

A 57-year-old man with end stage renal disease due to adult polycystic kidney disease, who had undergone renal transplantation, was admitted to another hospital with rapidly progressing jaundice for two months.
Liver function tests showed a picture of obstructive jaundice. Total bilirubin was 7.92 mg/dL, direct bilirubin was 5.82 mg/dL, alkaline phosphates was1050 IU /L, Alanine transaminase (ALT) was 80 IU/L, and Aspartate transaminase (AST) was 72 IU /L Serological test for viral hepatitis were negative. An abdominal ultrasonography was non-conclusive apart from showing the distorted anatomy of large polycystic areas in both native kidneys and liver.
Computed tomography demonstrated some cysts in the liver with a significant amount of normal residual parenchyma (figure 1). The patient was referred to our unit for further assessment. At fist in our unit endoscopic ultrasonography was done by linear and radial echoendosope (figure 2) and multiple large cystic lesions were found in the liver, two of which were measured more than 7 cm. The common bile duct (CBD) was compressed by a cyst in the distal part and in the hilum. The CBD was narrowed in distal and was dilated in proximal part, with no stone, sludge, or any lesion. Endoscopic retrograde cholangiopancreatography (ERCP) was performed, which showed that the CBD contained no stones, and the proximal part of the CBD, and right and left hepatic ducts were dilated due to narrowing induced in cystic lesions in distal part of the CBD and bifurcation, respectively. During ERCP cholangioscopy was performed. Filling defect was not seen, but external compression was seen in some areas. Sphincterotomy and brushing cytology were performed, with negative result for cholangiocarcinoma. One 14-cm plastic stent was inserted into the biliary duct. Post-ERCP the patient steadily recovered and jaundice was gradually resolved. He referred to liver transplantation unit.
Fig. 1.
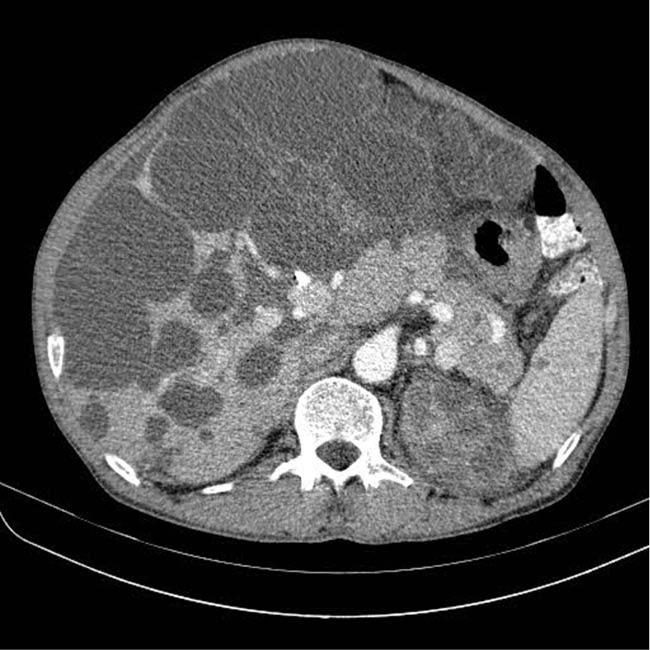
Computed tomogram of the abdomen shows massive polycystic liver disease with compression effect in the biliary tracts.
Fig. 2.
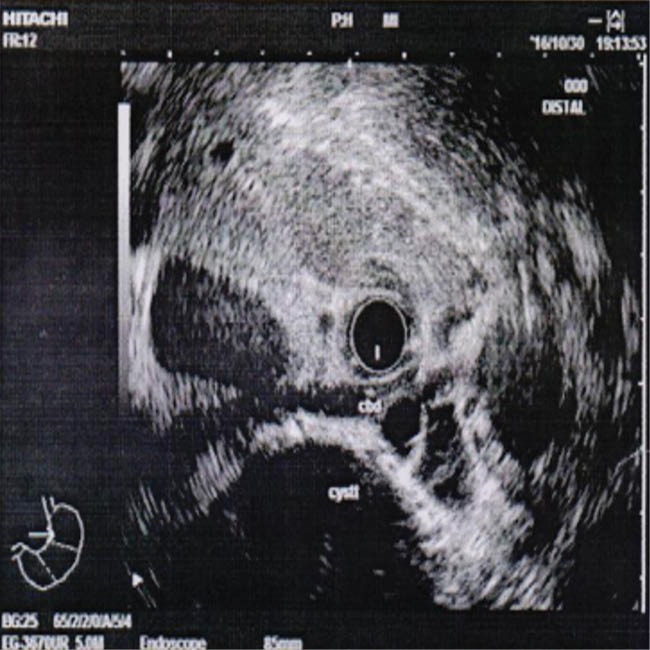
Endoscopic ultrasound shows liver cyst with compression effect in the common bile duct.
What is your diagnosis?
Autosomal dominant polycystic kidney disease (ADPKD) is a common disorder, occurring in approximately 1 in every 400 to 1000 live births.1 It is estimated that less than half of such cases will be diagnosed during the patients’ lifetimes, as the disease is often clinically silent.1 ADPKD is associated with cysts in the kidneys but patients may have a variety of other abnormalities, many of which are consistent with a generalized defect in epithelial cell differentiation and/or extracellular matrix function as a primary expression of the genetic abnormality in this disorder. In many cases asymptomatic cysts in the liver and pancreas can be helpful in confirming the diagnosis. The major extrarenal complications of ADPKD are cerebral aneurysms, hepatic and pancreatic cysts, cardiac valve disease, colonic diverticulitis, and abdominal wall and inguinal hernias.2
Liver involvement is the most frequent extrarenal manifestation in ADPKD.3 In ADPKD, liver cysts seem to develop later than renal cysts. And similar to the renal cysts, the prevalence of hepatic cysts increases with age. Earlier studies reported a prevalence of approximately 10-20% in patients aged below 30 years, and 50-70% in patients aged over 60 years.3 However, in a study of 230 patients with ADPKD aged between 15 and 46 years, magnetic resonance imaging showed hepatic cysts in 83%.4 Although the overall prevalence of a polycystic liver in patients with ADPKD is similar in men and women, women may develop cysts at an earlier age, and massive cysts occur almost exclusively in women, particularly those who have had several pregnancies.5 The cysts in Polycystic liver disease (PLD or PCLD) can also increase in size and number during pregnancy or simultaneously with the use of exogenous female steroid hormones.6 Most patients with liver cysts remain asymptomatic with preserved hepatic function. However, patients rarely develop pain,7 and other complication3 One of very rare complications is jaundice duo to bile duct compression, which has been reported in few cases.8-9 In the present report we described another patient with ADPKD who suffered from obstructive jaundice due to polycystic liver disease.
PLD is related to ADPKD and is linked either to PKD1 (the main locus responsible for the disease) or to PKD2.3 Davila and colleague,10 reported a second gene (SEC6) related to PLD. This gene encodes a component of the protein translocation machinery in the endoplasmatic reticulum. The prevalence of PLD in general population with an autopsy was 0.13%.11 And the incidence of PLD among patients with chronic renal failure due to polycystic kidneys exceeds 50% to 83%.4-12
PLD was regarded as a disorder of the development of small interlobular bile ducts. Embryological studies suggest an abnormality of remodeling of the ductal plate. Actually, liver cysts in patients with ADPKD arise from two different structures. Intrahepatic cysts originate from biliary microhamartomas. The latter represent overgrowth of bile ductules. While growing, they disconnect from the bile ducts from which they derive, although the epithelial lining retains the distinctive characteristics of biliary epithelia. These cysts are mostly located in the peripheral portions of the liver, exhibiting an intraparenchymatous location on contrast-enhanced computed tomography. Their diameter varies from less than 5 mm to more than 10 cm. It is surprising that peribiliary glands that surround large intrahepatic bile ducts also undergo cystic dilation in patients with polycystic disease.13
The risk of suffering from a complication related to PLD is ill-defined. No accurate assessment of its prevalence is available. Less than 5% of patients with PLD have acute medical complications.14 The complications depend on the number and amount of cysts. In single cyst related to cystic liver disease, complications include cyst infection, cyst hemorrhage (intracystic or hemoperitoneum), cyst rupture, cyst torsion, But in PLD, the complications are different and It is divided into two groups: 1. Related to cystic liver disease and 2. Unrelated to liver cysts. Complications related to cyst include abdominal mass, ascites, hepatic venous outflow obstruction, portal hypertension and variceal bleeding, inferior vena cava compression, jaundice duo to bile duct compression, Budd-Chiari syndrome, and progressive hepatic failure and unrelated complication to liver cysts include congenital hepatic fibrosis or biliary fibroadenomatosis, idiopathic dilation of the intra- or extrahepatic biliary tract (Caroli syndrome) and cholangiocarcinoma.3 Jaundice is rare and suggests bile duct compression in porta hepatis.3 This complication has been reported in few cases.7,8,15 Despite significant improvements in liver surgery and transplantation, choosing an appropriate treatment for liver cysts is still a challenge. Various treatment methods have been introduced, such as: percutaneous aspiration, stenting, cyst fenestration, liver resection, and liver transplantation.
Percutaneous aspiration with ethanol sclerotherapy is a safe, effective, and minimally invasive treatment for symptomatic congenital cysts. It is the initial treatment of choice for all patients with symptomatic congenital hepatic cysts.16 It is essential to note that aspiration combined with ethanol instillation to induce sclerosis of the cyst lining epithelium can be effective in patients with a few dominant cysts (type I PLD-few large cysts > 7 cm). However, long-term recurrence presents in 50% of cases.14 Sclerotherapy should be considered if a limited number (fewer than 5) of large cysts have to be treated.3 In selected patients, with compression in the portal or the caval circulation, percutaneous portal venous stenting and transhepatic portosystemic shunt (TIPS) or inferior vena cava stenting has also been used successfully.3
Cyst fenestration can be performed in patients with more diffuse PLD (type II PLD-multiple medium cysts 5-7 cm in diameter). Morbidity after laparoscopic fenestration ranges between 33% and 45%, with high recurrence rate. Patients with small cysts throughout the liver have a greater risk of persistence and recurrence of symptoms.14 Iwatsukiand colleague emphasized the expanding role of partial hepatectomy in the treatment of liver disease, as well as the need for considering, in some cases, the alternative mode of total hepatectomy and liver replacement.17 They reported that patients with benign disease who survived operation had minimal liability from recurrence of their original disease and none from the resection per surgery. Some reports suggest fenestration combined with segmental liver resection in patients with clearly defined cyst complexes.18-19 Candidates for combined resection/fenestration should have at least two adjacent liver segments not involved by cysts and with normal function.14
Liver transplantation is a drastic solution for the eradication of symptoms in massive PLD when the above-mentioned interventions were not an option.14 But it has been rarely used in the treatment of PLD, because patients typically present with preserved hepatocellular function.3 Isolated orthotopic liver transplantation should be considered in patients with ADPKD with severely disabling PLD when patients have diffuse cystic involvement and portal hypertension or have poor liver function or when liver resection has failed or cannot be used. Ideally, it should be performed before patients develop poor physical condition. Combined liver/kidney transplantation should be restricted to patients with advanced renal failure.3
Please cite this paper as:
Khoonsari MR, Zamani F, Asoubar M, Vafaeimanesh J. Obstructive Jaundice in a Patient with Polycystic Liver Disease. Middle East J Dig Dis 2018;10:117-120. doi: 10.15171/ mejdd.2018.101.
Footnotes
ETHICAL APPROVAL There is nothing to be declared.
CONFLICT OF INTEREST The authors declare no conflict of interest related to this work.
References
- 1.Davies F, Coles GA, Harper PS, Williams AJ, Evans C, Cochlin D. Polycystic kidney disease re-evaluated: a population-based study. Q J Med. 1991;79:477–85. doi: 10.1093/oxfordjournals.qjmed.a068568. [DOI] [PubMed] [Google Scholar]
- 2. M Bennett W, D Rose B, D Perrone R , M Sherian A. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Up to date 2016.6.
- 3.Chauveau D, Fakhouri F, Grünfeld JP. Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. J Am Soc Nephrol. 2000;11:1767–75. doi: 10.1681/ASN.V1191767. [DOI] [PubMed] [Google Scholar]
- 4.Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM. et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–9. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 5.Everson GT. Hepatic cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1993;22:520–5. doi: 10.1016/S0272-6386(12)80923-1. [DOI] [PubMed] [Google Scholar]
- 6.Everson GT, Taylor MR. Management of polycystic liver disease. Curr Gastroenterol Rep. 2005;7:19–25. doi: 10.1007/s11894-005-0061-6. [DOI] [PubMed] [Google Scholar]
- 7.Bajwa ZH, Sial KA, Malik AB, Steinman TI. Pain patterns in patients with polycystic kidney disease. Kidney Int. 2004;66:1561–9. doi: 10.1111/j.1523-1755.2004.00921.x. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld L, Abergel A, Bonny C, Poincloux L, Gayard P, Garcier JM. et al. [Complicated polycystic liver disease with intracystic hemorrhage and obstructive jaundice] Gastroenterol Clin Biol. 2001;25:818–22. [PubMed] [Google Scholar]
- 9.Dmitrewski J, Olliff S, Buckels JA. Obstructive Jaundice Associated with Polcystic Liver Disease. HPB Surg. 1996;10:117–20. doi: 10.1155/1996/83547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q. et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–7. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 11.Kwok MK, Lewin KJ. Massive hepatomegly in adult polycystic liver disease. Am J Surg Patho. 1998;12:321–4. doi: 10.1097/00000478-198804000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Thomsen HS, Thaysen JH. Frequency of hepatic cysts in adult Polycystic kidney disease. Acta Med Scand. 1998;224:381–4. doi: 10.1111/j.0954-6820.1988.tb19598.x. [DOI] [PubMed] [Google Scholar]
- 13.Kida T, Nakanuma Y, Terada T. Cystic dilatation of peribiliary glands in livers with adult polycystic disease and livers with solitary nonparasitic cysts: An autopsy study. Hepatology. 1992;16:334–40. doi: 10.1002/hep.1840160209. [DOI] [PubMed] [Google Scholar]
- 14.Delis SG, Bakoyiannis A, Triantopoulou C, Paraskeva K, Athanassiou K, Dervenis C. Obstructive Jaundice in Polycystic Liver Disease Related to Coexisting Cholangiocarcinoma. Case Rep Gastroenterol. 2008;2:162–9. doi: 10.1159/000129600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dumont PN, Rode A, Mabrut JY, Ducerf C, Golse N. Polycystic liver disease complicated by obstructive jaundice. J Visc Surg. 2016;153:149–51. doi: 10.1016/j.jviscsurg.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Tikkakoski T, Makela JT, Leinonen S, Päivänsalo M, Merikanto J, Karttunen A. et al. Treatment of symptomatic congenital hepatic cysts with single-session percutaneous drainage and ethanol sclerosis: Technique and outcome. J Vasc Interv Radiol. 1996;7:235–9. doi: 10.1016/S1051-0443(96)70767-4. [DOI] [PubMed] [Google Scholar]
- 17.Iwatsuki S, Starzl TE. Personal experience with 411 hepatic resections. Ann Surg. 1988;208:421–34. doi: 10.1097/00000658-198810000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newman KD, Torres VE, Rakela J, Nagorney DM. Treatment of highly symptomatic polycystic liver disease: Preliminary Experience with a Combined Hepatic Resection-Fenestration Procedure. Ann Surg. 1990;212:30–7. doi: 10.1097/00000658-199007000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farges O, Bismuth H:. Fenestration in the management of polycystic liver disease. World J Surg. 1995;19:25–30. doi: 10.1007/BF00316975. [DOI] [PubMed] [Google Scholar]