

Abstract
We update the status of research on succinic semialdehyde dehydrogenase (SSADH) deficiency (SSADHD), a rare disorder of GABA metabolism. An unusual disorder featuring accumulation of both GABA and its neuromodulatory analog, gamma-hydroxybutyric acid (GHB), recent studies have advanced the potential clinical application of NCS-382, a putative GHB receptor antagonist. Animal studies have provided proof-of-concept that enzyme replacement therapy could represent a long-term therapeutic option. The characterization of neuronal stem cells (NSCs) derived from aldehyde dehydrogenase 5a1−/− (aldh5a1−/−) mice, the murine model of SSADHD, has highlighted NSC utility as an in vitro system in which to study therapeutics and associated toxicological properties. Gene expression analyses have revealed that transcripts encoding GABAA receptors are down-regulated and may remain largely immature in aldh5a1−/− brain, characterized by excitatory as opposed to inhibitory outputs, the latter being the expected action in the mature central nervous system. This indicates that agents altering chloride channel activity may be therapeutically relevant in SSADHD. The most recent therapeutic prospects include mTOR (mechanistic target of rapamycin) inhibitors, drugs that have received attention with the elucidation of the effects of elevated GABA on autophagy. The outlook for novel therapeutic trials in SSADHD continues to improve.
Keywords: GABA, succinic semialdehyde dehydrogenase deficiency, gamma-hydroxybutyric aciduria, mTOR, Torin 2, enzyme replacement therapy
Introduction
Progress in understanding the pathophysiology and clinical features of succinic semialdehyde dehydrogenase deficiency (SSADHD) has continued since its description in the 1980s (Jakobs et al 1981; Gibson et al 1983). A major advance came in 1999 with the development of a murine knockout model, so-called aldehyde dehydrogenase 5a1 (aldh5a1−/−) mice (Hogema et al 2000). While the phenotype of this animal model is severe, with early lethality at 3 weeks of life, it has provided insights into GABAergic, glutamatergic, GHBergic, metabolic, oxidative stress, and other parameters otherwise unavailable (Gupta et al 2002; Cortez et al 2004; Buzzi et al 2006; Wu et al 2006; Jansen et al 2008; Pearl et al 2009; Vogel et al 2015, 2016, 2017a–f). Thus far, therapeutics that rescue this model from premature lethality include GABAB and GHB receptor antagonists, the non-physiological amino acid taurine, the antiepileptic agent vigabatrin, the ketogenic diet, and rapalog agents such as Torin 2, the latter an mTOR inhibitor. SGS742, a GABABR antagonist, is the subject of an ongoing clinical trial (www.clinicaltrials.gov; NCT02019667).
GABA, the major central inhibitory neurotransmitter (Schousboe and Waagepetersen 2007) and its related structural analog, gamma-hydroxybutyric acid (GHB), accumulate to supraphysiological levels in SSADHD (Malaspina et al 2016) (Fig. 1). The degree to which each contribute to pathophysiology remains unknown. However, emerging new roles for GABA exist beyond that of inhibitory neurotransmitter, including neuro-endocrine effects along the gut-brain axis, autophagy, circadian rhythms, and others (Kilb 2012; Lakhani et al 2014; Chellappa et al 2016; Mittal et al 2017). These roles provide novel opportunities to explore pathomechanisms in SSADHD. Here, we highlight new directions in SSADHD research over the last several years, and introduce pilot data supporting novel directions for research and preclinical drug development.
Fig. 1. GABA metabolism and intracellular interactions.
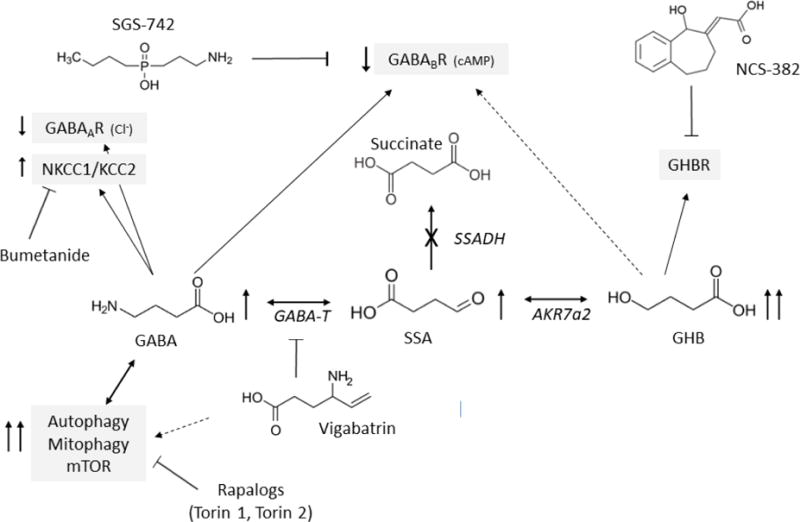
The site of the defect in patients with SSADHD is indicated by “X”. Abbreviations: GABA, γ-aminobutyric acid; GABAAR, ionotropic GABAA receptors; GABABR, metabotropic GABAB receptors. GABA-T, GABA-transaminase; SSA, succinic semialdehyde; AKR7a2, aldo-keto reductase 7a2; GHB, γ–hydroxybutyric acid; cAMP, cyclic AMP; NKCC1, sodium potassium chloride cotransporter 1; KCC2, neuronal potassium chloride cotransporter 2. In SSADHD, GABA, SSA and GHB accumulate (↑). Increased GABA and GHB activate GABAA and GABAB receptors and a putative GHB receptor (molecular nature unknown). However, a compensatory downregulation of GABA and GHB receptors (↓) has been reported in SSADHD suggesting excess GABA does not lead to increased inhibitory neurotransmission in vivo. NKCC1 and KCC2 control transmembrane chloride gradient and determine GABAA receptor directional transmembrane chloride flux. In experimental SSADHD, the expression ratio of NKCC1 and KCC2 (NKCC1/KCC2) is elevated (Vogel, 2017c), suggesting that activation of GABAA receptors causes chloride efflux from brain cells, membrane depolarization and activation of neurotransmission (see Fig. 3 for further details). NKCC1 inhibitors like bumetanide lower intracellular chloride concentration. In SSADHD, bumetanide may thus restore GABA inhibitory neurotransmission activity and efficiently suppress seizures. Finally, in SSADHD as well as in vigabatrin-treated animals, increased GABA activates the mTOR pathway with secondarily increased mitochondria number, oxidative stress, autophagy and mitophagy. mTOR inhibitors such as torin 1 and torin 2 improve GABA-induced, mTOR-pathway mediated intracellular defects and significantly prolong the lifespan of aldh5a1-deficient mice.
NCS-382, a Potential Novel Therapeutic for SSADHD
Pharmacological and Structural Considerations
NCS-382 is a putative GHB receptor (GHBR) antagonist with a Ki 14 times lower than that of GHB (Fig. 1; Maitre 1997; Vogensen et al 2013), and may be the only known antagonist of GHBRs (Bay et al 2014), whose molecular structure(s) remain undefined. NCS-382 was effective in rescuing aldh5a1−/− mice from premature lethality and in blocking the motor deficits induced by the GHB prodrug, gamma-butyrolactone (GBL; Ainslie et al 2016; Gupta et al 2002). NCS-382 exists as a racemic mixture (hydroxyl-carbon; Fig. 1). The R-isomer is twice as potent as the racemic mixture, and 13-fold more potent than the S-enantiomer (Castelli et al. 2004). Prior to our initial studies described below, preclinical pharmacokinetic/toxicological analyses of NCS-382, mandatory considerations prior to clinical intervention, had not been reported.
Pharmacokinetics and toxicology of NCS-382, and potential for drug-drug interactions
Limited earlier studies on the pharmacodynamic characteristics of NCS-382 were performed in baboon and pigeon, and were designed to exploit antagonist specificity for the GHBR in order to interrogate the central effects of GHB (Quang et al 2002; Castelli et al 2003; 2004). We obtained detailed pharmacokinetic measures following i.p. administration of NCS-382 (100 to 500 mg/kg) in C57/B6 mice (Ainslie et al 2016). The plasma elimination t1/2 for NCS-382 ranged from 0.25–0.68 h in a dose-dependent fashion. Brain residence for NCS-382 was longer, with t1/2 ranging from 0.76–0.97 h, and decreasing with increasing dose. The brain-to-plasma ratio based on area under the concentration-time curves (AUCs) ranged from 0.72–1.8. The trend for decreased brain t1/2 with increasing dose may reflect saturation of central GHB binding sites, leaving more unbound NCS‐382 for return to the systemic circulation. The fraction of total NCS-382 dose recovered in the urine was low (<4%), and undetectable in the feces (<150 ng/mg feces), suggesting metabolism as the primary route of elimination featuring glucuronidation (major product) and dehydrogenation (minor product) at the hydroxyl- moiety (Fig. 1). The intrinsic clearance of NCS‐382 (Clint) in the presence of NADPH and assessed by monitoring parent disappearance was 0.587 and 0.513 mL/min/mg protein in mouse and human liver microsomes (MLMs, HLMs), respectively. Calculated murine and human hepatic clearances were 5.2 and 1.2 L/h/kg body weight, respectively. The Michaelis constant (Km) for dehydrogenation was 29.5 ± 10 and 12.7 ± 4.9 μM in mouse and human, respectively. Glucuronide formation was linear in both species up to 100 μM. UGT2B7 (uridine 5′-diphosphoglucuronosyltransferase; UDP-glucuronosyltransferase 2B7) was suspected as the primary isoform of glucuronidating enzymes responsible for NCS-382 metabolism, based upon competition studies with the UGT2B7 inhibitor, diclofenac (Ainslie et al 2016). Co-administration of diclofenac (25 mg/kg) improved the efficacy of NCS-382 (300 mg/kg) to block the sedative and motor effects in animals treated with GBL. Plasma levels of glucuronides of NCS-382 and diclofenac decreased in combinatorial treatment relative to mice receiving either agent alone.
The identity of drug metabolizing enzymes active in the biotransformation(s) of NCS-382 also has not been reported, nor has the ability of NCS-382 to inhibit the typical enzymes, namely cytochrome P450s (CYP), involved in the metabolism of drugs. Accordingly, we employed HLMs and FDA-recommended probe substrates in reactions catalyzed by seven CYP isoforms (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4) (Vogel et al. 2017b). NCS-382 did not inhibit any of the tested enzymes at the highest tested dose (30 μM). Further, NCS-382 manifested minimal capacity to induce nuclear receptors involved in drug biotransformation and transport (aryl hydrocarbon, constitutive androstane, and pregnane X receptors) at supraphysiological doses (up to 500 μM). Collectively, these findings indicate a low-risk for CYP P450-mediated drug-drug interactions.
HepG2 cells were subsequently used to examine the cellular toxicity of NCS-382 at up to 1 mM. Multiple biomarkers assessing cellular integrity, survival, and organelle function revealed little evidence for NCS-382 cytotoxicity (Vogel et al 2017b). Gene expression studies using NCS-382 in HepG2 cells revealed only a minor number of genes (of 370 tested) showing dysregulation (Table 1). Additionally, high-dose NCS-382 demonstrated only minimal pharmacotoxicity in neural stem cells (NSCs, or neuronal progenitor cells) derived from aldh5a1−/− mice (e.g., aldh5a1−/− NSCs) (Vogel et al, 2017e). These cells were developed as an in vitro model of SSADHD, showing increased GHB content in culture medium, enhanced biomarkers of oxidative stress and increased mitochondrial number and highlighting the utility of NSCs as a useful preclinical screening tool for evaluating therapeutics for SSADHD (Vogel et al, 2017f). In sum, although a number of additional studies will be needed, pilot pharmacokinetic/safety/toxicological evaluations support the potential for clinical application of NCS-382 in SSADHD.
Table 1.
Genes altered >4-fold by NCS-382 (0.5 mM) in HepG2 cells.
| Gene | Gene product | Function | Gene grouping | Change |
|---|---|---|---|---|
| CD36 | CD36 Molecule | Involved in platelet activation, signaling and aggregation and metabolism | Steatosis | ↓ |
| HTRA4 | High-Temperature Requirement Factor A4 | Degrades misfolded secretory proteins | ER Stress & unfolded protein response | ↑ |
| SERPINA3 | Serpin Family A Member 3 | plasma protease inhibitor; deficiency has been associated with liver disease | Phospholipidosis | ↓ |
| SLC2A3 | Glucose Transporter Type 3, Brain | Facilitative glucose transporter | Phospholipidosis | ↓ |
| SLC51A | OST-Alpha | Transports the major species of bile acids | Cholestasis | ↓ |
| TNFRSF1A | Tumor Necrosis Factor Receptor Superfamily, Member 1A | Activates NF-κB, mediates apoptosis, and functions as a regulator of inflammation | Apoptosis & Necrosis | ↑ |
Preclinical efficacy of NCS-382 in aldh5a1−/− mice
We subsequently turned attention to the transport of NCS-382 in vitro. Our objective here was to investigate the potential of NCS-382 to block uptake of GHB. We demonstrated for the first time using Madin-Darby Canine Kidney (MDCK) cells that NCS-382 is actively transported and capable of inhibiting GHB transport (Vogel et al, 2017e). Following these in vitro assays with in vivo studies in aldh5a1−/− mice, we found the ratio of brain/liver GHB to be unaffected by chronic NCS-382 administration (300mg/kg; 7 consecutive days), which appeared paradoxical. This finding suggests that potential future application of NCS-382 may only be modestly beneficial since brain GHB levels do not appear to be modified with chronic treatment. We examined cortical regions from the NCS-treated mice and evaluated the expression of a number of solute carriers involved in neurotransmitter transport. As shown in Fig. 2., we found essentially all of these transporters down-regulated in aldh5a1−/− cortex in the absence of treatment. NCS-382 normalized the aberrant expression of seven of these carriers, including both glutamate and GABA transporters, but had no effect on six and actually induced significant down-regulation of the glutamate-cystine antiporter. This finding is of interest in view of the significant depletion of glutathione in this animal model, the fact that glutathione is composed of glutamate, cysteine and glycine, and the earlier finding that glutamate/glutamine levels are abnormal in aldh5a1−/− brain (Gupta, 2004; Chowdhury, 2007). These results provide modest preclinical support for the use of NCS-382 in SSADHD. Additional in vivo studies are in progress in aldh5a1−/− mice using NCS-382, assessing lifespan, body weight and neurobehavioral outcomes, and using both chronic and acute administration paradigms.
Fig. 2. Cortical gene expression profile of solute carriers (Slc) in aldh5a1−/− mice following NCS-382 administration (7 days, q.i.d., 300 mg/kg).
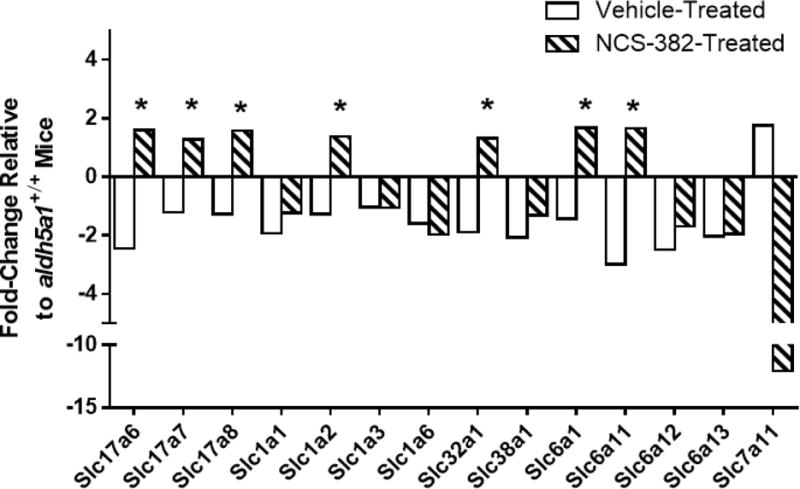
Relative levels are displayed, normalized to control aldh5a1−/− mice receiving vehicle. Functional role of carriers: *17a6, *17a7, *17a8, vesicular glutamate (glu) transporter, glu cotransporter, and glu transporter 3, respectively; 1a1, *1a2, 1a3, 1a4, excitatory amino acid transporters 3, 2, 1 and 4, respectively; *32a1, GABA vesicular transporter; 38a1, Na+-coupled amino acid transporter 1 (glutamine transport); *6a1, plasma membrane GABA transporter; *6a11, 6a12, 6a13, Na+-dependent GABA presynaptic terminal reuptake, Na+/Cl−-dependent betaine, and Na+/Cl−-dependent GABA 2 transporters, respectively; 7a11, glutamate-cysteine antiporter. Asterisked genes demonstrated correction of expression following chronic NCS-382 administration. Values represent pooled data of biological triplicates (n=3 animals each, NCS-382 and vehicle).
Enzyme Replacement Therapy for SSADHD
As a therapeutic approach, enzyme replacement therapy (ERT) has gained prominence in the lysosomal storage disorders, although it should be feasible in organic acidemias (Darvish-Damavandi et al 2016). We examined the feasibility of ERT in aldh5a1−/− mice using a GST(glutathione)-tagged human ALDH5A1 gene construct that overexpresses the GST-hSSADH fusion protein with accompanying ampicillin resistance in E. Coli (DNASU Plasmid Repository; Ramachandran et al. 2004). Crude extracts of E. coli were harvested that had been transfected and grown in standard LB broth supplemented with ampicillin at 37° C overnight, pelleted and lysed with Pefabloc (protease inhibitor; Sigma-Aldrich, St. Louis, MO USA) and lysozyme. GST-hSSADH resident in crude lysates of E. Coli was purified using Pierce™ GST spin purification kits, followed by dialysis and subsequent concentration using polyethersulfone (PES) columns. Resultant protein content was quantified using a standard BCA protein assay. The GST-tagged enzyme activity was cleaved with thrombin and the activity of ALDH5A1 determined employing spectrofluorometry based on the NAD/NADH couple (Gibson et al 1991).
The feasibility of ERT using bacterially-produced ALDH5A1 was subsequently evaluated in aldh5a1−/− mice. As endpoint, we employed rescue of this model from premature lethality (day of life (DOL) 21–23; endpoint, DOL 30, which is highly significant survival), the latter endpoint chosen in view of the limited availability of treatment protein. Purified ALDH5A1 was administered (i.p., 1 mg/kg in PBS, q.d.) beginning at day of life 10. Median survival of vehicle-treated aldh5a1−/− mice was 22 days compared to 80% survival rate (4 of 5 mice) to 30 days with ERT treatment (Logrank; p=0.04; Fig. 3 (inset)). Brain, liver and sera were collected from surviving ERT-treated subjects harvested at DOL 30. The expression of GABA receptor genes was contrasted between ERT (DOL 30) and untreated (DOL 21) aldh5a1−/− mice, with data normalization to DOL 21 aldh5a1+/+ mice. The expression of several GABAA receptor subunits (primarily gamma, epsilon and theta) were significantly corrected in aldh5a1−/− mice with enzyme intervention (Fig. 3). Since we hypothesized that parenterally administered SSADH would lower metabolites (GHB, GABA) in blood and other tissues, these intermediates were quantified (Fig. 4) in sham and enzyme-treated subjects using LC/MS-MS (Gibson et al 1990; Kok et al 1993). Although the numbers were low, we found a significant correction of GHB in the brain of enzyme treated animals, and a trend toward improved levels of GABA in blood. This promising trial was not sufficiently powered for biochemical measures, and more extensive evaluations are needed with larger n values. In particular, we need to assess the levels of SSADH activity in blood and organs, and, investigate the potential of PEGylation to increase protein t1/2 and reduce immunogenicity (Bell et al, 2017).
Fig. 3. Enzyme replacement therapy (ERT) in experimental SSADHD.
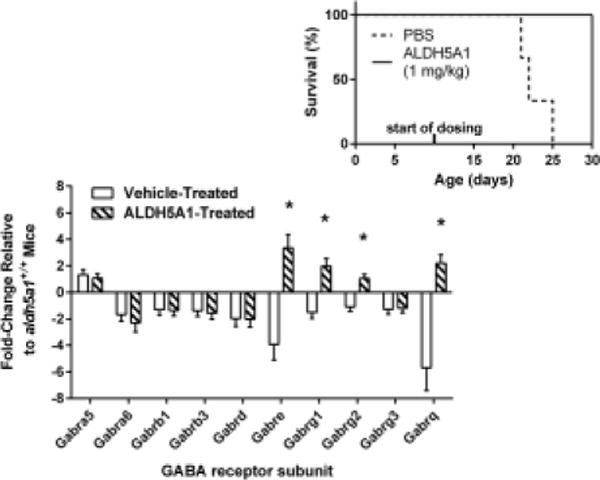
(insert) Aldh5a1−/− mouse survival-rate to day of life (DOL 30) as a function of enzyme replacement intervention. Purified human ALDH5A1 was administered daily (1 mg/kg/day), beginning at DOL 10, via i.p. injection. Also shown is the expression of GABA-related genes following ERT in aldh5a1−/− mice (fold change relative to aldh5a1+/+ mice; sagittal slices of 21 day old mice). Abbreviations: Gabra5, GABAA receptor subunit α5; Gabra6, α-6; Gabrb1, β-1; Gabrb3, β-3; Gabrd, δ; Gabre, ε; Gabrg1, γ-1; Gabrg2, γ-2; Gabrg3, γ-3; Gabrq, θ. Asterisked values represent directional correction of expression as a function of ERT.
Fig. 4. Metabolic measures in animals treated with ERT.
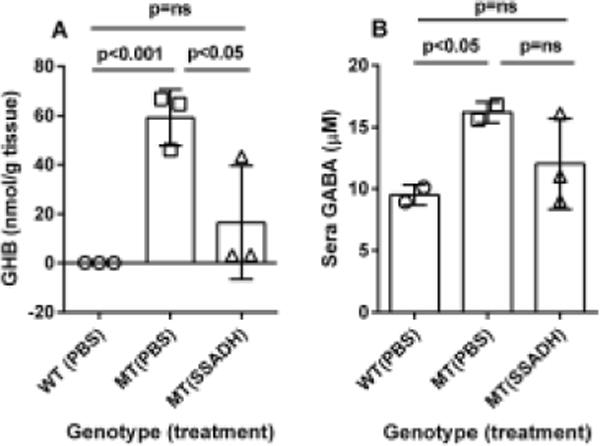
The treatment scheme is described in the legend to Fig. 3. (A) Brain GHB content as a function of genotype (WT=wild-type, aldh5a1+/+ mice; MT=mutant, aldh5a1−/− mice; PBS, phosphate-buffered saline; SSADH, recombinant SSADH. (B) GABA in sera. Data depicted as mean + SD. Statistical analyses employed one-way ANOVA with post-hoc analysis (t test).
The paradox of seizures in a SSADHD, a hyperGABAergic disorder
Chloride directional flux through the GABAA receptor in mammalian brain is regulated by the transmembrane chloride gradient which is itself controlled primarily by two transporters, the sodium-potassium-chloride symporter (NKCC1) and the potassium-chloride cotransporter (KCC2) (Fig. 5a; Kilb, 2012). Activation of the GABAA receptor when the activity of NKCC1 is increased and intracellular chloride concentrations are elevated results in chloride efflux, plasma membrane depolarization and paradoxical neurotransmission activation. Such a situation is observed in the fetal brain but is reversed postnatally when GABAA receptor activation consistently leads to increased chloride cellular uptake, hyperpolarization and neurotransmission inhibition. We hypothesized that in SSADHD, postnatal seizures were caused by overexpression of NKCC1 and a continuing excitatory capacity of GABAA receptors after birth. If confirmed, this mechanism would explain the paradox of seizures in a hyperGABAergic condition (Vogel et al 2017c). Furthermore, it would also suggest that inhibition of NKCC1 might have positive therapeutic effects in SSADHD. In support of these hypotheses, we found that NKCC1 was highly overexpressed in aldh5a1−/− brain (Fig. 5B). The expression of KCC2, which transports chloride ions out of the cell, was also increased but significantly less than that of NKCC1 (Fig. 5B). Hence, the NKCC1 to KCC2 ratio was significantly increased, suggesting that intracellular chloride concentrations might be increased in the aldh5a1−/− brain, favoring a depolarizing and excitatory activity as the predominant role for GABAA receptors. This remains to be quantitatively evaluated employing neurophysiological assessments in vitro via patch-clamp methodology, or perhaps in vivo using two-photon measurement with chloride ion probes.
Fig. 5. Intracellular chloride homeostasis, GABAergic neurotransmission and bumetanide in SSADHD (for abbreviations, see Fig. 1).
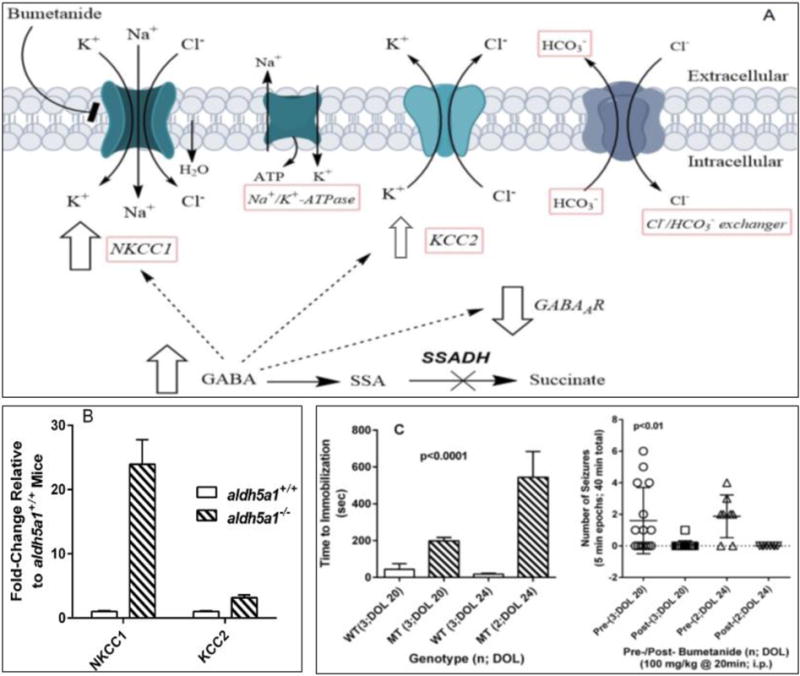
(A) Schematic diagram of membrane ion transport and mechanism of action of bumetanide. (B) NKCC1 and KCC2 gene expression in the hypothalamus of aldh5a1+/+ (n=5) and aldh5a1−/− (n=5) mice collected at postnatal day-of-life 20. Expression data were obtained using q-RT-PCR, validated pathway plates from Bio-Rad. (C) Time to sedation following acute dosing of 100 mg/kg bumetanide. Animals were evaluated at day of life (DOL) 20 and 24 for seizure events, and the time to immobilization (sedation) determined by visual recording using Noldus technology (see text). Bumetanide was administered intraperitoneally to the subject mice after 20 min. of initial observational recording, followed by an additional 20 minutes of recording. Total number of seizures was quantified in 5 min blocks of the total 40 min. recording period. Bumetanide was obtained from Enzo Life Sciences, Inc. (Farmingdale, NY, USA) and aseptically dissolved in DMSO vehicle. Vehicle-treated subjects did not show sedation. Aldh5a1−/− mice (mutant; MT) were significantly more resistant to the sedative effects of bumetanide as compared to aldh5a1+/+ (wild-type; WT) mice, the latter showing almost instantaneous immobilization. During the active period in aldh5a1−/− mice prior to immobilization (~3–8 min.), no seizure activity was noted. Additionally, this resistance increased significantly with age in aldh5a1−/− mice (t test, p<0.05). Abbreviations employed: n, number of animals studied; DOL, day of life; sec, seconds; min, minutes.
The potential therapeutic role of NKCC1 inhibition in SSADHD was next examined. In this context, we postulated that there would be resistance to the sedative activity of NKCC1 inhibitors in SSADHD, because of the increased expression of NKCC1. We treated aldh5a1+/+ and aldh5a1−/− mice with acute i.p. doses of bumetanide (25 and 100 mg/kg body weight) with video recording of seizure activity as well as assessment of the time to immobilization. Bumetanide is a known inhibitor of both NKCC1 and 2 originally approved for edema but with demonstrated antiepileptic efficacy in several neurological/epileptic disorders despite poor brain penetration (Levy et al 2013; Oliveros et al 2011; Rahmanzadeh et al 2016; Cleary et al 2013). We employed Pinnacle technology (https://www.pinnaclet.com) for recording of animal behavior in conjunction with a rubric developed by Noldus technology to evaluate seizure activity (http://www.noldus.com/animal-behavior-research). Our design was to quantify generalized tonic-clonic seizures in 5 minute epochs for 40 minutes in continuum. At the 20 minute mark, a single intraperitoneal administration of bumetanide (25 or 100 mg/kg) was given and the animals returned to the open field setting. A dose of 25 mg/kg bumetanide did not induce immobilization during the 40 minute recording period for either aldh5a1+/+ or aldh5a1−/− mice. Conversely, 100 mg/kg resulted in rapid induction of immobilization (Fig. 5C). Of interest, however, was the observation that aldh5a1−/− were significantly more resistant to the effect of bumetanide, at both DOL 20 and 24, with this resistance about 3-fold greater in the older aldh5a1−/− mice. In addition, no seizure activity was observed in the mutant mice in the time period between bumetanide injection and the onset of immobilization, an approximate period of 3–8 minutes. As well, the lower dose of bumetanide (25 mg/kg) also reduced seizure activity in aldh5a1−/− mice without sedation (data not shown). These preliminary data suggest the resistance of the aldh5a1−/− mice to the sedative effect of bumetanide is secondary to increased NKCC1 activity and disruption in the chloride gradient. This suggests that manipulation of NKCC1 and restoration of intracellular chloride homeostasis represents a potentially attractive therapeutic target in SSADHD.
GABA and mTOR: Treatment strategies and pathophysiological insights in SSADHD
Inhibition of mTOR, a therapeutic target in SSADHD
Increased mitochondrial numbers (including both the size and total number of organelles) were first documented in aldh5a1−/− mice in pyramidal hippocampal neurons (Nylen et al 2009). Subsequently, Lakhani and coworkers (2014) demonstrated that increased GABA levels in S. cerevisiae led to activation of mTOR (molecular target of rapamycin), manifesting as elevated mitochondrial numbers and enhanced oxidative stress. This same group of investigators further documented that mitochondrial numbers in brain and liver derived from aldh5a1−/− mice were increased and associated with enhanced oxidant stress, all of which could be normalized by the mTOR inhibitor, rapamycin.
These studies were the genesis of further preclinical interventional studies in aldh5a1−/− mice with mTOR inhibitors (rapamycin, temsirolimus), dual mTORC1/2 and PI3K inhibitors, as well as mTOR-independent autophagy inducing drugs (Vogel et al 2016; Fig. 6). Lifespan extension (rescue from premature lethality) for aldh5a1−/− mice was observed across a number of mTOR-specific and dual inhibitors, and findings with the dual inhibitor XL765 and Torin2 were striking. XL765 induced a modest weight improvement over 35 days until sacrifice at DOL 50 in aldh5a1−/− mice (Vogel et al 2016). Conversely, mTOR-independent inducers of autophagy were without benefit in mitigating premature lethality. These data suggest that induction of autophagy through mTOR-independent mechanisms was insufficient for rescue, suggesting that other functions associated with mTOR may be involved in the clinical efficacy of mTOR blockade in aldh5a1−/− mice.
Fig. 6. Simplified schematic diagram of the roles of AMPK and mTORC1 in the regulation of autophagy.
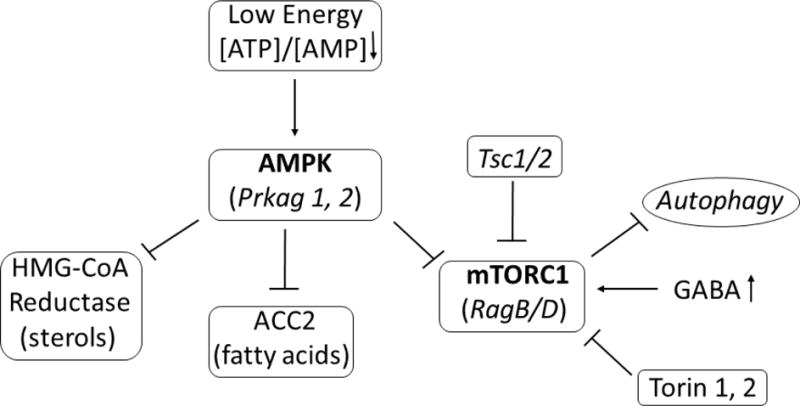
Both AMPK (adenosine monophosphate-activated protein kinase) and mTOR (mechanistic target of rapamycin) have key roles in the regulation of intracellular energetics, growth, and survival. Prkag1 and Prkag2 (protein kinase cAMP-activate γ subunits 1 and 2) represent components of the active AMPK trimeric structure. Conversely, RagB and D (ras-related GTP-binding proteins B and D) are involved in the regulation of mTORC1 function. Tsc1/2 (tuberous sclerosis proteins 1 and 2) are negative regulators of mTORC1. Abbreviations: HMG-CoA reductase, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; ACC, acetyl-CoA carboxylase; ATP, adenosine triphosphate; AMP, adenosine monophosphate.
Studies with Torin 2 administration in aldh5a1−/− mice, beginning at DOL10 under an escalating dose paradigm, were evaluated for seizures using a three channel recording electrocorticography (Pinnacle Technology; https://www.pinnaclet.com/eeg-emg-systems.html) and electrode-implanted aldh5a1+/+ and aldh5a1−/− mice. EEG was scored offline using semi-automated detection (Neuroscore, Data Sciences International, St. Paul, MN) for seizure frequency (total number of seizures), and total ictal time (cumulative time spent in seizures); where a seizure event was defined as a run of continuous spikes ≥ 3 s in duration on the EEG. Automatically detected events were verified by visual inspection (Dhamne et al 2017) Our prediction was that Torin 2 administration would lead to improvement in both parameters. We found that Torin 2 was ineffective at reducing seizure frequency in aldh5a1−/− mice (Table 2), and unexpectedly found that it significantly extended the total ictal time (Table 2). It is of interest that Torin 2 corrected (up-regulated) a number of GABA(A)ergic receptor subunits (Vogel et al 2016). If these receptors remain immature, consistent with our hypothesis for bumetanide (see above), then their subsequent up-regulation could conceivably exacerbate depolarization, and thus enhance cumulative epileptic outburst duration, as we observed (Table 2). On the other hand, others have provided evidence that mTOR inhibitors prevent the sprouting of mossy fibers, a form of synaptic reorganization which occurs in epilepsy and can lead to the formation of recurring excitatory circuits (Dudek et al 2017). Which mechanism explains enhanced seizure activity with Torin 2 remains under investigation.
Table 2.
Effect of Torin 2 on seizure frequency and total ictal time in aldh5a1+/+ and aldh5a1−/− mice
| Vehicle | Torin 2 (10 mg/kg) | t test (Veh. vs. Torin) | |
|---|---|---|---|
| Seizure Frequency (avg/hr) | |||
| aldh5a1+/+ | 5.4 ± 0.8 | 0.50 ± 0.59 | p=0.12 |
| aldh5a1−/− | 3.2 ± 0.8 | 9.6 ± 1.3 | p=0.11 |
| t test | p=0.41 | p=0.23 | |
| Total Ictal Time (seconds) (avg/hr) | |||
| aldh5a1+/+ | 22 ± 1.7 | 2.1 ± 1.2 | p=0.15 |
| aldh5a1−/− | 19 ± 2.0 | 282 ± 8.0 | p=0.03 |
| t test | p=0.90 | p=0.30 | |
Data depicted as mean + SEM. Seizure events included spike-trains, probable myoclonic as well as absence and tonic-clonic seizures
Future Directions
Dissecting the mTOR role using vigabatrin-treated aldh5a1+/+ and aldh5a1−/− mice
Vigabatrin (VGB)-treated aldh5a1+/+ mice represent a drug-induced form of GABA-transaminase deficiency, since VGB irreversibly inhibits this first enzyme of GABA catabolism (Fig. 1). Employing VGB, significantly elevated GABA in the CNS can be achieved at relatively low daily doses (~10 mg/kg), or via chronic subcutaneous delivery with calibrated osmotic minipumps (Vogel et al 2016; 2017b). A cardinal difference between these “models” resides in the absence of elevated GHB in VGB-treated aldh5a1+/+ mice, which enables us to begin to more clearly isolate the role of GABA and its effect on mTOR. We have begun exploring this aspect employing gene expression in these different models.
mTOR coordinates numerous intracellular bioenergetic cues that serve to control growth and catabolism (i.e. translation, autophagy) (Fig. 6). A number of gene expression changes correlated between the two animal models. Prkag1 was down-regulated in brain of both models (Vogel et al 2017b; 2017c). Prkag1 is a gamma regulatory subunit of the heterotrimeric AMP-activated protein kinase (AMPK), which also contains an alpha catalytic subunit and a non-catalytic beta subunit (Fig. 6). AMPK is an important energy-sensing enzyme that monitors cellular energy status. In response to cellular metabolic stresses, AMPK is activated, and thus phosphorylates and inactivates acetyl-CoA carboxylase (ACC) and 3-hydroxy 3-methylglutaryl-CoA reductase (HMGCR), key enzymes involved in regulating de novo biosynthesis of fatty acid and cholesterol. AMPK acts via direct phosphorylation of metabolic enzymes, and acts longer-term via phosphorylation of transcription regulators. During low energy conditions AMPK is a critical negative effector of mTOR activation.
Prkag2 was also down-regulated in aldh5a1−/− brain (and downregulated in VGB-treated mouse eye). Decreased expression of Prkag1 and 2 was normalized by the mTOR inhibitor Torin 1, and correction of AMPK’s activator (downregulated in aldh5a1−/− mice), Stk11, was normalized by Torin2. As well, Tsc1 and 2, signaling systems downstream of AMPK, also exhibited lower expression which was normalized by Torin 1 and Torin 2 in aldh5a1−/− mouse brain. Rag GTPases activate mTOR through its amino acid sensing pathway, and RagB/RagD expression was upregulated in aldh5a1−/− brain (RagB was upregulated in VGB-treated eye tissue, while RagD was upregulated in VGB-treated brain tissue). Together, these findings strongly suggest that correction of AMPK is involved in the pro-survival effects of Torin drugs in aldh5a1−/− mice. Understanding the potential clinical utility of mTOR inhibitors to treat disorders of GABA metabolism will continue to be a central theme in our laboratory.
Given the multisystem dysfunction in SSADHD, it is likely that a combination of therapies will be required to leverage incremental improvements in the phenotype. A logical starting point would be VGB, if the issue of ocular toxicity could be overcome. Indeed, with regard to this, inhibitors of mTOR might be added to mitigate the effects of additionally increased GABA associated with VGB. An antioxidant agent would also be of value, given the evidence for oxidative stress in this disorder (Gupta et al 2002). As shown in the current report, ERT may have therapeutic benefit, which could be combined with genetic manipulations in the future, such as CRISPRCas9 approaches. Nonetheless, our short-term goals remain the development of targeted therapy for SSADHD.
Supplementary Material
Suppl. Fig. Representative electrocorticographic recordings of seizure types in an aldh5a1−/− mouse. These data are representative of traces employed to generate the data of Table 2.
{kind=link}
Take home message.
We summarize recent advances in pharmacotherapeutic and enzyme-replacement approaches in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism.
Acknowledgments
Supported in part by NIH NS 82286, NS 98856, NS 85369 and EY27476. We remain grateful for the ongoing support of the SSADH Association (www.ssadh.net).
Details of Funding
Supported in part by NIH NS 82286, NS 98856, NS 85369 and EY27476.
Footnotes
Contributions
KRV, GRA, JBR, KMG-planning, conduct, data analysis, manuscript editing and drafting, reporting; DCW, AM, AR-conduct and data analysis, manuscript editing, reporting
Competing Interest Statement
The authors have no competing interests associated with the content of this manuscript
Conflicts of Interest
KRV, no conflict of interest; GRA, no conflict of interest; JBR, no conflict of interest; KMG, no conflict of interest; DCW, no conflict of interest; AM, no conflict of interest; AR, no conflict of interest
Ethics Approval
IACUC protocols ASAF 4232 and 4276 are active and approved for the use of vertebrate animals in research.
References
- Ainslie GR, Gibson KM, Vogel KR. A pharmacokinetic evaluation and metabolite identification of the GHB receptor antagonist NCS-382 in mouse informs novel therapeutic strategies for the treatment of GHB intoxication. Pharmacol Res Perspect. 2016;4:e00265. doi: 10.1002/prp2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay T, Eghorn LF, Klein AB, Wellendorph P. GHB receptor targets in the CNS: Focus on high-affinity binding sites. Biochem Pharmacol. 2014;87:220–8. doi: 10.1016/j.bcp.2013.10.028. [DOI] [PubMed] [Google Scholar]
- Bell SM, Wendt DJ, Zhang Y, et al. Formulation and PEGylation optimization of the therapeutic PEGylated phenylalanine ammonia lyase for the treatment of phenylketonuria. PLOS ONE. 2017;12:e0173269. doi: 10.1371/journal.pone.0173269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res. 2006;1090:15–22. doi: 10.1016/j.brainres.2006.02.131. [DOI] [PubMed] [Google Scholar]
- Castelli MP1, Ferraro L, Mocci I, et al. Selective gamma-hydroxybutyric acid receptor ligands increase extracellular glutamate in the hippocampus, but fail to activate G protein and to produce the sedative/hypnotic effect of gamma-hydroxybutyric acid. J Neurochem. 2003;87:722–32. doi: 10.1046/j.1471-4159.2003.02037.x. [DOI] [PubMed] [Google Scholar]
- Castelli MP, Pibiri F, Carboni G, Piras AP. A review of the pharmacology of NCS-382, a putative antagonist of the gamma-hydroxybutyric acid (GHB) receptor. CNS Drug Rev. 2004;10:243–260. doi: 10.1111/j.1527-3458.2004.tb00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappa SL, Gaggioni G, Ly JQ, et al. Circadian dynamics in measures of cortical excitation and inhibition balance. Sci Rep. 2016;6:33661. doi: 10.1038/srep33661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary RT, Sun H, Huynh T, et al. Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLOS ONE. 2013;8:e57148. doi: 10.1371/journal.pone.0057148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, Wu Y, Gibson KM, Snead OC. Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav. 2004;79:547–53. doi: 10.1016/j.pbb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Darvish-Damavandi M, Ho HK, Kang TS. Towards the development of an enzyme replacement therapy for the metabolic disorder propionic acidemia. Mol Genet Metab Rep. 2016;8:51–60. doi: 10.1016/j.ymgmr.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamne SC, Silverman JL, Super CE, et al. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol Autism. 2017;8:26. doi: 10.1186/s13229-017-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Shao L-R. Mossy fiber sprouting and recurrent excitation: direct electrophysiologic evidence and potential implications. Epilepsy Curr. 2004;4:184–187. doi: 10.1111/j.1535-7597.2004.04507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson KM, Sweetman L, Nyhan WL, et al. Succinic semialdehyde dehydrogenase deficiency: an inborn error of gamma-aminobutyric acid metabolism. Clin Chim Acta. 1983;133:33–42. doi: 10.1016/0009-8981(83)90018-9. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Aramaki S, Sweetman L, et al. Stable isotope dilution analysis of 4-hydroxybutyric acid: an accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. Biomed Environ Mass Spectrom. 1990;19:89–93. doi: 10.1002/bms.1200190207. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Lee CF, Chambliss KL, et al. 4-Hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta. 1991;196:219–21. doi: 10.1016/0009-8981(91)90076-o. [DOI] [PubMed] [Google Scholar]
- Gupta M, Greven R, Jansen EE, et al. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria) J Pharmacol Exp Ther. 2002;302:180–7. doi: 10.1124/jpet.302.1.180. [DOI] [PubMed] [Google Scholar]
- Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212–6. doi: 10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of γ-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–178. doi: 10.1016/0009-8981(81)90184-4. [DOI] [PubMed] [Google Scholar]
- Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol. 2008;8:112. doi: 10.1186/1471-213X-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilb W. Development of the GABAergic system from birth to adolescence. Neuroscientist. 2012;18:613–30. doi: 10.1177/1073858411422114. [DOI] [PubMed] [Google Scholar]
- Kok RM, Howells DW, van den Heuvel CC, et al. Stable isotope dilution analysis of GABA in CSF using simple solvent extraction and electron-capture negative-ion mass fragmentography. J Inherit Metab Dis. 1993;16:508–12. doi: 10.1007/BF00711667. [DOI] [PubMed] [Google Scholar]
- Lakhani R, Vogel KR, Till A, et al. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med. 2014;6:551–66. doi: 10.1002/emmm.201303356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy PD, Bellou A. Acute heart failure treatment. Curr Emerg Hosp Med Rep. 2013;1(2) doi: 10.1007/s40138-013-0012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina P, Roullet JB, Pearl PL, Ainslie GR, Vogel KR, Gibson KM. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem Int. 2016;99:72–84. doi: 10.1016/j.neuint.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitre M. The gamma-hydroxybutyrate signaling system in brain: organization and functional implications. Prog Neurobiol. 1997;51:337–361. doi: 10.1016/s0301-0082(96)00064-0. [DOI] [PubMed] [Google Scholar]
- Mittal R, Debs LH, Patel AP, et al. Neurotransmitters: the critical modulators regulating gut-brain axis. J Cell Physiol. 2017;232:2359–2372. doi: 10.1002/jcp.25518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylen K, Velazquez JL, Likhodii SS, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol. 2008;210:449–57. doi: 10.1016/j.expneurol.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros M, Pham JT, John E, Resheidat A, Bhat R. The use of bumetanide for oliguric acute renal failure in preterm infants. Pediatr Crit Care Med. 2011;12:210–4. doi: 10.1097/PCC.0b013e3181e912a7. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009;32:343–52. doi: 10.1007/s10545-009-1034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang LS, Shannon MW, Woolf AD, Desai MC, Maher TJ. Pretreatment of CD-1 mice with 4-methylpyrazole blocks toxicity from the gamma-hydroxybutyrate precursor, 1,4-butanediol. Life Sci. 2002;71:771–8. doi: 10.1016/s0024-3205(02)01744-7. [DOI] [PubMed] [Google Scholar]
- Rahmanzadeh R, Eftekhari S, Shahbazi A, et al. Effect of bumetanide, a selective NKCC1 inhibitor, on hallucinations of schizophrenic patients; a double-blind randomized clinical trial. Schizophr Res. 2017;184:145–146. doi: 10.1016/j.schres.2016.12.002. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Waagepetersen HS. GABA: homeostatic and pharmacological aspects. Prog Brain Res. 2007;160:9–19. doi: 10.1016/S0079-6123(06)60002-2. [DOI] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Jansen EE, Salomons GS, Gibson KM. Torin 1 partially corrects vigabatrin-induced mitochondrial increase in mouse. Ann Clin Transl Neurol. 2015;2:699–706. doi: 10.1002/acn3.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Gibson KM. mTOR inhibitors rescue premature lethality and attenuate dysregulation of GABAergic/glutamatergic transcription in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. J Inherit Metab Dis. 2016;39:877–886. doi: 10.1007/s10545-016-9959-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Pearl PL, Gibson KM. Aberrant mTOR signaling and disrupted autophagy: The missing link in potential vigabatrin-associated ocular toxicity? Clin Pharmacol Ther. 2017a;101:458–461. doi: 10.1002/cpt.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Roullet JB, McConnell A, Gibson KM. In vitro toxicological evaluation of NCS-382, a high-affinity antagonist of γ-hydroxybutyrate (GHB) binding. Toxicol In Vitro. 2017b;40:196–202. doi: 10.1016/j.tiv.2017.01.013. [DOI] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Schmidt MA, Wisor JP, Gibson KM. mTOR inhibition mitigates molecular and biochemical alterations of vigabatrin-induced visual field toxicity in mice. Pediatr Neurol. 2017c;66:44–52.e1. doi: 10.1016/j.pediatrneurol.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Jansen EE, Salomons GS, Gibson KM. Therapeutic relevance of mTOR inhibition in murine succinate semialdehyde dehydrogenase deficiency (SSADHD), a disorder of GABA metabolism. Biochim Biophys Acta. 2017d;1863:33–42. doi: 10.1016/j.bbadis.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, McConnell A, Roullet JB, Gibson KM. Toxicologic/transport properties of NCS-382, a γ-hydroxybutyrate (GHB) receptor ligand, in neuronal and epithelial cells: Therapeutic implications for SSADH deficiency, a GABA metabolic disorder. Toxicol In Vitro. 2017e Oct 11; doi: 10.1016/j.tiv.2017.10.015. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Jansen EE, Salomons GS, Roullet J-B, Gibson KM. In vitro modeling of experimental succinic semialdehyde dehydrogenase deficiency (SSADHD) using brain-derived neural stem cells. PLoS One. 2017f;12(10):e0186919. doi: 10.1371/journal.pone.0186919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogensen SB, Marek A, Bay T, et al. New synthesis and tritium labeling of a selective ligand for studying high-affinity γ-hydroxybutyrate (GHB) binding sites. J Med Chem. 2013;56:8201–8205. doi: 10.1021/jm4011719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Buzzi A, Frantseva M, Velazquez JPL, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol. 2006;59:42–52. doi: 10.1002/ana.20686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. Representative electrocorticographic recordings of seizure types in an aldh5a1−/− mouse. These data are representative of traces employed to generate the data of Table 2.