

SUMMARY
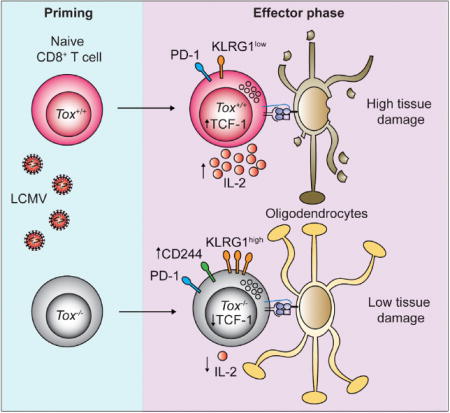
Infections are thought to trigger CD8+ cytotoxic T lymphocyte (CTL) responses during autoimmunity. However, the transcriptional programs governing the tissue-destructive potential of CTLs remain poorly defined. In a model of central nervous system (CNS) inflammation, we found that infection with lymphocytic choriomeningitis virus (LCMV), but not Listeria monocytogenes (Lm), drove autoimmunity. The DNA-binding factor TOX was induced in CTLs during LCMV infection and was essential for their encephalitogenic properties, and its expression was inhibited by interleukin-12 during Lm infection. TOX repressed the activity of several transcription factors (including Id2, TCF-1, and Notch) that are known to drive CTL differentiation. TOX also reduced immune checkpointsensitivity by restraining the expression of the inhibitory checkpoint receptor CD244 on the surface of CTLs, leading to increased CTL-mediated damage in the CNS. Our results identify TOX as a transcriptional regulator of tissue-destructive CTLs in autoimmunity, offering a potential mechanistic link to microbial triggers.
Graphical abstract
In Brief: Little is known about the transcriptional programs that drive the tissue destructive capacity of effector CD8+ T cells during autoimmunity. In an animal model of CNS inflammation, Page et al. demonstrate that expression of the DNA-binding factor TOX promotes the encephalitogenic potential of pathogen-primed CD8+ T cells and that TOX expression is determined by the microbial context of CTL priming.
INTRODUCTION
CD8+ cytotoxic T lymphocytes (CTLs) are important players in the body’s defense against infection and cancer and, in addition, contribute to the pathogenesis of several autoimmune diseases. Naive CTLs undergo clonal expansion and differentiate into cytotoxic effector T (Teff) cells upon encounter with their cognate antigen in secondary lymphoid organs. In the course of the immune response, CTLs generate distinct subsets of specialized Teff cells. So-called memory precursor effector cells (MPECs) show low expression of cytotoxic proteins but display a high potential to generate long-lived memory T cells with self-renewing capacity (Williams and Bevan, 2007). Conversely, short-lived effector T cells (SLECs) are terminally differentiated and express high amounts of cytotoxic effector molecules such as perforin and granzyme B but have a low capacity for memory formation (Kaech and Cui, 2012). Phenotypically, SLECs express the killer cell lectin-like receptor KLRG1 (Joshi and Kaech, 2008), MPECs express CD127 (Kaech et al., 2003), and double-positive effector cells (DPECs) are KLRG1hi CD127hi. CTL differentiation into SLECs and MPECs is orchestrated by various transcription factors. These include B lymphocyte-induced maturation protein 1, T-box transcription factor 21 (T-bet), and inhibitor of DNA binding 2 (Id2), which all drive SLEC differentiation (Joshi et al., 2007; Rutishauser et al., 2009; Yang et al., 2011), whereas eomesodermin (Eomes) and T Cell Factor 1 (TCF-1) support the generation of functional memory CTLs (Intlekofer et al., 2005; Zhou et al., 2010). However, little is known about the transcriptional programs regulating the tissue-destructive capacity of self-reactive CTLs in autoimmunity.
Multiple sclerosis (MS) is a chronic demyelinating autoimmune disease of the central nervous system (CNS) and results from a complex interplay between genetic and environmental factors (Friese and Fugger, 2009). Microbes have been associated with MS onset or relapses, but a causative link to specific infectious agents could not be established (Kurtzke, 1993). As supported by multiple independent lines of evidence, CTLs contribute to MS pathogenesis (Dendrou et al., 2015): (1) certain major histocompatibility complex (MHC) class I alleles are associated with the risk of developing MS (Friese et al., 2008), (2) CTLs represent a substantial fraction of T cells found in active MS lesions (Hauser et al., 1986), (3) CTLs are clonally expanded in MS lesions (Babbe et al., 2000) and persist in the cerebrospinal fluid and the peripheral blood (Skulina et al., 2004), and (4) CTLs can damage target cells in the CNS (Huseby et al., 2001).
Existing evidence suggests that the microbial context influences CTL differentiation (Obar et al., 2011). For instance, the cytokine microenvironment during CTL priming modulates the transcriptional landscape of the CTLs, giving rise to alternate fates of CTLs (Sad et al., 1995). Still, the molecular network that drives the tissue-destructive capacities of CTLs in autoimmunity remains largely unknown.
To address this, we exploited an animal model of CNS autoimmune disease (Cao et al., 2006). Adoptive CTL transfer and immunization experiments identified the nuclear DNA-binding factor TOX (thymocyte selection-associated HMG-box protein) as a transcriptional regulator of encephalitogenic CTLs. Specifically, TOX expression was determined by the microbial context of CTL priming. The capacity of TOX-expressing CTLs to inflict tissue damage in the CNS was due to relative immune checkpoint insensitivity. Moreover, TOX-expressing CTLs were observed in human MS lesions. These observations offer a mechanistic link between infectious triggers and autoimmune diseases.
RESULTS
The Encephalitogenic Potential of CTL Responses Is Determined by the Microbial Context
We exploited the mouse model ODC-OVA, which expresses ovalbumin (OVA) as a neo-self antigen in myelin-forming oligodendrocytes (Cao et al., 2006). To investigate how priming by distinct pathogens might affect the capacity of CTLs to precipitate autoimmune disease in these mice, we took advantage of two different pathogens expressing full-length OVA: lymphocytic choriomeningitis virus (LCMV-OVA) and Listeria monocytogenes (Lm-OVA). ODC-OVA mice lack an endogenous pool of OVA-reactive CD8+ T cells (Na et al., 2012), and we adoptively transferred OVA-specific OT-1 CD8+ T cell receptor (TCR) transgenic CTLs 1 day before their activation by the pathogens. Despite the capacity to induce distinct inflammatory environments inside lymphoid organs (Kolumam et al., 2005; Thompson et al., 2006), infection with the two microbes induced similar OT-1 cell expansion in wild-type (WT) and ODC-OVA mice (Figures S1A and S1B). In addition, kinetics of the OT-1 response in the blood, spleen, and lymph node of WT mice showed a similar clonal differentiation into SLECs and MPECs, whereas the fraction of KLRG1hi CD127hi DPECs was slightly increased over time after Lm-OVA priming (Figure S1A). On day five after LCMV-OVA infection, ODC-OVA mice developed ascending hind-limb paresis and paralysis (Figure 1A), similar to experimental autoimmune encephalomyelitis (EAE). In contrast, and in agreement with a previous report (Na et al., 2012), ODC-OVA mice infected with Lm-OVA remained largely clinically healthy (Figure 1A). Analogous findings were made in MOGiCre-GP mice, which express the LCMV glycoprotein in oligodendrocytes but, unlike ODC-OVA mice, harbor an endogenous repertoire of CD8+ T cells against the neo-self antigen expressed in oligodendrocytes (Figures S1C–S1E). Histopathological analysis of diseased ODC-OVA mice revealed a substantial loss of oligodendrocytes that was paralleled by the presence of apoptotic oligodendrocytes (Nogo A+ caspase-3+) in LCMV-OVA-but not Lm-OVA-infected ODC-OVA mice (Figure 1B). CTL infiltrate densities in the CNS were similar in LCMV-OVA- and Lm-OVA-infected mice (Figure 1C).
Figure 1. LCMV-OVA-but Not Lm-OVA-Primed OT-1 Cells Induce CD8+ T-Cell-Mediated CNS Disease in ODC-OVA Mice.
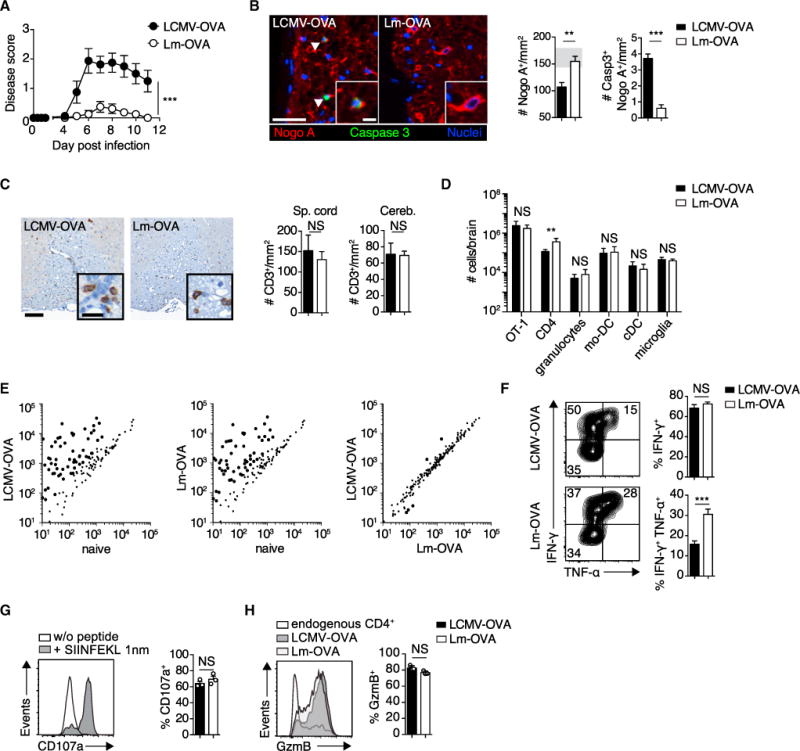
105 OT-1 cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with either 105 PFU LCMV-OVA or 5 3 103 CFU Lm-OVA.
(A) EAE disease course (n = 8 mice per group).
(B) Representative images of spinal cord sections (day 7) co-immunostained for Nogo A (oligodendrocytes) and activated caspase-3 (arrows indicate a cell positive for both caspase-3 and Nogo A) and quantification thereof (n = 6 mice per group). Gray zone in the bar graph indicates the range (mean ± SEM) of naive ODC-OVA mice (n = 3).
(C) Representative spinal cord section (day 7) immunostained with anti-CD3. Bar graphs show quantification of T cell infiltrates in spinal cord (Sp. cord) and cerebellum (Cereb.) (n = 3 mice per group).
(D) Flow-cytometric enumeration of CNS-infiltrating cells and microglia (day 7; n = 6 mice).
(E) Nanostring expression profiling of inflammatory genes in the spinal cord of naive or pathogen-challenged (day 7) ODC-OVA mice.
(F) Intracellular staining of IFN-γ and TNF-α in splenic OT-1 cells (day 7) after in vitro stimulation with cognate peptide (SIINFEKL; n = 6 mice).
(G) Degranulation as measured by surface expression of CD107a on CNS-infiltrating OT-1 cells after in vitro stimulation with SIINFEKL peptide.
(H) Intracellular staining for GzmB in CNS-infiltrating OT-1 cells (day 7; n = 3 mice).
Scale bars, 100 μm (B and C) or 20 μm (insets in (B and C). NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001 (two-way ANOVA with Bonferroni’s post-test for A; unpaired t test for B, D, and F–H). Data represent the pool of at least two independent experiments in (A), (B), (D), and (F) or one out of two representative experiments in (C), (G), and (H). Bars represent mean ± SEM.
See also Figure S1.
LCMV-OVA failed to trigger disease in ODC-OVA mice unless OT-1 cells were transferred (Figure S1F). Likewise, WT mice given an OT-1 cell transfer and challenged with LCMV-OVA did not develop disease (Figures S1F and S1G). Furthermore, intravenous infection with WT LCMV Armstrong (LCMV-Arm) did not induce CNS disease in ODC-OVA mice, even when they received naive or in-vitro-activated OT-1 cells, ruling out the possibility that the LCMV-induced inflammatory environment per se was sufficient for the observed disease outcome (Figure S1H). Neither OT-1 expansion nor the disease course was altered when we injected LCMV-OVA intraperitoneally (i.p.) instead of intravenously (i.v.) (Figure S1I). Given that we failed to recover either LCMV-OVA or Lm-OVA from the brain of i.v. infected mice (Figure S1J), differential microbial infection of the CNS was an unlikely cause of the clinical disease in ODC-OVA mice i.v. infected with LCMV-OVA.
We further quantified granulocytes, monocytes or macrophages, and dendritic cells in the brain of infected ODC-OVA mice. Besides slightly elevated CD4+ T cell infiltrates in Lm-OVA challenged mice, these analyses did not reveal substantial differences between the two microbes (Figure 1D). Furthermore, CNS inflammation in ODC-OVA mice resulted in elevated expression of numerous inflammatory genes irrespectively of the microbe used to trigger the T cell response (Table S1). These inflammatory signatures were largely overlapping and could not readily explain the differential disease outcome (Figure 1E). Furthermore, intracellular cytokine secretion assays showed similar functional avidity of OT-1 cells when activated by either microbe (Figures 1F and S1K). Judging from the ability of OT-1 cells to secrete both interferon-γ (IFN-γ) and tumor necrosis factor alpha (TNF-α), Lm-OVA-induced OT-1 responses were potentially more inflammatory than LCMV-OVA-induced responses (Figure 1F), which, however, inversely correlated with disease induction in vivo. Neither the analysis of GzmB expression in CNS-infiltrating OT-1 nor their capacity to degranulate in vitro revealed differences that could have explained the CTLs’ differential pathogenic potential in vivo (Figures 1G and 1H). Overall, these observations demonstrate that both LCMV-OVA and Lm-OVA induced comparable OT-1 cell expansion, OT-1 functionality, and CNS inflammatory infiltrates, but only LCMV-OVA-primed CTLs caused oligodendrocyte loss and precipitated disease in ODC-OVA mice.
Microbial Trigger Determines TOX Expression in CNS-Infiltrating OT-1 Cells
On the basis of the above observations, we speculated that the pathogen-associated inflammatory milieu could affect the transcriptional profile of CTLs. We performed RNA sequencing (RNA-seq) analysis and compared the transcriptional profile of fluorescence-activated cell sorting (FACS)-sorted OT-1 cells after LCMV-OVA or Lm-OVA infection in the spleen (days 3 and 7) and CNS (day 7; Figure 2A). On day 3 after infection, principal-component analysis (PCA) revealed distinct clustering of Lm-OVA-versus LCMV-OVA-primed OT-1 cells (Figure 2A), whereas the two types of OT-1 cells grouped more closely on day 7 of infection both in the spleen and brain (Figure 2A). Still, 191 and 189 genes were differentially expressed in OT-1 cells from the CNS and spleen (FC > 2), respectively, on day 7 of infection (Figure 2B and Tables S2 and S3). The gene encoding the DNA-binding factor TOX was strongly induced in LCMV-OVA-primed OT-1 cells infiltrating the CNS (Figure 2B), and among the transcription factors expressed in CTLs (Doering et al., 2012), Tox showed the largest difference in mRNA expression between these OT-1 cells in the CNS (Figure 2C). The same applied to their splenic counterparts (Figure S2A). Differential TOX expression was also observed at the protein level, most prominently in OT-1 cells infiltrating the CNS and to a lesser extent in the spleen (Figure 2D).
Figure 2. The DNA-Binding Factor TOX Is Induced in Brain-Infiltrating OT-1 Cells upon LCMV-OVA Challenge.
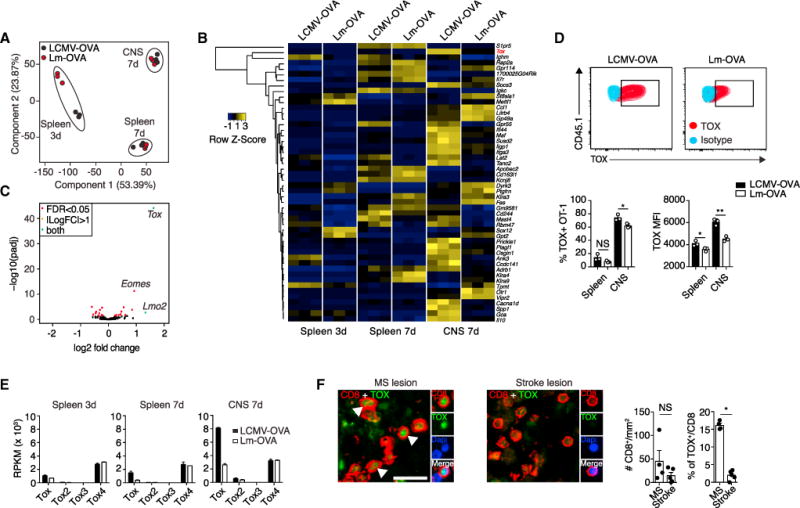
(A–E) 105 OT-1 cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with either 105 PFU LCMV-OVA or 5 3 103 CFU Lm-OVA.
(A) RNA-seq-based principal-component analysis (PCA) of FACS-sorted OT-1 cells from the spleen (days 3 and 7) and brain (day 7). Each dot represents an individual mouse.
(B) Heatmap analysis of the top 50 differentially expressed genes in CNS-infiltrating OT-1 after intravenous LCMV-OVA or Lm-OVA infection.
(C) Volcano plot depicting differential expression of 242 transcription factors (Doering et al., 2012) in CNS-infiltrating OT-1 cells (day 7) after intravenous priming with LCMV-OVA or Lm-OVA.
(D) Representative FACS plot and quantification of TOX expression in splenic and CNS-infiltrating OT-1 cells (day 7). MFI, mean fluorescence intensity. Data are representative of one out of five independent experiments. Bars represent mean ± SEM.
(E) Expression of the different Tox family members is presented as reads per kilobase per million reads (RPKM).
(F) Representative immunofluorescence co-staining for CD8 and TOX (counterstaining with DAPI for nuclei) in active MS lesions and stroke lesions and quantification thereof (n = 4 MS lesions and n = 5 stroke lesions).
Scale bars, 20 μm. NS, not significant; *p < 0.05; ***p < 0.001 (unpaired t test for D and F).
See also Figure S2.
Other genes encoding members of the TOX subfamily of HMG-box proteins, Tox2 and Tox4 (but not Tox3), were also detectable in activated OT-1 cells. However, only Tox was differentially expressed in OT-1 cells induced by Lm-OVA and LCMV-OVA (Figure 2E). In order to address whether the strength of antigenic stimulation provided to OT-1 cells was decisive for TOX induction, we generated recombinant LCMV-expressing low-affinity OT-1 peptide ligands (Q4H7; Zehn et al., 2009). Infection of mice with LCMV-expressing low-affinity OT-1 peptide reduced the magnitude of the OT-1 cell response (~20-fold) (Figure S2B). However, the few OT-1 cells that were generated had similar TOX expression to those that had been triggered by LCMV-OVA (cells expressing the WT, high-affinity OT-1 peptide ligand) (Figure S2C). This suggests that the strength of antigenic stimulation was decisive for the magnitude of the ensuing T cell response but was not a main determinant of TOX expression by the responding cells.
We next investigated whether TOX expression might also help us identify a subset of CTLs that accumulate during human CNS autoimmune diseases. We observed that a substantial fraction (16.1% ± 1.2%; n = 4) of CNS-infiltrating CTLs in MS lesions expressed TOX but that its expression was rare in CTLs inside post-ischemic brain lesions (2% ± 1.4%; n = 5) (Figure 2F). Although the histological findings in human biopsies remained correlative at this stage, they nevertheless were in line with our experimental observations in mice, suggesting a more general role of the transcriptional regulator TOX in licensing encephalitogenic CTLs.
IL-12 Represses TOX in a T-bet- and Eomes-Dependent Fashion
To gain insights into how the differential inflammatory microenvironment associated with LCMV-OVA and Lm-OVA infection might influence TOX induction in CTLs, we measured cytokine levels in the spleen. Among other differentially expressed cytokines (Figures 3A and S3A), we noted an induction of interleukin-12 (IL-12) in the spleen upon infection with Lm-OVA but not LCMV-OVA. IL-12 is known to inversely regulate the transcription factors Eomes and T-bet, which shape CD8+ T cell differentiation (Lazarevic et al., 2013). Moreover, analysis with the JASPAR database of transcription factor binding (Khan et al., 2018) revealed a putative Eomes as well as a T-bet binding site within 500 bp upstream of the Tox transcription start site (TSS). We therefore speculated that IL-12 modulates TOX expression in effector CTLs by regulating the aforementioned transcription factors. Indeed, the administration of recombinant IL-12 to LCMV-OVA-infected mice reduced TOX expression in transferred OT-1 cells (Figure 3B). However, exogenous IL-12 caused a wasting disease in LCMV-infected mice, as previously reported (Orange et al., 1995). This CNS-unrelated disease confounded the EAE disease score and rendered it unreliable. Thus, we further dissected the impact of IL-12 as a modulator of TOX expression by studying activated OT-1 cells in vitro. The addition of IL-12 reduced TOX expression in OT-1 cells in a dose-dependent manner that was paralleled by the reduction of Eomes and induction of T-bet (Figures 3C and S3B).
Figure 3. IL-12 Represses TOX in a T-bet- and Eomes-Dependent Fashion.
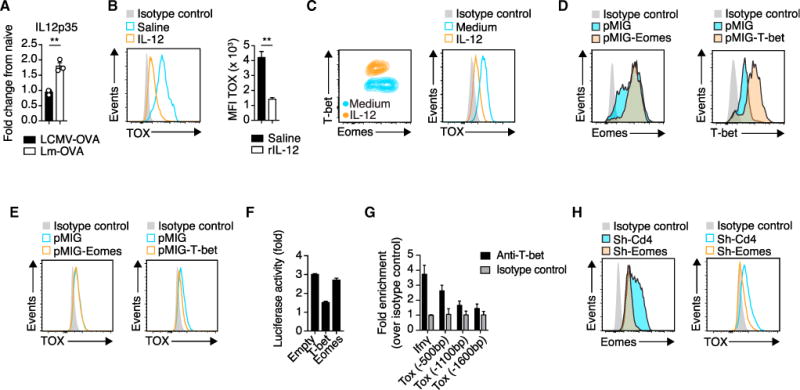
(A) Relative expression of IL12p35 (Gapdh used for normalization) measured by qRT-PCR in the spleen 24 hr after LCMV-OVA or Lm-OVA infection (n = 3 mice per group; data are normalized to naive mice).
(B) 105 OT-1 cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA, and IL-12 or vehicle (control) was administrated once a day for 5 consecutive days. Representative histogram and quantification of TOX expression in CNS-infiltrating OT-1 cells (day 7). MFI, mean fluorescence intensity.
(C) Flow-cytometric analysis of T-bet, Eomes, and TOX expression of in-vitro-activated OT-1 cells in the presence of IL-12 (10 ng/mL) or medium only (day 3 after activation).
(D) Expression of Eomes and T-bet in in-vitro-activated OT-1 cells transduced either with empty retrovirus or T-bet- or Eomes-expressing retrovirus.
(E) TOX expression in the transduced cells as in (D).
(F) Luciferase activity of EL-4 cells co-transfected with a TOX luciferase reporter (pGL3-TOX) together with either an empty vector or a T-bet- or Eomes-expressing vector. Data are expressed as the fold change between pGL3-TOX-transfected cells and cells transfected with the empty construct (pGL3-empty).
(G) T-bet ChIP analysis in splenic LCMV-OVA-activated OT-1 cells (day 7) followed by PCR amplification of regions located −0.5, −1.1, and −1.6 kb from the start codon of Tox and the promoter region of IFN-γ (positive control).
(H) Flow-cytometric analysis of Eomes and TOX expression in sh-Cd4 (control) and sh-Eomes retrovirally transduced in-vitro-activated OT-1 cells.
**p < 0.01 (unpaired t test for A and B). Data are representative of at least two independent experiments. See also Figure S3.
Next, we retrovirally overexpressed T-bet or Eomes in OT-1 cells in vitro (Figure 3D). T-bet but not Eomes overexpression repressed TOX expression (Figure 3E). We then transfected EL-4 mouse lymphoma cells with a luciferase reporter system containing a 2 kb Tox promoter region and determined whether T-bet repressed Tox transcription directly. EL-4 cells lack endogenous expression of T-bet and Eomes (Fukuoka et al., 2016), and retrovirally delivered T-bet but not Eomes reduced the activity of the Tox reporter construct by 50%–60% (Figure 3F). Furthermore, we analyzed LCMV-OVA-primed OT-1 cells by T-bet chromatin immunoprecipitation analysis followed by PCR (ChIP-PCR) of various Tox promoter regions. In accordance with a previous report (Dominguez et al., 2015), our analysis confirmed genomic T-bet binding within a region spanning the 500 bp upstream of the Tox start codon (Figure 3G).
To test whether Eomes repression as observed after IL-12 supplementation also modulated TOX expression, we performed short hairpin RNA (shRNA)-mediated knockdown of Eomes in in-vitro-activated OT-1 cells (Figure 3H). Indeed, knockdown of Eomes reduced TOX expression. In line with these observations, Eomes expression in splenic CTLs was lower after Lm-OVA than after LCMV-OVA infection (Figure S3C).
In addition to observing differential IL-12 induction between LCMV-OVA- and Lm-OVA-infected mice, we noted that IFN-α and IL-10 were more abundant in the spleens of LCMV-OVA-infected mice, but in vivo neutralization of these two cytokines did not reduce TOX expression in effector CTLs (Figure S3D).
In summary, these data suggest that IL-12 represses TOX by regulating T-bet and Eomes expression. Furthermore, they show that T-bet acts as a repressor of Tox by directly binding to its promoter.
Tox-Deficient CTLs Display Reduced Encephalitogenic Potential
We next investigated whether CTL-intrinsic TOX was required for the cells to cause disease in the ODC-OVA model. Adoptively transferred Tox−/−OT-1 cells caused only very mild clinical signs of EAE in LCMV-OVA-challenged ODC-OVA mice (Figure 4A). Accordingly, the density of oligodendrocytes in ODC-OVA mice that received Tox−/−OT-1 cells remained within normal ranges. Significantly fewer apoptotic (caspase-3+) oligodendrocytes were noted in the spinal cords of ODC-OVA mice that received Tox−/−OT-1 cells than in recipients of Tox+/+ OT-1 cells (Figure 4B). However, the total number of CNS-infiltrating OT-1 cells was similar in the two groups (Figure 4C). Tox−/−and Tox+/+ OT-1 cells were equally abundant in the blood, spleen, and lymph nodes (Figures S4A and S4B), and comparable percentages of these cells expressed the proliferation marker Ki67 when they infiltrated the CNS (Figure S4C). Similar results were obtained when knockdown of Tox was performed in mature Tox+/+ OT-1 cells by means of a retrovirally delivered shRNA (Figures S4D–S4G). These results argued against the possibility that a disturbed ontogenic differentiation of Tox−/−OT-1 donor cells rather than a role of Tox in the antiviral CTL response could have accounted for the profound impact of Tox deficiency on disease in ODC-OVA mice. To investigate whether impaired cytolytic activity was underlying the failure of Tox−/−CTLs to mediate EAE, we compared degranulation, GzmB expression, and primary ex vivo cytotoxicity of CNS-infiltrating Tox+/+ and Tox−/−OT-1 cells. Upon in vitro restimulation with OVA peptide, Tox−/−OT-1 cell surface expression of the degranulation marker CD107a was modestly reduced (Figure 4D). Conversely, GzmB was slightly more abundant in splenic and CNS-infiltrating Tox−/−OT-1 cells (Figure 4E), and primary ex vivo cytotoxicity of OT-1 cells on peptide-pulsed EL-4 target cells was independent of Tox (Figure 4F).
Figure 4. Tox-Deficient CTLs Are Less Encephalitogenic.
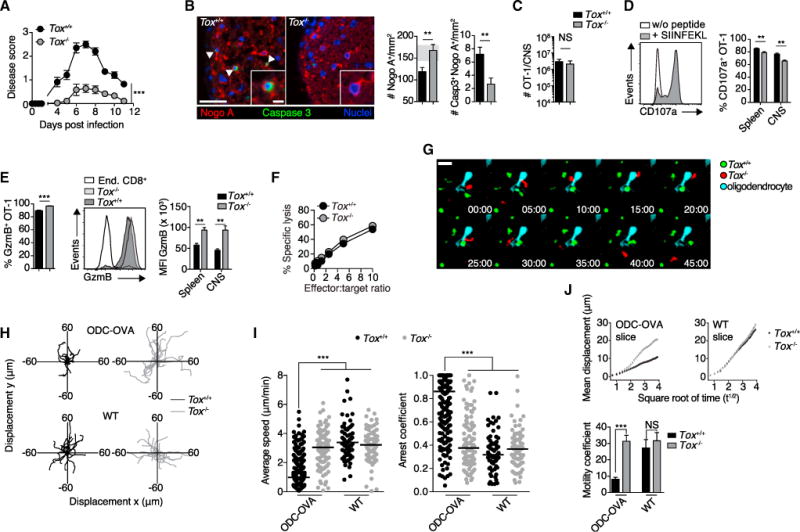
105 Tox+/+ or OT-1 Tox−/−cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA.
(A) EAE disease course (n = 10 mice per group).
(B) Representative images of spinal cord section (day 7) co-immunostained for Nogo A (oligodendrocytes) and activated caspase-3 (arrows indicate a cell positive for both caspase-3 and Nogo A) and quantification thereof (n = 8 mice per group). Gray zone in the bar graph indicates the range (mean ± SEM) of naive ODC-OVA mice (n = 3 mice).
(C) Flow-cytometric enumeration of CNS-infiltrating OT-1 cells (day 7, n = 6 mice).
(D) Degranulation as measured by CD107a surface expression on isolated splenic and CNS-infiltrating OT-1 cells after in vitro stimulation with SIINFEKL peptide.
(E) Flow-cytometric enumeration of GzmB-expressing OT-1 cells isolated from the CNS (left graph). GzmB expression in splenic and CNS-infiltrating OT-1 cells is as indicated in (A) (day 7, n = 6 mice, right graph). MFI, mean fluorescence intensity.
(F) Cytolytic activity of CNS-infiltrating Tox+/+ and Tox−/−OT-1 cells (day 7) measured in co-culture with SIINFEKL-pulsed EL-4 target cells.
(G) Representative time-lapse imaging of CNS-isolated Tox+/+ (green) and Tox−/−(red) OT-1 cells co-incubated onto organotypic hippocampal slice cultures of ODC-OVA mice. Oligodendrocytes were transduced with eGFP-expressing Adeno-associated vector under the control of myelin basic promotor (AAV-MBP-eGFP; see also STAR Methods and Video S1); bottom right corners indicate time (min:s).
(H) Superimposed 15 min individual tracks (lines) in the xy plane of 20 randomly selected OT-1 cells (starting coordinate set as 0). Data are pooled from two independent experiments.
(I) Average speed (μm/min) and arrest coefficient of Tox+/+ and Tox−/−OT-1 individual cells incubated onto WT or ODC-OVA slices. Horizontal lines indicate the median.
(J) Mean displacement of Tox+/+ and Tox−/−OT-1 cells in WT and ODC-OVA slices is plotted against the square root of time (min1/2). Motility coefficient was calculated as M = D2/4t, where M is the motility coefficient, D is displacement, and t is time.
Scale bars represent 100 μm (B) or 20 μm (G and inset in B). NS, not significant; **p < 0.01; ***p < 0.001 (two-way ANOVA with Bonferroni’s post-test for A, unpaired t test for B–E, one-way ANOVA with Tukey’s post-test for I, and Kruskal-Wallis ANOVA for J). Data represent the pool of at least two independent experiments in (A)–(E), (I), and (J) or one out of two representative experiments in (F). Bars represent mean ± SEM. See also Figure S4 and Videos S1 and S2.
We next investigated whether TOX influences the movement of CTLs in the complex tissue architecture of the brain. Immunological synapse formation with TCR engagement delivers a “stop signal” to CTLs, which decreases their motility and enables stable conjugate formation with target cells (Miller et al., 2002). Thus, we performed time-lapse imaging of CNS-derived Tox+/+ and Tox−/−effector OT-1 cells in hippocampal organotypic slice cultures in order to investigate the immunological synapse formation with antigen-positive oligodendrocytes (Figure 4G and Video S1). Time-lapse tracking of fluorescently labeled Tox+/+ OT-1 cells in ODC-OVA brain slices demonstrated an “arrested” phenotype (Figure 4H), whereas the tissue patrolling behavior of Tox−/−OT-1 cells was largely unaffected by antigenic encounters with OVA-expressing oligodendrocytes (Figure 4H). The median stop time of Tox+/+ OT-1 cells was 12 min, whereas it was 2.5 min for Tox−/−OT-1 cells (p < 0.0001, Kolmogorov-Smirnov test) (Figure S4H). Tox+/+ OT-1 cells more frequently stopped their movements as measured by an increased arrest coefficient as well as an overall reduced speed and motility coefficient (Figures 4I and 4J). In contrast, Tox+/+ and Tox−/−OT-1 cells showed similar motility parameters when studied in WT brain slices, indicating that in the absence of cognate antigen, the intrinsic motility of brain-infiltrating CTLs was unaffected by TOX (Figures 4H–4J and Video S2). Together, these data suggest that TOX is essential for autoreactive CTLs to form stable immunological synapses with antigen-expressing oligodendrocytes.
TOX Modulates Transcriptional Programs Related to Effector CTL Differentiation
To identify TOX-regulated genes in CTLs, we analyzed the global gene expression profiles of CNS-infiltrating Tox+/+ and Tox−/−OT-1 cells, which clustered separately according to PCA (Figure S5A and Table S4). Interestingly, genes such as Klrg1, Gzma, and Klra5, which are associated with terminal effector differentiation, were repressed in the presence of TOX. Conversely, the expression of genes encoding TCF-1, CCR7, and lymphotoxin-β (Tcf7, Ccr7, and Ltb, respectively) was higher in Tox-sufficient OT-1 cells (Figure 5A). In addition, the gene Cd244, encoding the inhibitory receptor CD244 (also known as 2B4), was more highly expressed by Tox−/−OT-1 cells. To get insights into how TOX regulates gene expression in CTL differentiation, we performed chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) in CNS-infiltrating OT-1 cells. Among the 7,579 TOX binding sites detected, 5,121 showed a specific enrichment in Tox+/+ as compared with Tox−/−OT-1 cells (Figure 5B). Further, analysis of peak distribution revealed around 75% binding in regions proximal to TSSs. To evaluate which altered genes identified in our RNA-seq analysis could be direct targets of TOX, we compared significantly altered genes (padj < 0.01) with ChIP-seq TOX binding events. 81 genes showed an enrichment of TOX binding sites proximal to their TSS (Figure 5B and Table S5), revealing a significant enrichment of GO terms associated with immune system processes (Figure 5C). These altered genes were either up- or downregulated in our RNA-seq, suggesting that TOX can exert transactivator and repressor functions (Figure 5D). Among those genes, we identified a TOX binding site 2.5 kb upstream of the TSS of the gene Id2 (Figure 5E). qRT-PCR analysis revealed 3-fold lower expression of Id2 in CNS-infiltrating Tox+/+ OT-1 cells than in Tox−/−OT-1 cells (Figure 5F). Subsequent gene-set enrichment analysis (GSEA) showed that the TOX-dependent transcriptional signature was inversely correlated with terminal-effector-associated genes known to be induced by Id2 (Masson et al., 2013) (Figure 5G). We identified a TOX binding site upstream of Nrarp, an additional gene known to drive terminal differentiation of CTLs (Backer et al., 2014) and a component of the Notch signaling pathway. GSEA of a Notch-dependent gene signature corroborated an inverse correlation between genes induced by Notch in CTLs and expression of Tox (Figure S5B).
Figure 5. TOX Represses Expression Signature of Terminal Differentiation in CTLs.
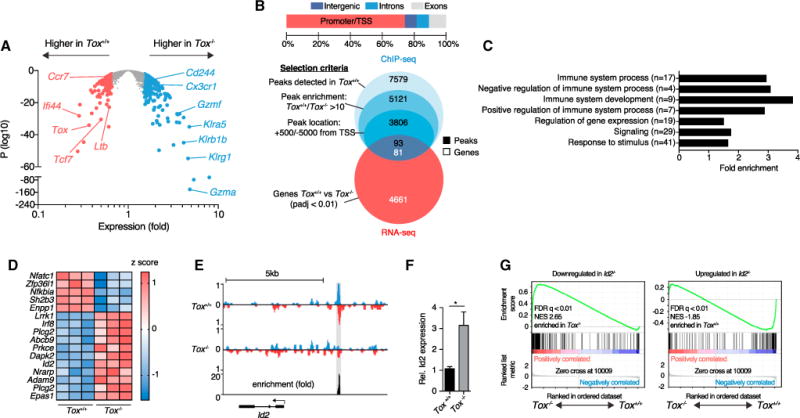
105 Tox+/+ or Tox−/−OT-1 cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA. CNS-infiltrating OT-1 cells were sorted 7 days later for (A) RNA-seq gene expression, (B–E) ChIP-seq analysis of TOX binding site, or (F) qRT-PCR.
(A) Volcano plot comparing differential gene expression in Tox+/+ versus Tox−/−OT-1 cells (n = 3 mice per group).
(B) Distribution of the 5,121 peaks between promoters (from −5,000 bp to +500 bp from transcription starting sites [TSSs]), introns, exons, and intergenic regions. Venn diagrams show the selection of ChIP-seq peaks and their overlap with RNA-seq data.
(C) Selected GO terms enrichment of selected peaks identified in (B); numbers in brackets represent total number of genes in that specific GO term.
(D) Heatmap showing relative expression of a selection of genes belonging to immunological processes and identified in (B); each column represents an individual mouse.
(E) ChIP-seq analysis of TOX binding in the Id2 upstream region. We determined fold enrichment of TOX binding by dividing the number of reads in Tox+/+ OT-1 cells by the number of reads in Tox−/−OT-1 cells at the Id2 binding site. Detected peak is highlighted in gray.
(F) Relative expression of Id2 (Gapdh used for normalization) measured by qRT-PCR in Tox+/+ and Tox−/−OT-1 cells (n = 5 mice per group).
(G) Gene-set enrichment analysis of Id2-dependent gene sets in the gene expression profile (RNA-seq analysis as in A shows genes with lower expression in Id2−/−[left] or higher expression in Id2+/+ [right] CTLs; Masson et al., 2013). FDR, false-discovery rate; NES, normalized enrichment score.
*p < 0.05 (unpaired t test in F). Bars represent mean ± SEM. See also Figure S5.
Next, we determined whether Lm-OVA-primed Tox+/+ OT-1 cells (TOXlo phenotype) share a gene expression profile with Tox−/−OT-1 cells generated in LCMV-OVA infection. We cross-referenced differentially expressed genes emerging from the comparison of LCMV-OVA-induced Tox+/+ OT-1 cells (TOXhi phenotype) with either LCMV-OVA-induced Tox−/−OT-1 cells or Lm-OVA-primed Tox+/+ OT-1 cells (TOXlo phenotype). Of the set of genes that were upregulated in Lm-OVA-primed CNS-infiltrating Tox+/+ OT-1 cells, about 30% were also enriched in CNS-infiltrating LCMV-induced Tox−/−OT-1 cells (Figure S5C). This included gene products that are known to be induced in terminally differentiated effector CTLs, such as Klrg1, Gzma, S1pr5, and Cx3cr1 but not Cd244. Similarly, about 30% of the genes expressed at higher levels in LCMV-OVA-induced Tox+/+ OT-1 cells than in Lm-OVA-primed Tox+/+ OT-1 cells were also upregulated in LCMV-OVA-triggered Tox+/+ OT-1 cells compared with LCMV-OVA-induced Tox−/−OT-1 cells (Figure S5D). In addition, we noted a pathogen-dependent but TOX-independent signature when comparing LCMV-OVA against Lm-OVA-primed CTLs.
Together, these data identify TOX as a regulator of CTL differentiation by repressing Notch- and Id2-dependent gene signatures.
TOX Inhibits Terminal Differentiation of CTLs and Represses the Inhibitory Receptor CD244
Gene expression profiles of Tox−/−OT-1 cells were reminiscent of a terminal differentiation signature. Hence, we compared clonal differentiation of adoptively transferred OT-1 cells in blood, spleen, and lymph nodes in WT recipients. In Tox−/−OT-1 cells, the frequency of KLRG1hi CD127lo SLECs was increased and the frequency of KLRG1lo CD127hi MPECs was reduced throughout the various time points assessed (Figures 6A and 6B), whereas the overall number of OT-1 cells in blood and spleen was unaffected by Tox deficiency (see Figure S4B). In analogous experiments, we co-transferred congenically marked Tox+/+ (CD45.1+) and Tox−/−(CD45.1/2+) OT-1 cells. Throughout the observation time of >40 days, KLRG1hi CD127lo SLECs and KLRG1hi CD127hi DPECs were more abundant in Tox−/−OT-1 cells than in Tox+/+ cells inside the same recipients (Figure S6A). Conversely, the amount of KLRG1lo CD127hi MPECs remained lower in Tox−/−OT-1 cells (Figure S6A). Similarly, splenic and CNS-infiltrating Tox−/− OT-1 cells differentiated mainly into SLECs (Figure 6C). We further assessed whether the rather high number of 105 adoptively transferred OT-1 cells accounted for the differentiation of Tox−/−OT-1 cells into SLECs. However, biased SLEC differentiation of Tox−/−OT-1 cells was observed when as few as 102 cells were transferred (Figure S6B). In addition, analogous results were obtained when the differentiation of Tox−/−P14 cells (CD8+ TCR transgenic cells specific to the LCMV gp33–41 peptide) was studied upon LCMV-Arm challenge (Figure S6C).
Figure 6. TOX Reduces the Generation of Terminally Differentiated CTLs.
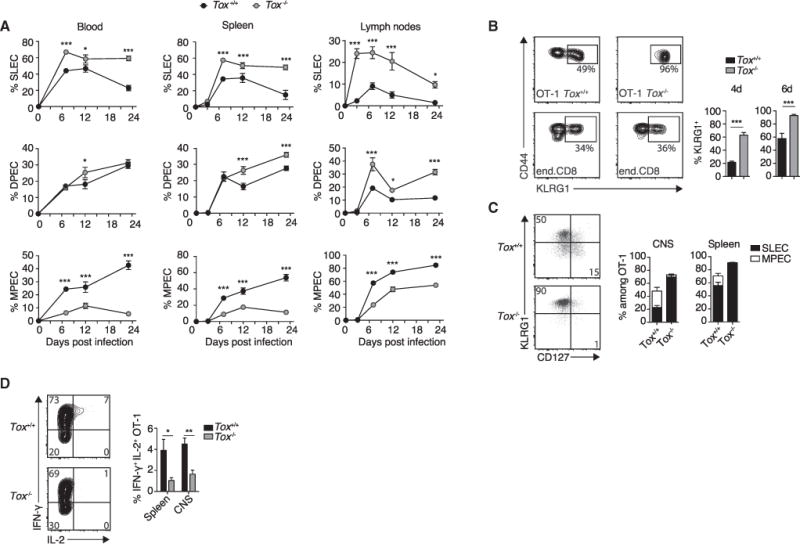
105 Tox+/+ or Tox−/−OT-1 cells were adoptively transferred into WT (A) or ODC-OVA (B–D) mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA.
(A) Time-course analysis of clonal differentiation of OT-1 cells into SLECs (KLRG1hi CD127lo), DPECs (KLRG1hi CD127hi), and MPECs (KLRG1lo CD127hi) in the blood, spleen, and lymph nodes.
(B) Representative flow-cytometry plot showing surface expression of CD44 and KLRG1 on OT-1 cells or endogenous pool of CD8+ T cells in the blood (day 6). Numbers in the plot indicate the percentage of KLRG1hi cells among effector Tox+/+ or Tox−/−OT-1 cells (CD44+, CD45.1+) or endogenous effector CD8+ T cells (CD44+, CD45.2+). Bar graphs show quantification thereof at the indicated time points (n = 6 mice per group).
(C) Representative flow-cytometry plot and quantification of the frequency of SLECs and MPECs in Tox+/+ and Tox−/−OT-1 cells isolated from the spleen and the CNS (day 6; n = 6 mice per group).
(D) Intracellular staining for IFN-γ and IL-2 in CNS-infiltrating Tox+/+ and Tox−/−OT-1 cells after in vitro stimulation with SIINFEKL peptide (day 6; n = 6 mice per group).
*p < 0.05; **p < 0.01; ***p < 0.001 (two-way ANOVA with Sidak’s post-test for A and unpaired t test for B and D). Data are representative of at least two independent experiments (B–D). Bars represent mean ± SEM. See also Figure S6.
Next, we tested whether Tox deficiency altered the cytokine production of CTLs. CNS-infiltrating and splenic Tox+/+ and Tox−/−OT-1 cells produced similar amount of IFN-γ (Figure S6D). However, OT-1 cells deficient in Tox expressed modestly lower amounts of TNF-α in the spleen and were poor producers of IL-2 in both the spleen and CNS (Figures 6D and S6D), which was consistent with the inability of SLECs to produce IL-2 (Sarkar et al., 2008).
We assessed TCF-1, IRF4, T-bet, and Eomes expression at the protein level in LCMV-OVA-primed Tox+/+ and Tox−/−OT-1 cells. Tox−/−OT-1 expressed less TCF-1, whereas IRF-4, Eomes, and T-bet expression was unaltered in the CNS (Figure 7A). RNA-seq analysis (see Table S4) had indicated that Tox deficiency was associated with increased expression of the inhibitory receptor SLAM family member Cd244 (Waggoner and Kumar, 2012). We observed the same at the protein level when studying OT-1 cells isolated from the CNS (Figure 7B), blood, and secondary lymphoid organs (Figure 7C). Similarly, CD244 was induced in Tox−/−P14 cells upon LCMV-Arm infection (Figure S7A). In contrast, surface expression of other inhibitory receptors, such as lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin mucin-3 (TIM-3), and programmed death-1 (PD-1), was unaffected by Tox deficiency (Figure 7B). Similarly to Tox−/−OT-1 cells, Tcf7−/−P14 cells showed increased CD244 surface expression on day 7 after LCMV infection (Figure 7D), corroborating the role of TCF-1 as a negative regulator of Cd244 (Utzschneider et al., 2016). Tox deficiency did not affect the expression of KLRG1 or CD127 on naive splenic OT-1 cells. Also, the inhibitory receptors CD244, LAG-3, TIM-3, and PD-1 (Figure S7B) were expressed at comparable levels, and Tox−/−OT-1 mice had normal levels of naive splenic CD8+ T cells (Figure S7C).
Figure 7. Inhibitory Receptor CD244 Is Induced on Tox-Deficient Effector CTLs and Blockade of CD244-CD48 Signaling Restores Their Encephalitogenic Properties.
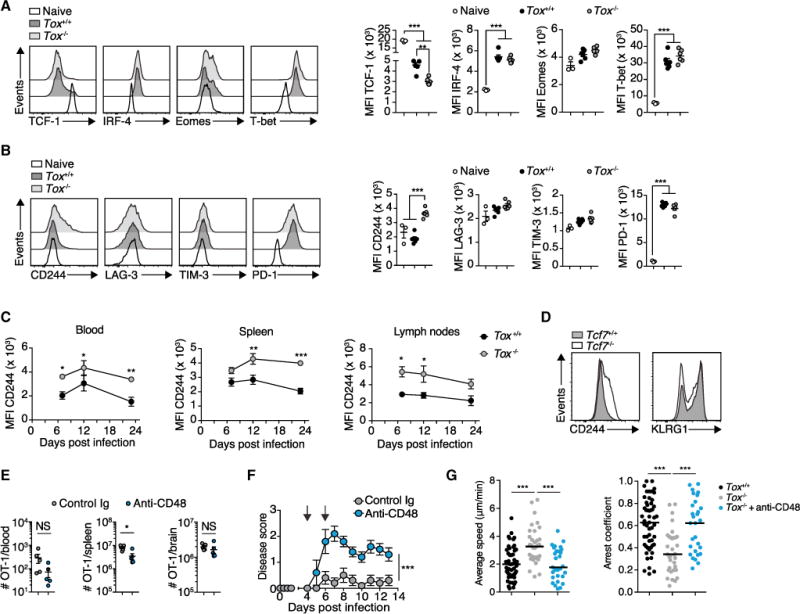
(A and B) 105 Tox+/+ or Tox−/−OT-1 cells were adoptively transferred into ODC-OVA mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA. CNS-infiltrating OT-1 cells were isolated at day 6. (A) Representative histograms and quantification of TCF-1, IRF-4, Eomes, and T-bet expression. (B) Representative histograms and quantification of surface expression of CD244, LAG-3, TIM-3, and PD-1 (n = 6 mice per group; naive splenic OT-1 cells served as control; n = 3 mice).
(C) 105 Tox+/+ or Tox−/−OT-1 cells were adoptively transferred into WT mice. One day later (day 0), mice were challenged i.v. with 105 PFU LCMV-OVA. Time-course analysis of CD244 expression in OT-1 cells from the blood, spleen, and lymph nodes. MFI, mean fluorescence intensity.
(D) Flow-cytometric analysis of surface expression of CD244 and KLRG1 on adoptively transferred Tcf7+/+ and Tcf7−/−P14 cells in the blood 7 days after LCMV-Arm infection of the WT recipient.(E and F) Mice received Tox−/−OT-1 cells and were challenged with LCMV-OVA as in (A). Anti-CD48-neutralizing antibody (HM48-1) or its respective isotype control antibody was administered i.p. (300 μg) 4 and 6 days after infection (n = 5 mice per group). (E) Flow-cytometric enumeration of Tox−/−OT-1 cells in the blood, spleen, and brain (day 7; n = 6 mice per group). (F) EAE disease course.
(G) Average speed (μm/min) and arrest coefficient of Tox+/+ and Tox−/−OT-1 cells that were treated with and without anti-CD48 and incubated onto ODC-OVA slices. Horizontal lines indicate the median.
NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001 (unpaired t test for F, two-way ANOVA with Bonferroni’s post-test for E, and one-way ANOVA with Tukey’s post-test for A, B, and G). Data represent one out of at least two independent experiments (A–G). Bars represent mean ± SEM. See also Figure S7.
Together, these data strongly suggest that in primed CTLs, TOX induces TCF-1 (potentially through repression of Id2, as shown in Masson et al., 2013; see Figures 5F and 5G) and thereby averts terminal differentiation of CTLs that highly express the inhibitory receptor CD244. We thus speculated that blocking the inhibitory CD244 signaling would exacerbate disease induced by Tox−/−OT-1 effector cells. Four days after LCMV-OVA challenge, we started the administration of neutralizing antibody against CD48, one of the main ligands of CD244. This did not affect the expansion of Tox−/−OT-1 cells, but it restored their capability to cause CNS disease (Figures 7E and 7F). Time-lapse tracking of fluorescence-labeled Tox−/−OT-1 cells on organotypic slice culture showed that neutralization of CD48 restored their tissue-patrolling behavior, as evidenced by a normalized “average speed” and “arrest coefficient” (Figure 7G). In contrast, antibody-mediated PD-1 blockade did not affect OT-1 cell expansion, differentiation, or GzmB expression. Besides lower PD-1 surface detection, anti-PD-1 antibody treatment did not reduce the expression of other inhibitory receptors (Figures S7D–S7H), but it precipitated disease in recipients of Tox−/−OT-1 cells (Figure S7I).
Next, we investigated whether CD48-neutralizing antibody accentuated the pathogenicity of Tox+/+ OT-1 cells in LCMV-OVA-infected ODC-OVA mice. Unlike in recipients of Tox−/−OT-1 cells, CD48 neutralization in recipients of Tox+/+ OT-1 cells did not further aggravate CNS disease (Figure S7J). Neither did CD48 neutralization lead to substantial disease in Lm-OVA-infected mice (Figure S7J). The latter finding suggests that additional mechanisms besides CD244 expression curtail the potential of Lm-OVA-induced OT-1 cells to cause CNS disease. CD48 neutralization did not affect expansion, CNS infiltration, GzmB expression, degranulation, cytokine production, or differentiation of Tox+/+ OT-1 cells (Figures S7K–S7P), which was in line with unaltered disease.
In addition, the CD48-CD244 axis has been shown to play a major role in regulating natural killer (NK) cell function (Waggoner and Kumar, 2012). However, we failed to detect a substantial impact of CD48 blockade on NK cell expansion, CNS infiltration, or GzmB and inhibitory receptor expression (Figures S7Q–S7S). The only effect of CD48 neutralization on NK cells we found was increased NK cell surface CD244 expression, which was most likely due to reduced CD48-mediated internalization of its receptor CD244, as previously described (Sandusky et al., 2006) (Figure S7S). Together, these data suggest that the DNA-binding factor TOX renders self-reactive CTLs less responsive to CD244 ligand inhibition and thereby bolsters their encephalitogenic potential independently of PD-1 signaling.
DISCUSSION
Here, we exploited an animal model of neuroinflammation and identified TOX as a transcriptional regulator of tissue-destructive CTLs. TOX attenuated the terminal differentiation of CTLs by modulating Id2-, TCF-1-, and Notch-driven pathways (Backer et al., 2014; Yang et al., 2011; Zhou et al., 2010). Although transcriptional regulators of terminal differentiation, type 1 and type 2 cytokine profiles, and exhaustion have been investigated, the factors and programs governing tissue-destructive capacity in autoimmunity have remained unknown.
Primary CTL expansion was TOX independent, and Tox−/−OT-1 effector cells exhibited primary ex vivo cytotoxic activity comparable to that of their WT counterparts. In contrast, CTL-mediated death of oligodendroglia in vivo and resulting disease were largely dependent on TOX. These differences between ex vivo and in vivo effector function suggest that the tissue microenvironment is decisive for the cytotoxic effector function of CTLs. In the context of cancer immunosurveillance, analogous concepts are supported by several lines of convergent evidence (Pardoll, 2012). Therefore, our findings suggest that, analogous to activity of tumor-infiltrating lymphocytes, the tissue-destructive activity of autoreactive CTLs is guided by immune checkpoints such as CD244 ligands (Waggoner and Kumar, 2012). Accordingly, an important evolutionary purpose of CTL checkpoint inhibition could be limiting autoimmune tissue damage. TOX restrains CD244 expression upon Teff cell differentiation into SLECs, thus licensing them to cause autoimmune destruction. In concert with this proposal, reduced CD244 expression has been observed in patients suffering from autoimmune diseases such as systemic lupus erythematosus (Kim et al., 2010). Thus, this signaling pathway could play a role in pathogenesis of human autoimmune disorders when deregulated. In line with this observation, blockade of CD244-CD48 ligand interactions restored the encephalitogenic properties of Tox-deficient CTLs, providing a mechanistic explanation for the in vivo dependency of encephalitogenic CTLs on TOX. Our data provide evidence that supports the role of TOX and its repression of CD244 in encephalitogenic CTLs. Conversely, the absence of disease in anti-CD48-blocked Lm-OVA-infected mice suggests that besides CD244, additional immune checkpoints curtail the encephalitogenic potential of Lm-induced CTLs. In addition, blocking CD48 might also affect the CD2-LFA-3 axis (Makgoba et al., 1989). PD-1, TIM-3, and LAG-3 are frequently found on effector CTLs (Pardoll, 2012), but their similar expression levels in Tox-deficient and -sufficient CTLs suggest that other, less commonly studied pathways could be accountable.
Our study further suggests that the microbial context of CTL induction could imprint qualitative differences in CTL responses and thereby determine their encephalitogenic potential. Such imprints might consist of epigenetic modifications known to occur during acquisition of CTL effector differentiation (Scott-Browne et al., 2016). Consistent with this hypothesis, CD8+ T cells of MS patients were found to be abnormally hypermethylated (Bos et al., 2015). Earlier reports have demonstrated that pathogen-derived signals (Kaech and Wherry, 2007) and the inflammatory milieu (Obar et al., 2011) influence the differentiation and fate of antigen-specific CTLs that could be in part mediated by IL-12 (Joshi et al., 2007). Furthermore, our data do not exclude the possibility that additional IL-12-induced pathway(s), including STAT4 signaling (Thieu et al., 2008) together with or independently of Eomes and/or T-bet, could contribute equally to regulation of TOX expression. Interestingly, Eomes has recently been shown to contribute to the pathogenic potential of autoreactive CD4+ T cells in EAE (Stienne et al., 2016). Given that the knockdown of Eomes reduced TOX expression in CTLs, Eomes might thus also modulate encephalitogenic properties of CTLs, but formal proof needs to be provided in future work. TOX expression was particularly pronounced in CNS-infiltrating CTLs. Although pathogen imprint inside secondary lymphoid organs seems likely too, it remains possible that yet-unknown signals in the CNS microenvironment could fine-tune CTL polarization and responsiveness to self-structures.
TOX has originally been identified as a factor required for thymic development of CD4+ T-lineage cells (Aliahmad and Kaye, 2008) and bone marrow development of innate lymphoid cells (Seehus et al., 2015), the latter of which include NK cells (Aliahmad et al., 2010), but was not essential for CD8+ T cell development (Aliahmad and Kaye, 2008). With regard to CTLs, Tox was considered a central hub gene in differential network analyses comparing memory CTLs emerging from acute and chronic infection (Doering et al., 2012). Our study focuses on the role of TOX during primary expansion of CTLs and provides evidence that distinct pathogens induce CTLs with differential TOX expression. Further, the genetic ablation of Tox impairs their functional responsiveness when they encounter self-antigens in the CNS. Additional studies will, however, be needed to decipher whether TOX expression is sufficient to confer CTLs with encephalitogenic capacity.
We observed that brain-infiltrating Tox−/−effector CTLs are poor producers of IL-2, which is indicative of a reduced recall response capacity upon antigen re-encounter (Sarkar et al., 2008). This Tox−/−phenotype exhibits some similarity to the phenotype of Tcf7-deficient memory CTLs in the context of chronic infection (Utzschneider et al., 2016) and is consistent with our finding that TOX affects Tcf7-dependent transcriptional networks. However, the putative role of TOX in memory T cells remains to be addressed in future studies.
Collectively, our findings reveal that upon CTL priming, the DNA-binding factor TOX acts as a transcriptional regulator of differentiation, which affects the cells’ encephalitogenic properties. A refined understanding of the microbial imprint on transcriptional networks of CTLs could be beneficial for the development of novel interventions in autoimmune diseases such as MS.
STAR+METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Armenian hamster anti-CD48 (clone HM48-1) neutralizing Ab | BioXcell | Cat# BE0147 |
| Rat anti-CD3 (clone 17A2) | BioLegend | Cat# 100220 |
| Armenian hamster anti-CD3 (clone 145.2C11) | BioLegend | Cat# 100331 |
| Rat anti-CD4 (clone RM4-5) | BioLegend | Cat# 116016 |
| Rat anti-CD4 (clone YTS191) depleting Ab | BioXcell | Cat# BE0119 |
| Rat anti-CD8a (clone 53-6.7) | BioLegend | Cat# 100725 |
| Rat anti-CD11b (clone M1/70) | BioLegend | Cat# 101212 |
| Armenian hamster anti-CD11c (clone N418) | BioLegend | Cat# 117310 |
| Armenian hamster anti-CD28 (clone 37.51) | BioLegend | Cat# 102112 |
| Goat anti-hamster IgG (H+L) | Jackson Immunoresearch | Cat# 127-005-099 |
| Rat anti-CD44 (clone IM7) | BioLegend | Cat# 103028 |
| Rat anti-CD45 (clone 30-F11) | BioLegend | Cat# 103116 |
| Mouse anti-CD45.1 (clone A20) | BioLegend | Cat# 110722 |
| Mouse anti-CD45.2 (clone 104) | BioLegend | Cat# 109822 |
| Rat anti-CD45R/B220 (clone RA3-6B2) | BioLegend | Cat# 103236 |
| Rat anti-CD49b (clone DX5) | BioLegend | Cat# 108909 |
| Rat anti-CD107a (clone 1D4B) | BioLegend | Cat# 121606 |
| Rat anti-CD127 (clone A7R34) | BioLegend | Cat# 135014 |
| Mouse anti-CD244 (clone m2B4 (B6)458.1) | BioLegend | Cat# 133508 |
| Mouse anti-granzyme B (clone GB11) | BioLegend | Cat# 515406 |
| Rat anti-IFN-γ (clone XMG1.2) | BioLegend | Cat# 505829 |
| Rat anti-IL-2 (clone JES6-5H4) | BioLegend | Cat# 503808 |
| Rat anti-IL-10 (clone JES5-2A5) neutralizing Ab | BioXcell | Cat# BE0049 |
| Armenian hamster anti-IFN-a (clone TIF-3C5) neutralizing Ab | Leinco Technologies | Cat# I-1183 |
| Rat anti-IRF4 (clone IRF4.3E4) | BioLegend | Cat# 664406 |
| Mouse anti-TIGIT (clone 1G9) | BioLegend | Cat# 142107 |
| Rat anti-Ki-67 (clone SolA15) | eBioscience | Cat# 12-5698-82 |
| Syrian hamster anti-KLRG1 (clone 2F1/KLRG1) | BioLegend | Cat# 138410 |
| Armenian hamster anti-NKG2D (clone C7) | BioLegend | Cat# 115705 |
| Rat anti-NKp46 (clone 29A1.4) | BioLegend | Cat# 137611 |
| Mouse anti-LAG-3 (clone 11C3C65) | BioLegend | Cat# 369306 |
| Mouse anti-T-bet (clone 4B10) | BioLegend | Cat# 644814 |
| Mouse anti-T-bet (clone 4B10) for ChIP | Santa Cruz Biotechnology | Cat# sc-21749 |
| Mouse anti-TIM-3 (clone B8.2C12) | BioLegend | Cat# 134008 |
| Rat anti-PD-1 (clone RMP1-30) | BioLegend | Cat# 109103 |
| Rat anti-PD-1 (clone 29F.1A12) neutralizing Ab | BioXcell | Cat# BE0273 |
| Rat anti-TNF-α (clone MP6-XT22) | BioLegend | Cat# 506323 |
| Rat anti-Eomes (Dan11mag) | eBioscience | Cat# 25-4875-80 |
| Rabbit anti-TCF-1 (C63D9) | Cell Signaling Technology | Cat# 6709S |
| Rat anti-TOX (clone TXRX10) | eBioscience | Cat# 12-6502-82 |
| Rat anti-CD3 (clone CD3-12) | Biorad | Cat# MCA1477A488 |
| Biotin-labeled rabbit anti-rat antibody | Dako | Cat# E0468 |
| Mouse anti-Nogo A (clone 11C7) | Oertle et al., 2003 | N/A |
| Rabbit anti-cleaved caspase-3 | Cell Signaling Technology | Cat# 9661 |
| Alexa555-labeled-goat anti-mouse | Invitrogen | Cat# A21127 |
| Goat-anti-mouse Fab fragment | Jackson Immunoresearch | Cat# 115-006-006 |
| Rabbit anti-GFP (clone D5.1) | Cell Signaling Technology | Cat# 2956 |
| Mouse-anti-human CD8 (clone C8/144B) | Dako | Cat# IR62361-2 |
| Alexa647-labeled-donkey anti-rat | Jackson Immunoresearch | Cat# 712-605-153 |
| Rabbit anti-TOX1 | AbCam | Cat# ab155768 |
| Bacterial and Virus Strains | ||
| Listeria-OVA | Zehn et al., 2009 | N/A |
| Listeria-GP33 | Oberle et al., 2016 | N/A |
| pAAV-MBP-eGFP | von Jonquieres et al., 2013 | N/A |
| LCMV-OVA | Kallert et al., 2017 | N/A |
| LCMV-Q4H7 | Zehn et al., 2009 | N/A |
| LCMV WT (Armstrong) | Flatz et al., 2006 | N/A |
| LCMV-GP33 | Kallert et al., 2017 | N/A |
| Biological Samples | ||
| MS lesions | University of Göttingen | N/A |
| Post ischemic lesion | University of Göttingen | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Molecular Probes | Cat# D1306 |
| CellTracker Red CMPTX | Molecular Probes | Cat# C34552 |
| Green CFSE | BioLegend | Cat# 423801 |
| Collagenase A | Roche | Cat# 11088793001 |
| DNaseI | Roche | Cat# 10104159001 |
| Monensin | BioLegend | Cat# 420701 |
| SIINFEKL peptide | Polypeptide Group | N/A |
| Recombinant murine IL-12 | Peprotech | Cat# 210-12 |
| Recombinant murine IL-15 | Peprotech | Cat# 210-15 |
| Critical Commercial Assays | ||
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-075 |
| AccuCheck Counting Beads | Molecular Probes | Cat# PCB100 |
| Alexa Fluor 488 Tyramide Reagent | Molecular Probes | Cat# B40953 |
| FoxP3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Fixation buffer | BioLegend | Cat# 420801 |
| Intracellular Staining Perm Wash Buffer | BioLegend | Cat# 421002 |
| CytoTox 96 NonRadioactive Cytotoxicity Assay | Promega | Cat# G1780 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| TruSeq ChIP Sample Preparation Kit | Illumina | Cat# IP-202-1012 |
| TruSeq RNA Sample Prep Kit v2 | Illumina | Cat# RS-122-2001 |
| Dual Glo Luciferase Assay Kit | Promega | Cat# E2920 |
| Nucleofector kit L | Lonza | Cat# VACA-1005 |
| iScript cDNA synthesis kit | Biorad | Cat# 1708891 |
| iQ SYBR Green Supermix | Biorad | Cat# 1708864 |
| nCounter GX Mouse Inflammation Kit | Nanostring technologies | Cat# XT-GXA-MIN2-24 |
| Deposited Data | ||
| RNA-seq OT-1 LCMV-OVA/Lm-OVA | This paper | GEO: GSE93805 |
| RNA-seq OT-1 Tox+/+/Tox−/− | This paper | GEO: GSE93804 |
| ChIP-seq OT-1 Tox+/+/Tox−/− | This paper | GEO: GSE93953 |
| Id2-dependent CTL signature upon HK×31 Influenza infection | Masson et al., 2013 | GEO: GSE44140 |
| Notch-1-2-dependent CTL signature upon HK×31 Influenza infection | Backer et al., 2014 | E-MTAB-2999 |
| Microarray data T cell exhaustion signature | Doering et al., 2012 | GEO: GSE41867 |
| Experimental Models: Cell Lines | ||
| MC57G | ATCC | ATCC CRL-2295 |
| EL4 | ATCC | ATCC TIB-39 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Charles River | Stock#: 000664 |
| Mouse: ODC-OVA | Laboratory of Thomas Hunig | N/A |
| Mouse: MOGiCre-GP | This paper | Generated at Cyagen Biosciences |
| Mouse: Tox−/− | Laboratory of Jonathan Kaye | N/A |
| Mouse: OT-1 | Charles River | Stock#: 003831 |
| Mouse: P14Tcf7−/− | Laboratory of Daniel Pinschewer | N/A |
| Oligonucleotides | ||
| Id2 PrimePCR SYBR Green Assay | Bio-Rad | Cat# qMmuCED0044963 |
| Mm_Gapdh_3_SG QuantiTect Primer Assay | QIAGEN | Cat# QT01658692 |
| Il12p35: forward primer 5′-CATCAACGCAGCACTTCAGAA-3′; reverse primer 5′-TCGATGGCCACCAGCAT-3′ | Microsynth | N/A |
| Ifna: forward primer 5′-CGGAATTCTCTCCTGCCTGAAGGAC-3′; reverse primer 5′-AAGGGTACCACACAGTGATCCTGTGGAA-3′ | Microsynth | N/A |
| Il10: forward primer 5′-GGTTGCCAAGCCTTATCGGA-3′; reverse primer 5′-ACCTGCTCCACTGCCTTGCT-3′ | Microsynth | N/A |
| Tox ChIP PCR (−0.5 kb upstream of start codon): forward primer 5′-GGGGTACTGGGCGTTTTATT-3′; reverse primer 5′-GATCTTGATCTCCCCTGCAA-3′ | Microsynth | N/A |
| Tox ChIP PCR (-1.1 kb upstream of start codon): forward primer 5′-AGGGACCCCAGTTGATCTCT-3′; reverse primer 5′-AGCACCCCTGGAGTCTTTTT-3′ | Microsynth | N/A |
| Tox ChIP PCR (-1.6 kb upstream of start codon): forward primer 5′-GGGTGGGCAATGTTTACCTT-3′; reverse primer 5′-TGACGCCAGAAACAGATACCT-3′ | Microsynth | N/A |
| ShRNA targeting sequence for Eomes: 5′ -TCAGGAGGAACTAATCTCTTCT-3′ | This paper | N/A |
| ShRNA targeting sequence for Cd4: 5′-AAACGATCCTTTCTCCCATGCC-3′ | Laboratory of Matthew Pipkin | Chen et al., 2014 |
| ShRNA targeting sequence for Tox: 5′-TATAGACCTGTTTCTGTTCTTC-3′ | This paper | N/A |
| Recombinant DNA | ||
| pMSCV IRES-GFP | Addgene | N/A |
| pMSCV Eomes-IRES-GFP | Laboratory of Gabrielle Belz | Mackay et al., 2015 |
| pMSCV T-bet-IRES-GFP | Laboratory of Gabrielle Belz | Joshi et al., 2007 |
| pGL3-Enhancer | Promega | N/A |
| pGL3-Enhancer-Tox-promoter | This paper | N/A |
| pGL4.74 (encoding Renilla luciferase) | Promega | N/A |
| pMSCV miR30 backbone | Laboratory of Matthew Pipkin | Chen et al., 2014 |
| Software and Algorithms | ||
| Imaris 5.7.2 ×64 | Bitplane | N/A |
| OsiriX 8 | OsiriX | N/A |
| Definiens Developer 2.5 | Definiens | N/A |
| FlowJo 10.2 | TreeStar | N/A |
| R language and environment for statistical computing and graphics | https://www.r-project.org | N/A |
| STAR | https://github.com/alexdobin/STAR | N/A |
| SAMtools | http://samtools.sourceforge.net/ | N/A |
| HTSeq | http://www-huber.embl.de/users/anders/HTSeq/ | N/A |
| Bioconductor and packages | https://www.bioconductor.org | N/A |
| Bowtie | http://bowtie-bio.sourceforge.net/index.shtml | N/A |
| MACS2 | https://pypi.python.org/pypi/MACS2 | N/A |
| geNorm | https://genorm.cmgg.be | N/A |
| Pannoramic Viewer software | 3DHistech | N/A |
| Other | ||
| HiSeq 2000 | Illumina | N/A |
| HiSeq 2500 | Illumina | N/A |
| AutoMACS | Miltenyi Biotec | N/A |
| SpectraMax L reader | Molecular devices | N/A |
| Pannoramic Digital Slide Scanner 250 FLASH II | 3DHistech | N/A |
| Gallios cytometer | Beckman-Coulter | N/A |
| BD FACSAria II Cell Sorter | BD Biosciences | N/A |
| CFX 96 instrument | Bio-Rad | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Doron Merkler (doron.merkler@unige.ch).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT C57BL/6J were obtained from Charles River (France). Tox−/−(Aliahmad and Kaye, 2008) were kindly provided by J. Kaye and crossed with OT-1 TCR transgenic and P14 TCR transgenic mice with a different CD45 allele to perform adoptive transfer experiments. ODC-OVA mice were kindly provided by T. Hünig and bred in our animal facility. Tcf7−/−(Verbeek et al., 1995) mice were crossed to P14 TCR transgenic mice and bred in house. Tcf7+/+ P14 mice were used as control donor mice. Stop-GPflox mice were generated by inserting a cDNA cassette downstream of a CAG promoter encoding for the LCMV Glycoprotein (GP) of LCMV, together with an IRES-YFP into the ROSA26 locus, and preceded by a loxP-flanked stop sequence (see also Figure S1C). MOGiCre-GP (MOGiCre/+:Stop-GPflox/+) mouse line was generated by crossing mice expressing the Cre-recombinase under the control of the oligodendrocyte-specific promoter (MOGiCre; Hövelmeyer et al., 2005) with Stop-GP mice. All mice were lodged under specific-pathogen-free P2 conditions in the animal facilities of the University Medical Center of Geneva and Basel. Male and female sex and age-matched mice between six weeks and twelve weeks of age were used for experiments. All animal experiments were authorized by the cantonal veterinary office of Geneva and performed in agreement with the Swiss law for animal protection.
Human MS and stroke lesions
For histopathological analysis, brain sections derived from biopsies of MS lesions (n = 4 lesions from 4 different female and male cases [46.6 ± 20.8 years, mean ± SD]). All lesions classified as early active lesions as described previously (Lucchinetti et al., 2000) or stroke lesions (n = 5 lesions from 4 different cases, 63.8 ± 7.6 years, mean ± SD) were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin according to standardized protocols. Their use for scientific purposes was in accordance with institutional ethical guidelines and approved by the ethics committee of the University of Göttingen (Germany). Informed consent was obtained from all subjects.
METHODS DETAILS
Pathogen infection
Recombinant Listeria monocytogenes expressing the LCMV GP33-41 peptide KAVYNFATC (Lm-GP33) or the chicken ovalbumin (OVA) containing the native ligand SIINFEKL257–264 (Lm-OVA) were previously described (Oberle et al., 2016; Zehn et al., 2009). The following virus variants were used: Lymphocytic Choriomeningitis Virus wild-type (LCMV, Armstrong strain) or following replication competent LCMV-variants encoding for i) the signal sequence of LCMV glycoprotein harboring the GP33-41 epitope (LCMV-GP33) instead of the full-length LCMV GP ii) the full-length OVA as a transgene (LCMV-OVA) (Kallert et al., 2017) or iii) the low OVA affinity variant Q4H7 (LCMV-Q4H7) as a transgene. Viruses were produced, titrated, and administered to mice as previously described (Flatz et al., 2006). For peripheral challenge, mice were i.v. injected into the tail vein with 5,000 colony-forming units (CFU) of log-phase Listeria or i.v. or i.p. injected with 105 plaque-forming units (PFU) of LCMV. Infected animals were monitored daily for occurrence of classical experimental autoimmune encephalomyelitis (EAE) symptoms and scored as follows. 1, flaccid tail; 2, impaired righting reflex and hind limb weakness; 3, complete hind limb paralysis; 4, complete hind limb paralysis with partial fore limb paralysis; 5, moribund. Severely diseased animals were immediately sacrificed. For depletion of CD4+ T cells, mice were treated i.p. twice over a 3-day interval with 250 mg anti-CD4 antibody (YTS191). Depletion of CD4+ T cells was confirmed by flow cytometry showing a CD4+ T cell frequency < 0.1% during LCMV-OVA challenge. Anti-CD48-neutralizing antibody (HM48-1, BioxCell) or anti-PD-1-neutralizing antibody (29F.1A12) or their respective isotype control were administered i.p. (300 μg) 4 and 6 days after LCMV-OVA infection. For blockade of IL-10, mAb to IL-10 (JES5-2A5; BioxCell) was administered i.p. (500 μg) 0, 2, 4 and 6 days after LCMV-OVA infection. For blockade of IFN-α mAb to IFN-α (TIF-3C5; Leinco Technologies) was administered i.p. (1mg) 0, 2 and 4 days after LCMV-OVA infection. Where specified in the text, mice were in addition injected daily i.p. with 100ul of vehicle, or vehicle containing 200ng rIL-12 for 5 consecutive days starting at the same day of LCMV-OVA infection.
Purification of naive CD8+ T cells and in vitro stimulation
Naive OT-1 or P14 cells, respectively, from spleen were purified using naive CD8a+ T Cell Isolation Kit (Miltenyi Biotec) and subsequently separated by AutoMACS (Miltenyi Biotec). Purity of the cells (95%) was confirmed by flow cytometry. For in vitro stimulation, 2×105 naive CD8+ T cells were resuspended in D-MEM supplemented with 10% FCS (fetal calf serum), 2 mM GlutaMAX (GIBCO), 10 mM HEPES (GIBCO), 100 U/mL Penicillin/Streptomycin (GIBCO), and 50 μM 2-Mercaptoethanol and stimulated with anti-CD3 (clone 145.2C11, Biolegend), anti-CD28 (clone 37.51, Biolegend) (1 mg/ml) and recombinant murine IL-12 (Peprotech) in 96-well plates pre-coated with 100 μg/mL goat anti-hamster IgG (H+L) secondary antibody (Jackson Immunoresearch).
FACS analysis and sorting
For staining, the following antibodies were used: anti-CD3 (17A2, Biolegend), anti-CD4 (RM4-5, Biolegend), anti-CD8a (53-6.7, Biolegend), anti-CD11b (M1/70, Biolegend), anti-CD11c (N418, Biolegend), anti-CD44 (IM7, Biolegend), anti-CD45 (30-F11, Biolegend), anti-CD45.1 (A20, Biolegend), anti-CD45.2 (104, Biolegend), anti-CD45R/B220 (RA3-6B2, Biolegend), anti-CD49b (DX5, Biolegend), anti-CD107a (1D4B, Biolegend), anti-CD127 (A7R34, Biolegend), anti-CD244 (m2B4 (B6)458.1, Biolegend), anti-Eomes (Dan11mag, Ebioscience), anti-granzyme B (GB11, Biolegend), anti-IFN-γ (XMG1.2, Biolegend), anti-IL-2 (JES6-5H4, Biolegend), anti-IRF4 (IRF4.3E4, Biolegend), anti-Ki-67 (SolA15, Ebioscience), anti-KLRG1 (2F1/KLRG1, Biolegend), anti-LAG-3 (11C3C65, Biolegend), anti-NKG2D (C7, Biolegend), anti-NKP46 (29A1.4, Biolegend), anti-PD-1 (RMP1-30, Biolegend), anti-T-bet (4B10, Biolegend), anti-TCF-1 (C63D9, Cell Signaling Technology), anti-TIGIT (1G9, Biolegend), anti-TIM-3 (B8.2C12, Biolegend), anti-TNF-α (MP6-XT22, Biolegend), anti-TOX (TXRX10, Ebioscience). Peripheral blood samples and splenocytes were collected in FACS-Buffer (10% FCS, 10 mM EDTA, 0.01% NaN3 in PBS). For the preparation of CNS-infiltrating leukocytes, brains were minced, digested with CollagenaseA/DNaseI (Roche) and homogenized using 70 μm cell strainers (BD Biosciences). Leukocytes were separated using a discontinuous percoll gradient (30/70%). Surface staining was carried out with directly labeled antibodies in FACS buffer. Peripheral blood erythrocytes were lysed using BD FACS lysing solution (BD Biosciences). Isolated cells were quantified using AccuCheck Counting Beads (Invitrogen). Intracellular staining of TCF-1, Eomes, IRF4, Ki67, T-bet, and TOX was carried out using FoxP3/Transcription Factor Staining Buffer Set (eBioscience) according to manufacturer’s instructions. For ex vivo staining of Granzyme B, cells were fixed and permeabilized using commercial permabilization buffer set (Biolegend). To assess degranulation and intracellular cytokine production, brain leukocytes and splenocytes were cultured for 5h in the presence of 5 μg/ml FITC labeled anti-CD107a antibody and monensin. Cells were stimulated in vitro with SIINFEKL peptide or left unstimulated. Cells were fixed and permeabilized using commercial fixation/permabilization buffer set (Biolegend) followed by intracellular staining for cytokines. Flow cytometric samples were acquired on a Gallios cytometer (Beckman-Coulter) equipped with three lasers (blue: 488 nm, red: 633 nm and violet: 405 nm) using appropriate filter sets and compensation controls. Gates were assigned according to appropriate control populations. In experiments which required high purity, OT-1 cells were sorted by FACS into 4°C sorting buffer using Aria II flow cytometer (BD Biosciences).
Immunohistochemistry
For immunohistochemical bright field staining of mouse tissue, brains and spinal cords were prepared and fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Antigen retrieval was performed according to standardized protocols by heating with citrate buffer (pH6). Endogenous peroxidases (peroxidase blocking reagent, Dako) were neutralized and unspecific binding blocked for 5 min (PBS/1% BSA/2% FCS). Tissue sections were incubated with rat anti-CD3 (CD3-12, Biorad). Bound primary antibodies were visualized with biotin-labeled anti-rat antibody and streptavidin-peroxidase staining method using polymerized 3,3′-diaminobenzidine (all reagents from Dako; haemalaun counterstaining of nuclei). For immunofluorescence staining of mouse tissue, PFA-fixed paraffin sections were blocked for 5 min (PBS/1% BSA/2% FCS). Sections were incubated with mouse anti-Nogo A (11C7) (Oertle et al., 2003), rabbit anti-cleaved caspase 3 (Cell Signaling Technology) or rabbit anti-GFP (D5.1, Cell Signaling Technology). Bound antibodies were visualized by using goat-anti-rabbit-HRP and tyramide signal amplification (TSA) (Alexa488) and Alexa555-labeled-goat anti-mouse antibody (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen). Immunostained sections were scanned using Pannoramic Digital Slide Scanner 250 FLASH II (3DHistech) in 200× magnification. Oligodendrocyte quantifications were performed manually using Pannoramic Viewer software (3DHistech) and apoptotic oligodendrocytes were quantified automatically by using Tissue Studio software (Definiens ®). Total number of oligodendrocytes and Capsase3-positive oligodendrocytes was calculated and expressed as cells/mm2. For representative images, white balance was adjusted and contrast was linearly enhanced using the tools “levels,” “curves,” “brightness” and “contrast” in Photoshop CS6 (Adobe).
PFA-fixed human brain sections were deparaffinised and antigen retrieval was performed according to standardized protocols by heating with citrate buffer (pH6). Endogenous peroxidases (peroxidase blocking reagent, Dako) were neutralized and unspecific binding blocked (PBS/10% FCS). To eliminate potential free binding sites on anti-mouse secondary antibodies, sections were blocked (PBS/1% normal mouse serum) followed by goat-anti-mouse Fab fragment (Jackson Immunoresearch) and incubated with mouse-anti-human CD8 (C8/144B, Dako) and rat-anti-TOX (TXRX10, Ebioscience). Bound mouse and rat antibodies were visualized with Alexa555-labeled goat-anti-mouse (Invitrogen) and Alexa647-labeled-donkey anti-rat antibody (Jackson Immunoresearch), respectively. Nuclei were stained with DAPI (Invitrogen). Slides were scanned at high resolution (0.325 um/px) using a slide scanner (Pannoramic Flash II 250, 3D-Histech). Nuclear TOX fluorescence within CD8 cells was quantified using a custom algorithm in Definiens Developer XD (Definiens ®). In short, all tissue was detected automatically and CD8+ T cells were identified based on their respective spectral characteristics. CD8+ cell nuclei were detected based on DAPI signal. The mean intensity of the TOX channel was exported for each CD8 cell nucleus. Based on a fixed threshold for TOX intensity, CD8+ cells were assigned into TOX+ and TOX−groups.
Retrovirus production and transduction of CD8+ T cells
For T-bet and Eomes overexpression murine stem cell virus (MSCV)–T-bet–internal ribosome entry site pMIG-T-bet has been previously described (Joshi et al., 2007), and pMIG-Eomes and pMIG-empty (provided by Professor Gabrielle Belz (WEHI) and described previously (Mackay et al., 2015)). Eomes-specific shRNA and Cd4-shRNA (provided by Professor Matthew Pipkin (Scripps Research Institute)) were cloned into the retroviral vector containing the miR30 backbone (Chen et al., 2014). The following 22-nt shRNA ‘guide sequences’ were used for cloning: 5′-TCAGGAGGAACTAATCTCTTCT 3′−(Eomes); 5′-AAACGATCCTTTCTCCCATGCC 3′−(Cd4). Stable silencing of Tox was achieved by cloning the following 22-nt shRNA ‘guide sequence’ in the optimized miR-E backbone: 5′-TATAGACCTGTTTCTGTTCTTC 3′−(Tox) (Fellmann et al., 2013). In vitro activated CD8+ T cells were transduced by spin-infection for 90min at 800 g with retroviral supernatant 18h following activation in the presence of 8 μg/mL polybrene (Sigma-Aldrich). Transduction efficacy was determined by GFP (pMIG vector) or mAmetrine (miR30 and miR-E vectors) expression. For adoptive transfer experiment, retrovirally transduced cells were maintained for a further week in IL-15-containing medium (10ng/ml) and transduced cells (CD45.1+) were transferred into ODC-OVA recipients by i.v. injection.
Time-lapse imaging of CTLs
Hippocampal organotypic slice cultures from postnatal 4-5 day ODC-OVA or WT mice were prepared with a 400 μm thickness, as previously described (Stoppini et al., 1991). Slices were cultured on membrane confetti (Millipore) placed on an insert (Millipore) and maintained in a CO2 incubator at 37°C until day in vitro 4 (DIV4) and then at 33°C. Hippocampal organotypic slices were infected at DIV7 with an adeno-associated viral vector AAV-MBP-eGFP (von Jonquieres et al., 2013). Seven days after LCMV-OVA infection, CNS-infiltrating OT-1 cells were sorted and labeled with CellTracker Red CMPTX (Invitrogen) or Green CFSE (Biolegend). 2 × 105 Tox+/+ and Tox−/−OT-1 lymphocytes were incubated 2 h before imaging onto hippocampal organotypic slice cultures on DIV14. In some experiments, CNS-infiltrating Tox−/−OT-1 isolated from anti-CD48-treated ODC-OVA mice were used. XYT dual channel sequential imaging was performed in culture medium at 32°C for 1 h at 1 frame per 30 s under a 20× objective with a Fluoview 300 confocal system (Olympus, Japan) using a 488 nm solid state laser for AAV-MBP-eGFP and CFSE labeled Tox+/+ OT-1 lymphocytes and a 543 nm laser for CMPTX labeled Tox−/−OT-1 lymphocytes. Each xy plane spanned 800 μm 3 600 μm at a resolution of 0.8 μm per pixel. Cell tracking data was generated with Imaris 5.7.2 ×64 software (Bitplane). A custom R script was used to superimpose individual T cell tracks. Motility coefficient was calculated from slope of displacement against the square root of time (t1/2) (Miller et al., 2002). OT-1 cell contact to oligodendrocyte was analyzed with OsiriX software using home-made plugins. A contact was considered effective when a lymphocyte was co-localized in xyz with an oligodendrocyte and moved less than 1 μm for at least 2 consecutive frames.
In vitro killing assay
CytoTox 96 nonradioactive cytotoxicity assay (Promega) was used to measure the cytotoxic activities of OT-1 cells, according to the manufacturer’s instructions. Briefly, CNS-infiltrating Tox+/+ or Tox−/−OT-1 cells (2 × 106) were FACS-sorted from LCMV-OVA-infected ODC-OVA mice 7 days after infection. OT-1 cells (from 10:1 to 0.6:1 effector to target ratios) were incubated in presence 104 SIINFEKL-pulsed EL-4 cells per well on 96-well U-bottomed plates. Cells were then incubated for 4 h in a humidified 5% CO2 chamber at 37°C, and centrifuged at 500 × g for 5 min. Aliquots (50 μL) were then transferred from all wells to fresh 96-well flat-bottomed plates, and equal volumes of reconstituted substrate mix were added to each well. Plates were then incubated in the dark at room temperature for 45 min. Stop solution (50 μL) was then added, and absorbance values were measured at 492 nm. Cell death percentages at each effector-to-target cell ratio were calculated using [A (experimental) - A (effector spontaneous) - A (target spontaneous)] 3 100/[A (target maximum) - A (target spontaneous)].
Quantitative RT-PCR
Total RNA was extracted from spleen, brain tissue or FACS-sorted CNS-infiltrating OT-1 cells using RNeasy Mini Kit (QIAGEN), cDNA was synthesized using iScript cDNA synthesis kit (Biorad) and relative expression of Id2 was determined with gene-specific primer pair (PrimePCR SYBR® Green Assay, BioRad, qMmuCED0044963). The oligonucleotide sequences used for murine IL-12p35 were: sense primer, 5′-CATCAACGCAGCACTTCAGAA-3′ and antisense primer, 5′-TCGATGGCCACCAGCAT-3′, for murine IFN-α were: sense primer, 5′-CGGAATTCTCTCCTGCCTGAAGGAC-3′ and antisense primer, 5′-AAGGGTACCACACAGTGATCCTGTGGAA-3′, for murine IL-10 were sense primer, 5′-GGTTGCCAAGCCTTATCGGA-3′ and antisense primer, 5′-ACCTGCTCCACTGCCTT GCT-3′. qPCR was performed on a CFX 96 instrument (BioRad) and gene expression levels were normalized to Gapdh as housekeeping gene (QIAGEN) using iQ SYBR® Green Supermix (Biorad).
NanoString nCounter Expression analysis
100 ng of RNA isolated from spinal cord of ODC-OVA were hybridized with multiplexed Nanostring probes for expression profiling of 248 inflammation-related genes (nCounter GX Mouse Inflammation Kit V2). Samples were processed according to published procedure (Geiss et al., 2008) (Barcodes were counted for – 1150 fields of view per sample. Background correction was done by substracting the mean + 2 standard deviations of the negative controls for each sample. Values < 1 were fixed to 1. Positive controls were used as quality assessment: we checked that the ratio between the highest and the lowest positive control averages among samples was below 3. Then counts for target genes were normalized with the geometric mean of 4 reference genes (Hprt, Gapdh, Pgk1 and Cltc) selected as the most stable using the geNorm algorithm (Vandesompele et al., 2002). Data are depicted as scatterplot of the normalized counts of individual genes from different experimental conditions.
RNA-seq analysis
For sequencing, the RNA-samples were prepared with the “TruSeq RNA Sample Prep Kit v2” according to the manufacturer’s protocol (Illumina). Single read (50 bp) sequencing was conducted using a HiSeq 2000 (Illumina). Three independent biological replicates were analyzed for each condition. Sequencing quality was checked and approved via the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence images were transformed to BCL files with the Illumina BaseCaller software and samples were demultiplexed to FASTQ files with CASAVA (version 1.8.2). Sequences were aligned to the genome reference sequence of Mus musculus (UCSC genome assembly version mm10). Alignment was performed using the STAR alignment software (version 2.3.0e) allowing 2 mismatches within 50 bases. Subsequently, conversion of resulting SAM files to sorted BAM files, filtering of unique hits and counting was conducted with SAMtools (version 0.1.18) and HTSeq (version 0.6.1p1). Data was preprocessed and analyzed in the R/Bioconductor environment (www.bioconductor.org) using the DESeq2 package (version 1.8). Specifically, the data were normalized and tested for differentially expressed genes based on a generalized linear model likelihood ratio test assuming negative binomial data distribution. Candidate genes were filtered to a minimum of 2 (LCMV-OVA versus Lm-OVA) or 1.5-fold change (Tox+/+ v.s. Tox−/−) and FDR-corrected p < 0.05. Gene annotation was performed using Mus musculus entries from Ensembl (www.ensembl.org) via the biomaRt package (version 2.18.0).
For gene set enrichment analysis (GSEA), a ranked list of the change (fold values) in RNA-sequencing read values between Tox+/+ and Tox−/−OT-1 cells isolated from CNS of ODC-OVA mice was generated. The java GSEA Desktop Application v2.2.1 was used to run the analysis. Two Id2-dependent gene-sets (up and downregulated) were generated by filtering the up-and downregulated genes (FC > 1.5, p < 0.05) separately in Id2+/+ DbNP366 CTLs versus to Id2−/−DbNP366 CTLs (GSE44140) (Masson et al., 2013). Notch1-2-dependent gene-set was generated by filtering downregulated genes (FC > 2, p < 0.05) separately in Notch1-2+/+ DbNP366 CTLs versus to Notch1-2−/−DbNP366 CTLs (E-MTAB-2999) (Backer et al., 2014). Normalized Enrichment Scores were calculated using the function GseaPreranked.
ChIP-seq and ChIP-PCR analysis
Chromatin was purified from 20 3 106 CNS-infiltrating Tox+/+ or Tox−/−OT-1 cells. ChIP was performed using a rabbit polyclonal anti-TOX1 antibody (Abcam, ab155768). Immunoprecipitated DNA was prepared for sequencing using the Illumina ChIP-seq sample preparation protocol. In brief, 5 ng of ChIP-enriched DNA was end repaired and an adenosine overhang added to the 3′ends. Paired-end Illumina adapters were ligated to the DNA before enrichment using 18 cycles of amplification with Illumina PCR primers. Libraries were validated on the Bioanalyzer 2100 (Agilent) and Qubit fluorimeter (Invitrogen) and sequenced using single-end (100nt-long) on Illumina HiSeq2500. FastQ reads were mapped to the ENSEMBL reference genome (GRCm38.80) using bowtie version 0.12.7 with standard settings, except that any reads mapping to more than one location in the genome (ambiguous reads) were discarded (m = 1). Duplicated reads were removed using Samtools version 0.1.18. Peak calling was done with MACS2 version 2.0.10.20130520 with standard parameters using the input as control. 7579 peaks were reported. The coverage per base for each peak (in Reads per Million, RPM) was then calculated in both, Tox+/+ and Tox−/−CTLs. Peaks having a maximal value of Tox+/+/Tox−/−coverage-ratio higher than 10 were kept for further analysis (n = 5121). Peaks overlapping a promoter (5000 bp upstream and 500 downstream of the annotated TSS) by at least 1bp were considered as localized in promoter regions (n = 3806). The peaks which were overlapping exonic regions were assigned to exons (n = 547) and peaks overlapping intronic regions were assigned to introns (n = 392). Remaining peaks were assigned to intergenic regions (n = 376). Peaks localized in promoter regions were then crossed with RNA-seq results. Only peaks in promoter regions of differentially expressed genes with an adjusted p value lower than 0.01 were kept for downstream analysis. Gene ontology (GO) analysis on selected gene sets was performed using PANTHER (protein analysis through evolutionary relationship) Classification System version 11.1, 2016 (http://pantherdb.org). The PANTHER Statistical overrepresentation test with GO biological process complete annotation data was used; GO terms with p < 0.05 were considered significant.
ChIP followed by PCR (ChIP-PCR) analysis was performed on splenic OT-1 cells isolated from the spleen 6 days after LCMV-OVA infection to evaluate T-bet binding in the promoter region of Tox. Mouse monoclonal antibody to T-bet (4B10; Santa Cruz Biotechnology) or its respective isotype control was used for ChIP. Primers for amplification of the region upstream of Tox were as follows: – 0.5kb, 5′- GGGGTACTGGGCGTTTTATT −3′ (forward) and 5′-GATCTTGATCTCCCCTGCAA –3′ (reverse); −1.1kb, 5′-AGGGACCCCAGTTGATCTCT −3′ (forward) and 5′-AGCACCCCTGGAGTCTTTTT −3′ (reverse); −1.6kb 5′ GGGTGGGCAATGTTTACCTT −3′ (forward) and 5′-TGACGCCAGAAACAGATACCT −3′ (reverse). ChIP primers for Ifng have have been described previously (Dominguez et al., 2015).
Tox promoter activity by luciferase assay
A 2kb fragment (upstream of the start codon) corresponding to the Tox promoter was cloned into the pGL3-Enhancer vector to drive firefly luciferase expression (Tox reporter) (Promega). EL4 cells (lacking endogenous T-bet and Eomes) were transfected with the Nucleofection kit (Amaxa-Lonza) as described previously (Beima et al., 2006). EL4 cells were co-transfected with the Tox-reporter and an empty (pMIG) or T-bet (pMIG-T-bet) or Eomes-encoding vector (pMIG-Eomes), as described above. Transfected cells were sorted based on GFP expression and luciferase activities were measured using the Dual-Glo Luciferase Assay kit (Promega). Firefly luciferase activities in each sample were first normalized against Renilla luciferase activities in the same sample and then normalized against that in cells transfected with the empty vector (pGL3-Enhancer).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, the dispersion, the precision of measures (mean ± s.e.m or mean ± s.d.) and the statistical significance are reported in the figures and figure legends. In figures, asterisks denote statistical significance as calculated by Student’s t test, two-way ANOVA with Bonferroni’s or Sidak’s post-test, Kruskal–Wallis ANOVA and one-way anova with Tukey’s post-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Statistical analysis was performed in GraphPad Prism 7.
DATA AND SOFTWARE AVAILABILITY
Software
All software is freely or commercially available and is listed in the STAR Methods.
Data Resources
The raw and processed RNA-seq data have been deposited to the Gene Expression Omnibus (GEO) under accession numbers GEO: GSE93804 and GSE93805. The ChIP-seq data have been deposited under accession number GEO: GSE93953.
Supplementary Material
Highlights.
LCMV-primed but not Listeria-primed CD8+ T cells exert encephalitogenic activity
LCMV infection induces the DNA-binding factor TOX in CNS-infiltrating CD8+ T cells
Loss of TOX abrogates the tissue-destructive ability of CD8+ T cells
TOX restrains terminal differentiation of effector CD8+ T cells
Acknowledgments
D.M., D.D.P., and M.S. are supported by the Klaus-Tschira Foundation. D.M. and D.D.P. are supported by the Swiss National Science Foundation (310030_173010 to D.M. and CRSII3_160772 to D.P. and D.M.). D.M., N.P., and K.S. are supported by the Swiss MS Society. D.M. is supported by the Helmut Horten Foundation, Gebert-Rüf Foundation (GRS-049/13), and Fondation Ernst et Lucie Schmidheiny. R.M. is supported by the Swiss National Science Foundation (PP00P3_150712). The plasmids for pMIG-T-bet and pMIG-Eomes were kindly provided by Gabrielle Belz (WEHI, Australia). ODC-OVA mice were kindly provided by T. Hünig (University of Würzburg).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, five tables, and two videos and can be found with this article online at https://doi.org/10.1016/j.immuni. 2018.04.005.
AUTHOR CONTRIBUTIONS
N.P. performed and analyzed experiments and wrote the manuscript. B.K. and I. W. assisted in histological processing and analysis. M.D.R. performed time-lapse imaging. K.S., G.D.L., and I.V. assisted in flow-cytometry analysis. H.S., R.M., and S.L. assisted and performed ChIP-seq analysis. M.K. performed image-analysis and cell tracking. T.L. and G.S. performed RNA-seq analysis. W.B. assisted in analysis of human samples. M.S., D.Z., and D.D.P. conceived experiments and assisted in manuscript writing. D.M. conceived and designed the project and wrote the manuscript.
DECLARATION OF INTERESTS
M.K., D.M., D.D.P., and N.P. have a patent related to this work: PCT/EP2015/076458, “Tri-segmented arenaviruses as vaccine vectors.”
References
- Aliahmad P, Kaye J. Development of all CD4 T lineages requires nuclear factor TOX. J Exp Med. 2008;205:245–256. doi: 10.1084/jem.20071944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliahmad P, de la Torre B, Kaye J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat Immunol. 2010;11:945–952. doi: 10.1038/ni.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schröder R, Deckert M, Schmidt S, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer RA, Helbig C, Gentek R, Kent A, Laidlaw BJ, Dominguez CX, de Souza YS, van Trierum SE, van Beek R, Rimmelzwaan GF, et al. A central role for Notch in effector CD8(+) T cell differentiation. Nat Immunol. 2014;15:1143–1151. doi: 10.1038/ni.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, Weinmann AS. T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J Biol Chem. 2006;281:11992–12000. doi: 10.1074/jbc.M513613200. [DOI] [PubMed] [Google Scholar]
- Bos SD, Page CM, Andreassen BK, Elboudwarej E, Gustavsen MW, Briggs F, Quach H, Leikfoss IS, Bjølgerud A, Berge T, et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS ONE. 2015;10:e0117403. doi: 10.1371/journal.pone.0117403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Toben C, Na SY, Stark K, Nitschke L, Peterson A, Gold R, Schimpl A, Hünig T. Induction of experimental autoimmune encephalomyelitis in transgenic mice expressing ovalbumin in oligodendrocytes. Eur J Immunol. 2006;36:207–215. doi: 10.1002/eji.200535211. [DOI] [PubMed] [Google Scholar]
- Chen R, Bélanger S, Frederick MA, Li B, Johnston RJ, Xiao N, Liu YC, Sharma S, Peters B, Rao A, et al. In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity. 2014;41:325–338. doi: 10.1016/j.immuni.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–558. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, Kaech SM. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med. 2015;212:2041–2056. doi: 10.1084/jem.20150186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Flatz L, Bergthaler A, de la Torre JC, Pinschewer DD. Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proc Natl Acad Sci USA. 2006;103:4663–4668. doi: 10.1073/pnas.0600652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Fugger L. Pathogenic CD8(+) T cells in multiple sclerosis. Ann Neurol. 2009;66:132–141. doi: 10.1002/ana.21744. [DOI] [PubMed] [Google Scholar]
- Friese MA, Jakobsen KB, Friis L, Etzensperger R, Craner MJ, McMahon RM, Jensen LT, Huygelen V, Jones EY, Bell JI, Fugger L. Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med. 2008;14:1227–1235. doi: 10.1038/nm.1881. [DOI] [PubMed] [Google Scholar]
- Fukuoka N, Harada M, Nishida A, Ito Y, Shiota H, Kataoka T. Eomesodermin promotes interferon-γ expression and binds to multiple conserved noncoding sequences across the Ifng locus in mouse thymoma cell lines. Genes Cells. 2016;21:146–162. doi: 10.1111/gtc.12328. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Hövelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F, von Hoch L, Kramer D, Minichiello L, Kollias G, et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–5884. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. 2008;180:1309–1315. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- Kallert SM, Darbre S, Bonilla WV, Kreutzfeldt M, Page N, Müller P, Kreuzaler M, Lu M, Favre S, Kreppel F, et al. Replicating viral vector platform exploits alarmin signals for potent CD8+ T cell-mediated tumour immunotherapy. Nat Commun. 2017;8:15327. doi: 10.1038/ncomms15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46:D260–D266. doi: 10.1093/nar/gkx1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JR, Mathew SO, Patel RK, Pertusi RM, Mathew PA. Altered expression of signalling lymphocyte activation molecule (SLAM) family receptors CS1 (CD319) and 2B4 (CD244) in patients with systemic lupus erythematosus. Clin Exp Immunol. 2010;160:348–358. doi: 10.1111/j.1365-2249.2010.04116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13:777–789. doi: 10.1038/nri3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR. T-box transcription factors combine with the cytokines TGF-b and IL-15 to control tissue-resident memory T cell fate. Immunity. 2015;43:1101–1111. doi: 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Makgoba MW, Sanders ME, Shaw S. The CD2-LFA-3 and LFA-1-ICAM pathways: relevance to T-cell recognition. Immunol Today. 1989;10:417–422. doi: 10.1016/0167-5699(89)90039-X. [DOI] [PubMed] [Google Scholar]
- Masson F, Minnich M, Olshansky M, Bilic I, Mount AM, Kallies A, Speed TP, Busslinger M, Nutt SL, Belz GT. Id2-mediated inhibition of E2A represses memory CD8+ T cell differentiation. J Immunol. 2013;190:4585–4594. doi: 10.4049/jimmunol.1300099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296:1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- Na SY, Hermann A, Sanchez-Ruiz M, Storch A, Deckert M, Hünig T. Oligodendrocytes enforce immune tolerance of the uninfected brain by purging the peripheral repertoire of autoreactive CD8+ T cells. Immunity. 2012;37:134–146. doi: 10.1016/j.immuni.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham QM, Zickovich JM, Lefrançois L. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J Immunol. 2011;187:4967–4978. doi: 10.4049/jimmunol.1102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle SG, Hanna-El-Daher L, Chennupati V, Enouz S, Scherer S, Prlic M, Zehn D. A minimum epitope overlap between infections strongly narrows the emerging T cell repertoire. Cell Rep. 2016;17:627–635. doi: 10.1016/j.celrep.2016.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brösamle C, et al. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci. 2003;23:5393–5406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Salazar-Mather TP, Opal SM, Spencer RL, Miller AH, McEwen BS, Biron CA. Mechanism of interleukin 12-mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995;181:901–914. doi: 10.1084/jem.181.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, Kaech SM. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sad S, Marcotte R, Mosmann TR. Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity. 1995;2:271–279. doi: 10.1016/1074-7613(95)90051-9. [DOI] [PubMed] [Google Scholar]
- Sandusky MM, Messmer B, Watzl C. Regulation of 2B4 (CD244)-mediated NK cell activation by ligand-induced receptor modulation. Eur J Immunol. 2006;36:3268–3276. doi: 10.1002/eji.200636146. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, Pereira RM. Dynamic changes in chromatin accessibility occur in CD8+ T cells responding to viral infection. Immunity. 2016;45:1327–1340. doi: 10.1016/j.immuni.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehus CR, Aliahmad P, de la Torre B, Iliev ID, Spurka L, Funari VA, Kaye J. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat Immunol. 2015;16:599–608. doi: 10.1038/ni.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulina C, Schmidt S, Dornmair K, Babbe H, Roers A, Rajewsky K, Wekerle H, Hohlfeld R, Goebels N. Multiple sclerosis: brain-infiltrating CD8+ T cells persist as clonal expansions in the cerebrospinal fluid and blood. Proc Natl Acad Sci USA. 2004;101:2428–2433. doi: 10.1073/pnas.0308689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienne C, Michieletto MF, Benamar M, Carrié N, Bernard I, Nguyen XH, Lippi Y, Duguet F, Liblau RS, Hedrick SM, et al. Foxo3 transcription factor drives pathogenic T helper 1 differentiation by inducing the expression of eomes. Immunity. 2016;45:774–787. doi: 10.1016/j.immuni.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008;29:679–690. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol. 2006;177:1746–1754. doi: 10.4049/jimmunol.177.3.1746. [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, Oosterwegel M, Wilson A, MacDonald HR, Clevers H. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. 1995;374:70–74. doi: 10.1038/374070a0. [DOI] [PubMed] [Google Scholar]
- von Jonquieres G, Mersmann N, Klugmann CB, Harasta AE, Lutz B, Teahan O, Housley GD, Fröhlich D, Krämer-Albers EM, Klugmann M. Glial promoter selectivity following AAV-delivery to the immature brain. PLoS ONE. 2013;8:e65646. doi: 10.1371/journal.pone.0065646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner SN, Kumar V. Evolving role of 2B4/CD244 in T and NK cell responses during virus infection. Front Immunol. 2012;3:377. doi: 10.3389/fimmu.2012.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. 2009;458:211–214. doi: 10.1038/nature07657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.