

Abstract
Objective:
The objective of this to find the prevalence of skeletal and visceral changes in thalassemics and its relationship with variation in blood components.
Methodology:
This is a cross-sectional study conducted in tertiary care hospital in Karachi among patients diagnosed with thalassemia major who require regular blood transfusions.
Result:
Among 200 individuals, 95 were females, whereas 105 were males. 96.5% of the study sample showed normocytic normochromic blood picture. Mean pre-transfusion hemoglobin (Hb) for 200 patients was 8.91 g/dl, while the mean post-transfusion Hb was 12.07 g/dl. Among all the variables, some strong predictors of change were age and HbA which were found associated with the development of cardiac change in transfusion-dependent thalassemia patients. Hepatomegaly was observed in 66.5% of the patients while normal liver span was observed in 33.5%. 6% of patients showed evidence of skeletal changes on X-ray. Splenomegaly was observed in 26.5%, while in 4.5% of the patients, spleen was not visualized due to splenectomy. Cardiac involvement was observed in 8% of the patients on T2*magnetic resonance imaging.
Conclusion:
Visceral changes particularly hepatomegaly is very common among transfusion-dependent thalassemia patients. Blood studies for Hb and ferritin levels help to detect these changes when conventional investigations are not possible.
Keywords: Alpha thalassemia, beta thalassemia, complete blood picture, hepatomegaly, skeletal changes, splenomegaly, thalassemia, visceral changes
Introduction
Thalassemia is a blood disorder which is passed on to offspring’s by the parents with defected gene for blood hemoglobin (Hb) production. This disorder results from a quantitative deficiency of specific globin chain for Hb production.[1] This disorder takes up two clinical forms, thalassemia minor and thalassemia major, later one being transfusion dependent due to decreased Hb level which is not enough to meet with the daily circulatory demand of the body. This increased demand of red cell production leads to accelerated bone turnover which causes bone marrow expansion and skeletal changes which is evident as osteoporotic and osteopenic radiological findings in the bones of thalassemia patients. A study showed increased serum levels for bone resorption marker and decreased levels for bone-forming markers in these patients.[2,3] Persistent anemia and continued destruction of red blood cells (RBCs) render these patients to depend on the blood transfusions. The transfusion cycles have positive effects in the linear growth specially if started in the first decade of life. A study demonstrated that normal growth was observed in the group of thalassemia patients who underwent regular blood transfusion with chelation therapy when compared to the group who were transfused infrequently were found to have average height below 50th centile.[4]
Transfusion therapy is the mainstay of management of thalassemia major. Since thalassemia is growing in populations globally, it is estimated that there will be around 900,000 new clinically significant thalassemia patients by the year 2025.[5] Hence, the burden of transfusion and its complications will also be increased. A similar rise is expected in Pakistan where carrier frequency for thalassemia is about 5.4%.[6]
Transfusion therapy is life support for severely anemic thalassemic patients, but it comes with its side effects, the most prominent being iron overload related complications. Not only the amount of blood transfused but also the duration of iron exposure gives the best estimate of organ dysfunction as observed in the study which showed iron overload cardiomyopathy, increased liver iron, and endocrine organopathy in transfusion-dependent thalassemia patients which were due to iron deposition in these organs.[7] The greatest effect of transfusion-related iron overload complication in thalassemic patients is observed in cardiac tissues where it contributes as the most common cause of death in the form of arrhythmia and heart failure.[8] Liver injury due to iron overload is one of the worrisome side effects of continuous transfusion without chelation therapy which is evident as the lysosomal hemosiderin in hepatocyte and Kupffer cells of biopsy samples from liver of thalassemia patients.[9] Serum ferritin analysis and electrocardiographic study of thalassemia major patients at the Department of Pathology, Islamabad Medical and Dental College, have pointed toward the direct relationship between serum ferritin levels and abnormalities observed in electrocardiogram of these patients.[9]
Factors other than increased iron level which might be responsible for the development of complications in such patients are still not documented and what is the effect of changing blood levels of different components in the progression of complication is unknown. This study is conducted to find the relationship of variation in different blood components with skeletal and visceral changes among thalassemic patient, which will help in using changing levels of different components of blood such as serum ferritin, mean corpuscular volume (MCV), pre- and post-transfusion Hb, and peripheral blood smear as a tool for the assessment of risk factors in the development of cardiac, hepatic, and skeletal complications among these patients long before the appearance of their clinical symptoms.
Methodology
Study design
The study design was a cross-sectional study.
Setting
This study was conducted at the Outpatient department of Jinnah Postgraduate Medical Centre (JPMC), and its constituent National Institute of Child Health (NICH), Karachi.
Duration
The study duration was from August 2016 to October 2016.
Sample technique
Nonprobability convenient sampling.
Duration and sample size
The study was conducted from August 1 to October 30 during which 200 thalassemic children were enrolled for transfusion. These were selected according to the inclusion and exclusion criteria mentioned as follows.
Sample selection
Inclusion criteria
Patients who were proven cases of thalassemia by clinical evaluation, blood smear, and Hb electrophoresis and were on regular transfusion were approached in this study. Among them, those giving informed consent were included in this study.
Those in between the age group of 10 to 32 years were also included.
Exclusion criteria
Patients on transfusion with any other blood disorder but thalassemia were not included in this study.
Patients whose parents were not willing to participate in this study were also excluded from the study.
Measuring of variables
Dependent variables
Changing levels of the components of blood picture (such as Hb, MCV, serum iron, serum ferritin, HbA2, and HbF level) were considered as dependent variables.
Independent variables
Age, gender, residence, ethnicity, skeletal changes, hepatosplenomegaly, and cardiomyopathy was considered as independent variables.
Data collection procedure
Data were collected through interviews by filling a pro forma having variables such as demographics (age, gender, residence, and ethnicity), details of blood picture (Hb, MCV, peripheral blood smear, serum iron, serum ferritin, HbA2, and HbF levels), and details of body changes such as skeletal changes documented on the basis of radiological evidence, hepatosplenomegaly which was evaluated on clinical examination, iron overload, cardiomyopathy as observed on T2* magnetic resonance (MR), and liver involvement for hepatitis on the basis of hepatitis profile test. Data were collected by principal investigator. Interviews were carried out in a separate room. Informed consent was obtained from the participants in accordance with JPMC and the National Institute of Child Health’s guidelines for consent. Consent from parents was obtained for the participants who fell in the age group below 12 years. Participants were assured that their confidentiality will be maintained throughout the research process and their identity or any other information obtained during interview will not be shared with anyone and will only be used for research purpose. Participants were allowed to withdraw from the survey at any point. Data were rechecked for any missing variables and were given identification numbers.
Data analysis plan
Data were entered in Microsoft Office Excel 2010 and then imputed in software for SPSS version 19 for analysis. Frequency and percentages were calculated for age, gender, skeletal changes, liver changes, splenic changes, and cardiac involvement, while mean and standard deviation for Hb, MCV, age, and serum ferritin.
Ethics approval and consent to participate
We took approval from the Ethical Review Committee of JPMC and NICH, Karachi. Before enrolling the study participants, key stakeholders operating in the study area were informed about the nature and objectives of the research. Participants were informed about study objective and procedures, and informed consent was obtained from the participant. The interviews were conducted in privacy; there was no sharing of information and even name not mentioned in questionnaire, counseling was done after interview, there was no direct benefit to participant, and study participants were free to withdraw from the study at any time. Properly, the aims and objectives of this study were explained to the participants, and they were also told that we will use the information when the study will be published.
Result
Tables 1 and 2 show that from the study sample of 200 thalassemia patients majority belonged to the age group of <15 years of age (90%), and out of those 200 patients, 95 patients (47.5%) were female while 105 patients (52.5%) were male.
Table 1.
Age (years)

Table 2.
Gender

Since no statistically significant difference was observed among the two genders, hence data are not separated on the basis of gender.
Table 3 shows the comparison between pre-transfusion Hb and MCV. It shows that mean pre-transfusion Hb for 200 patients was 8.91 g/dl (+1.235), while the mean post-transfusion Hb was 12.07 g/dl (+1.47) and the mean pre-transfusion MCV was 81.82 fl (+9.46) while the mean post-transfusion MCV was 83.84 fl (+4.013). Mean serum ferritin for 200 patients was 6559.52 ng/ml (+3602.70). The prevalence of different Hb levels was found as mean HbA2 (3.18%), mean HbF (74.07%), and mean HbA (40.51%).
Table 3.
Descriptive statistics

In our study, 96.5% of the study sample showed the normocytic normochromic type of blood picture, and 3.5% of the patients showed microcytic anemia on blood picture. 100% of the patients had evidence of iron overload as increased serum ferritin levels.
Charts 1-3 show that HbA2 was in the range of 2.1–4 for 31.5% (N = 63), while only 0.5% had HbA2 in the range of 6.1–8 (N = 1), for HbA, we found that it was in the range of 0–20 for 12.5% (N = 25), while only 2% had HbA in the range of 21–40 (N = 4), similarly, for HbF, we found that 47% of the patient had HbF in the range of 65–100 (N = 94), while only 2% had HbF in the range of 0.2–3.0 (N = 4).
Chart 1.

HbA2
Chart 2.

HbA
Chart 3.

HbF
Table 4 shows the prevalence of body and visceral changes among 200 thalassemia patients. It shows that of those 200, 94% had no skeletal changes while only 6% showed evidence of skeletal changes on X-ray (which involved sclerotic changes of long bone in 3 patients, 1 patient had fracture of tibia with widening of bones of feet, 1 patient showed reduced bone mass density in vertebrae and bones of the forearm, while 1 patient had evidence of growth retardation). Splenomegaly was observed in 26.5% of the patients, and the rest of 69% showed normal spleen size while in 4.5% of the patients spleen was not visualized due to splenectomy. Cardiac involvement was observed in 8% of the patients while 92% had no cardiac involvement on T2*MR imaging (MRI).
Table 4.
Body changes

Chart 4 shows that hepatomegaly was observed in 66.5% of the patients while normal liver span was observed in 33.5%.
Chart 4.

Liver changes
All variables had an association with the incidence of body and visceral changes, and these changes were found associated with the increasing age limits. Cardiac changes were associated with increased age [Table 5] and increasing blood levels of HbA2, HbF, and especially with HbA as shown in Table 6. Hepatomegaly and splenomegaly were related to increased age and HbA2 levels in blood, but no definite pattern was observed for increasing or decreasing blood levels of HbA and HbF [Tables 7 and 8. Skeletal changes showed association with increased blood HbF levels and age. Among all the variables, some strong predictors of change in these thalassemic patients were age and HbA which were found associated with the development of cardiac change in transfusion-dependent thalassemia patients. Although these levels of different types were found associated, there was no specific limit of the value above which these changes were found. Hb Table 6 shows a positive association between age and the development of cardiac changes (P = 0.005) with 11 patients of 180 having cardiac involvement in age group of <15 years, while 5 patients of 18 in age group of 15–25 years showed evidence of cardiac changes on T2* MRI. Table 7 shows a positive association between increasing levels of HbA and cardiac changes (P = 0.032).
Table 5.
Association between age and cardiac changes
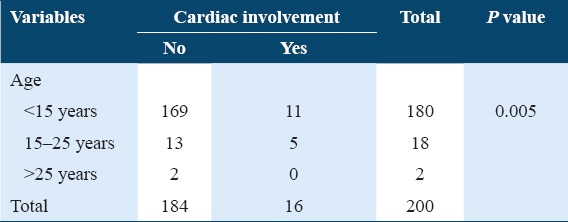
Table 6.
Association between cardiac changes and different types of Hb

Table 7.
Association between splenic changes and different types of hemoglobin

Table 8.
Association between liver changes and different types of hemoglobin

Negative association was found between splenomegaly and HbA2 (P = 0.753) and between skeletal changes and HbA2 (P = 0.907).
Discussion
In our study of 200 patients, we found that the evidence of iron overload was present in all the thalassemic patients who were being chronically transfused, as observed by the increased level of serum ferritin. The development of skeletal and visceral changes (cardiac changes and hepatosplenomegaly) was associated with the decreased levels of HbA and HbA2 and increased levels of HbF. Cardiac changes had a strong association with HbA levels and age. The most frequent visceral change observed among these patients in our study was hepatomegaly which was present in 67% of them.
Therapies increasing HbF levels have shown to improve clinical outcome in patients with sickle cell disease and thalassemia major. In rare forms of thalassemia major, there is a hereditary persistence of HbF. This form is associated with relatively benign clinical outcome. There have been different therapeutic agents for inducing HbF, use of which have reported approximately 30 g/L increase in Hb levels. The most common agent used for inducing HbF expression is hydroxyurea. The exact mechanism of action for hydroxyurea is not known; however, it is associated with cytotoxic effects resulting in stress erythropoiesis with increased levels of HbF. Hydroxyurea increases gamma RNA expression up to 9 folds which improve alpha/non-alpha chain imbalance and hence increase in erythropoiesis. Other beneficial outcomes of hydroxyurea therapy include a reduction in circulating leukocyte levels, increased reticulocyte counts, and decreased hemolysis. It also increases the release of NO due to soluble guanylyl cyclase and cGMP dependent protein kinase pathway activity. Hydroxyurea therapy also decreases phosphatidylserine externalization on RBC surface which improves red cell survival and decreases thrombin generation which reduces hypercoagulable state. Ultimately, there is overall improved endocrine, vascular, and cardiac function. Other agents used for HbF induction include 5-azacitidine and decitabine.[10]
Cardiac changes are the leading cause of death in thalassemics. Iron deposition in cardiac muscles can increase chances for congestive heart failure, sudden cardiac death, and arrhythmias. There are several mechanisms accounting for these risks. One mechanism is direct iron toxicity. Chronic elevation of cardiac output due to anemia can also lead to cardiac failure. Increased afterload (due to vascular pathologies because of metabolic and endocrine dearrangements) further deteriorates the clinical picture. Detterich et al. report cardiac MRI findings on T2 imaging done among 78 thalassemics. Iron deposition was evident in 45 patients. Detterich et al. have further hypothesized that in modalities where MRI imaging is not available, 12-lead echocardiography can be predictive for cardiotoxicity due to iron overload. Electrocardiography findings include QT prolongation, left shift of T-waves, and ST/T-wave abnormalities.[11]
In our study, 11 patients (6.1%) of 169 from the age group of <15 years old demonstrated cardiac changes, while 5 patients (27.7%) of 13 from the age group of 15–25 years old showed evidence of cardiac changes on T*2 MRI. A study conducted in hematology clinics in Qena and Menia University hospitals from May 2013 to September 2014 among pediatric age group of thalassemia patients showed correlation between age and increased carotid artery intima thickness (CIMT) secondary to iron overload.[12] Another study done at hospitals at Cagliari and Los Angeles showed similar results where 77 thalassemia major patients on transfusion underwent serial cardiac MR screening for the detection of iron overload cardiomyopathy, and it was found that none of patients from age group below 9.5 years showed cardiac changes while the ones from age group 15–18 years showed detectable cardiac iron by 1.28 (28%) per year.[13] Along with chelation therapy, HbF induction therapy has also been considered to be effective in improving cardiac function. Lowry et al. in his clinical trial reports case of 50-year-old male patient with thalassemia intermedia with Hb level 6.5 g/dL without transfusion. Patient developed iron overload at the mid-40s associated with hepatic iron content exceeding 6000 micrograms of iron per gram of liver, cardiac hemochromatosis evidenced by ventricular ectopy and abnormal left ventricular function, and testicular failure. The patient was administered daily intravenous infusions of deferoxamine; however, there was no clinical improvement. The patient developed congestive heart failure and was shifted to intensive care unit. The patient was started with azacitidine therapy. After the first cycle of azacitidine therapy, this patient no longer required transfusions. There were twice weekly cycles of azacitidine therapy which was reduced to one cycle per week due to recurrent neutropenia. At the start of therapy, patient’s ejection fraction was 16% which improved to nearly normal after 2 years of therapy. The patient had normal exercise response and was able to return to work. Azacitidine therapy was then stopped. This study supports the use of HbF induction therapy in improving clinical outcome and survival rates.[14]
A study conducted in Italy in the year 2002 among thalassemia major patients who presented with atypical symptoms of heart failure demonstrated very high ferritin levels before their death and suggested that rigorous chelation therapy may prove beneficial in the improvement of cardiac function in cases of transfusion-induced cardiomyopathy.[15] In our study, we found a positive association between cardiac changes and age, in conjunction with this, evidence of iron overload in the form of increased ferritin level is present among 100% of our sample population which is very alarming, raised ferritin levels may prove to be helpful investigation tool for early detection of cardiac dysfunction, especially in our setup, where screening for cardiomyopathy through cardiac MRI is very expensive.
Our study has shown a strong correlation between increased mean serum ferritin (6559.52 ng/ml) of the sample population of 200 patients and the presence of hepatomegaly among 67% of them. This finding coincides well with the results of a study in which P Mazza compared serum ferritin levels, MRI, and liver iron concentration to find their relationship for the development of hemochromatosis. His study demonstrated very strong association of increased serum ferritin levels with the development of hemochromatosis as evident on MRI and liver iron concentration.[16] Under normal circumstances, hepcidin regulates normal levels of iron by its effect on gastrointestinal iron absorption. In patients with ineffective erythropoiesis, hepcidin levels are insufficient which lead to iron deposition in various organs. Mouse models have shown a decrease in levels of ferritin with the administration of synthetic hepcidin. However, its administration in humans is difficult. The bioactive form of hepcidin is rapidly excreted by the kidney. The structure of hepcidin includes disulfide bridging which is responsible for its high cost and thus lowers its availability.[17]
Our study shows 12 patients with skeletal changes. Skeletal changes observed on X-ray include reduced bone density marked in distal radius, carpals, and base of metacarpals with widened metaphysis in two patients, two patients with low bone age, growth retardation in one patient, and bilateral fusion of lunate and triquetral bones in representing transverse carpal coalition in one patient. One patient showed the altered morphology of both ulna, one had sclerotic lesions in phalangeal bones of the hand, one patient showed low bone mass in all lumbar vertebrae, especially in L2 and L3 vertebrae, and one patient showed widening of distal humerus, whereas one patient demonstrated distal tibial fracture. One patient showed sclerotic lesions in distal femur and widening of metaphysis of distal femur. The mean ferritin among patients of this study was found to be 6559 ± 3602 ng/ml, whereas mean ferritin among patients with skeletal changes was found to be 6650 ± 2989 ng/ml. Giuzio et al. report his study, results among which were 16.6% of patients with lower limb length discrepancy, 5.5% with upper limb length discrepancy, 8.3% axial deviation of limbs, 2.7% osteochondrosis, and 25% osteopenia.[18] Aging of the bone starts earlier in patients with thalassemia major, due to an imbalance between osteoclastic and osteoblastic mechanisms. Iron overload, chronic anemia, and endocrine disturbances cause alterations in RANK/RANKL/OPG system and lead to enhanced osteoclastic activity. It has been seen that 50% of cases with osteopenia in thalassemics are due to endocrine complications mainly hypogonadotropic hypogonadism. Growth hormone disturbances also contribute to osteopenia and osteoporosis.[19] Other mechanisms include hyperplasia of bone marrow and increase in intramedullary space due to extramedullary hematopoieses eventually leading to osteoporosis. Vogiatzi reports bone mineral density measured using DEXA scan in 18 pre-pubertal children with thalassemia major on transfusions and iron chelation therapy. Normal Z-scores, i.e., >1 was found in 38.8% of patients, Z-scores between −1 and 2.5 were of 38.8% of patients, and 22.2% had Z-scores <−2.5. The mean decline in Z-scores was found to be −0.38% per year.[20] A study conducted in Iran from February 2002 to October 2004 demonstrated that decreased bone mineral density in thalassemia patients correlates well with low Hb levels.[20] A similar finding is present in our study where skeletal changes were found to occur in patients with low level of HbA and HbA2. Management options include induction of puberty using estrogens and testosterone in girls and boys, respectively. Treatment of hypogonadism has shown good outcomes. Use of bisphosphonates has also shown promising results in the fate of osteoporotic changes.
A study conducted in India in 2003 showed high frequency of hepatitis B virus infection in multi-transfused patients with B thalassemia.[21] Contrary to it, the sample population of 200 thalassemia patients in our study screened negative (100%) for HbV infection.
In our study, we concluded that skeletal changes were present in 6% of the population. While observing for visceral changes, we found that splenomegaly was present in 26.5%, cardiac changes in 8%, while hepatomegaly being the most prominent viscera to be affected showed its prevalence in 66.5% of the sample population. We also found that prevalence of different type of Hb affected viscera differently, where cardiac changes were found with increased HbA2 and HbF levels, and especially, HbA while hepatomegaly had shown its prevalence with increased HbA2 levels.
This study included only single measurement of serum ferritin levels which might not be an accurate indicator of iron overload, serial monitoring of serum ferritin is needed to know about the actual picture of iron overload. Due to the short duration of the study and lack of financial resources, we were not able to follow-up the enrolled patients. Organ changes take years to develop, and hence, follow-up is needed to monitor the clinical changes of the involved system and to compare it with the complete blood picture as the organ changes progress. This study was conducted in one transfusion center of the city; similar study needs to be conducted in more transfusion centers of different areas to know about the true prevalence of association of the given findings in thalassemia patients across the country.
Conclusion
Body and visceral changes after regular transfusion take years to develop. These changes usually go unnoticed in our region until the development of full-blown symptoms due to lack of screening programs and compliance to these programs because of the high cost of investigations. Skeletal and visceral changes such as splenomegaly, cardiomyopathy, and hepatomegaly are very prevalent among these patients and are related to changing Hb levels and increased serum ferritin levels. Blood studies for Hb and iron may help to detect these changes years before the development of complication where detection using investigations such as MRI and biopsy is not possible.
References
- 1.Housman D, Forget BG, Skoultchi A, Benz EJ., Jr Quantitative deficiency of chain-specific globin messenger ribonucleic acids in the thalassemia syndromes. Proc Natl Acad Sci U S A. 1973;70:1809–13. doi: 10.1073/pnas.70.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salama OS, Al-Tonbary YA, Shahin RA, Sharaf Eldeen OA. Unbalanced bone turnover in children with β-thalassemia. Hematology. 2006 Jun 1;11(3):197–202. doi: 10.1080/10245330600702851. [DOI] [PubMed] [Google Scholar]
- 3.Scutellari PN, Orzincolo C, Andraghetti D, Gamberini MR. Anomalies of the masticatory apparatus in beta-thalassemia. The present status after transfusion and iron-chelating therapy. Radiol Med. 1994;87:389–96. [PubMed] [Google Scholar]
- 4.Viprakasit V, Tanphaichitr VS, Mahasandana C, Assteerawatt A, Suwantol L, Veerakul G, et al. Linear growth in homozygous beta-thalassemia and beta-thalassemia/hemoglobin E patients under different treatment regimens. J Med Assoc Thai. 2001;84:929–41. [PubMed] [Google Scholar]
- 5.Vichinsky EP. Changing patterns of thalassemia worldwide. Ann N Y Acad Sci. 2005;1054:18–24. doi: 10.1196/annals.1345.003. [DOI] [PubMed] [Google Scholar]
- 6.Khan SN, Riazuddin S. Molecular characterization of beta-thalassemia in Pakistan. Hemoglobin. 1998;22:333–45. doi: 10.3109/03630269809071528. [DOI] [PubMed] [Google Scholar]
- 7.Vichinsky E, Butensky E, Fung E, Hudes M, Theil E, Ferrell L, et al. Comparison of organ dysfunction in transfused patients with SCD or beta thalassemia. Am J Hematol. 2005;80:70–4. doi: 10.1002/ajh.20402. [DOI] [PubMed] [Google Scholar]
- 8.Wu HP, Lin CL, Chang YC, Wu KH, Lei RL, Peng CT, et al. Survival and complication rates in patients with thalassemia major in Taiwan. Pediatr Blood Cancer. 2017;64:135–8. doi: 10.1002/pbc.26181. [DOI] [PubMed] [Google Scholar]
- 9.Thakerngpol K, Fucharoen S, Boonyaphipat P, Srisook K, Sahaphong S, Vathanophas V, et al. Liver injury due to iron overload in thalassemia:Histopathologic and ultrastructural studies. Biometals. 1996;9:177–83. doi: 10.1007/BF00144623. [DOI] [PubMed] [Google Scholar]
- 10.Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood. 2013;121:2199–212. doi: 10.1182/blood-2012-10-408021. [DOI] [PubMed] [Google Scholar]
- 11.Detterich J, Noetzli L, Dorey F, Bar-Cohen Y, Harmatz P, Coates T, et al. Electrocardiographic consequences of cardiac iron overload in thalassemia major. Am J Hematol. 2012;87:139–44. doi: 10.1002/ajh.22205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abdelsamei HA, El-Sherif AM, Ismail AM, Abdel Hakeem GL. The role of the carotid doppler examination in the evaluation of atherosclerotic changes in β-thalassemia patients. Mediterr J Hematol Infect Dis. 2015;7:e2015023. doi: 10.4084/MJHID.2015.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood JC, Origa R, Agus A, Matta G, Coates TD, Galanello R, et al. Onset of cardiac iron loading in pediatric patients with thalassemia major. Haematologica. 2008;93:917–20. doi: 10.3324/haematol.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowrey CH, Nienhuis AW. Brief report:Treatment with azacitidine of patients with end-stage beta-thalassemia. N Engl J Med. 1993;329:845–8. doi: 10.1056/NEJM199309163291205. [DOI] [PubMed] [Google Scholar]
- 15.Forni GL, Derchi G. Typical manifestation of acute congestive heart failure in patients with Thalassaemia major causing diagnostic delay in the emergency room. Eur J Heart Fail. 2003;5:607–8. doi: 10.1016/s1388-9842(03)00102-8. [DOI] [PubMed] [Google Scholar]
- 16.Mazza P, Giua R, De Marco S, Bonetti MG, Amurri B, Masi C, et al. Iron overload in thalassemia:Comparative analysis of magnetic resonance imaging, serum ferritin and iron content of the liver. Haematologica. 1995;80:398–404. [PubMed] [Google Scholar]
- 17.Finberg KE. Striking the target in iron overload disorders. J Clin Invest. 2013;123:1424–7. doi: 10.1172/JCI68889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yazigi A, Maalouf G, Inati-Khoriati A, Tamim H, Saab C. Bone mineral density in beta - thalassemic lebanese children. J Musculoskelet Neuronal Interact. 2002;2:463–8. [PubMed] [Google Scholar]
- 19.Skordis N, Toumba M. Bone disease in thalassaemia major:Recent advances in pathogenesis and clinical aspects. Pediatr Endocrinol Rev. 2011;8(Suppl 2):300–6. [PubMed] [Google Scholar]
- 20.Vogiatzi MG, Macklin EA, Fung EB, Cheung AM, Vichinsky E, Olivieri N, et al. Bone disease in thalassemia:A frequent and still unresolved problem. J Bone Miner Res. 2009;24:543–57. doi: 10.1359/jbmr.080505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh H, Pradhan M, Singh RL, Phadke S, Naik SR, Aggarwal R, et al. High frequency of hepatitis B virus infection in patients with beta-thalassemia receiving multiple transfusions. Vox Sang. 2003;84:292. doi: 10.1046/j.1423-0410.2003.00300.x. [DOI] [PubMed] [Google Scholar]