. 2012;42:774–81. doi: 10.1111/j.1872-034X.2012.00979.x. [DOI] [PubMed] [Google Scholar]
- 12.Tung HD, Wang JH, Tseng PL, et al. Neither diabetes mellitus nor overweight is a risk factor for hepatocellular carcinoma in a dual HBV and HCV endemic area: community cross-sectional and case-control studies. Am J Gastroenterol. 2010;105:624–31. doi: 10.1038/ajg.2009.711. [DOI] [PubMed] [Google Scholar]
- 13.White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol. 2008;49:831–44. doi: 10.1016/j.jhep.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Bisceglie AM, Shiffman ML, Everson GT, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med. 2008;359:2429–41. doi: 10.1056/NEJMoa0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lok AS, Everhart JE, Wright EC, et al. Maintenance peginterferon therapy and other factors associated with hepatocellular carcinoma in patients with advanced hepatitis C. Gastroenterology. 2011;140:840–9. doi: 10.1053/j.gastro.2010.11.050. quiz e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–2. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sangiovanni A, Manini MA, Iavarone M, et al. The diagnostic and economic impact of contrast imaging techniques in the diagnosis of small hepatocellular carcinoma in cirrhosis. Gut. 2010;59:638–44. doi: 10.1136/gut.2009.187286. [DOI] [PubMed] [Google Scholar]
- 18.Khalili K, KT JH, Haider MA, et al. Implementation of AASLD hepatocellular carcinoma practice guideline in North America: two years of experience. Hepatology. 2008;48:362A. [Google Scholar]
- 19.Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–52. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 20.Jee SH, Ohrr H, Sull JW, et al. Fasting serum glucose level and cancer risk in Korean men and women. JAMA. 2005;293:194–202. doi: 10.1001/jama.293.2.194. [DOI] [PubMed] [Google Scholar]
- 21.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 22.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–91. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 23.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–6. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 24.Sakurai T, Maeda S, Chang L, et al. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci USA. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hui L, Zatloukal K, Scheuch H, et al. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–53. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F, Castranova V. Beyond apoptosis of JNK1 in liver cancer. Cell Cycle. 2009;8:1145–7. doi: 10.4161/cc.8.8.8200. [DOI] [PubMed] [Google Scholar]
- 27.Arase Y, Kobayashi M, Suzuki F, et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology. 2013;57:964–73. doi: 10.1002/hep.26087. [DOI] [PubMed] [Google Scholar]
- 28.Ohata K, Hamasaki K, Toriyama K, et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer. 2003;97:3036–43. doi: 10.1002/cncr.11427. [DOI] [PubMed] [Google Scholar]
- 29.Hui JM, Sud A, Farrell GC, et al. Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression [corrected] Gastroenterology. 2003;125:1695–704. doi: 10.1053/j.gastro.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 30.Petrides AS, Vogt C, Schulze-Berge D, et al. Pathogenesis of glucose intolerance and diabetes mellitus in cirrhosis. Hepatology. 1994;19:616–27. doi: 10.1002/hep.1840190312. [DOI] [PubMed] [Google Scholar]
- 31.Petrides AS, Stanley T, Matthews DE, et al. Insulin resistance in cirrhosis: prolonged reduction of hyperinsulinemia normalizes insulin sensitivity. Hepatology. 1998;28:141–9. doi: 10.1002/hep.510280119. [DOI] [PubMed] [Google Scholar]
- 32.Perseghin G, Mazzaferro V, Sereni LP, et al. Contribution of reduced insulin sensitivity and secretion to the pathogenesis of hepatogenous diabetes: effect of liver transplantation. Hepatology. 2000;31:694–703. doi: 10.1002/hep.510310320. [DOI] [PubMed] [Google Scholar]
- 33.Deschenes M, Somberg KA. Effect of transjugular intrahepatic portosys-temic shunt (TIPS) on glycemic control in cirrhotic patients with diabetes mellitus. Am J Gastroenterol. 1998;93:483. doi: 10.1111/j.1572-0241.1998.481_4.x. [DOI] [PubMed] [Google Scholar]
- 34.Hickman IJ, Macdonald GA. Impact of diabetes on the severity of liver disease. Am J Med. 2007;120:829–34. doi: 10.1016/j.amjmed.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 35.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–32. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 36.Svegliati-Baroni G, Ridolfi F, Di Sario A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology. 1999;29:1743–51. doi: 10.1002/hep.510290632. [DOI] [PubMed] [Google Scholar]
- 37.Paradis V, Dargere D, Vidaud M, et al. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology. 1999;30:968–76. doi: 10.1002/hep.510300425. [DOI] [PubMed] [Google Scholar]
- 38.Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–44. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- 39.Flemming JA, Yang JD, Vittinghoff E, et al. Risk prediction of hepatocellular carcinoma in patients with cirrhosis: the ADRESS-HCC risk model. Cancer. 2014;120:3485–93. doi: 10.1002/cncr.28832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang JD, Kim WR, Coelho R, et al. Cirrhosis is present in most patients with hepatitis B and hepatocellular carcinoma. Clin Gastroenterol Hepatology. 2011;9:64–70. doi: 10.1016/j.cgh.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.