

Abstract
Despite advances in the field, colorectal cancer (CRC) remains a leading cause of cancer-related mortality worldwide. Research into bioactive sphingolipids over the past two decades has played an important role in increasing our understanding of the pathogenesis and therapeutics of CRC. In the complex metabolic network of sphingolipids, ceramidases (CDases) have a key function. These enzymes hydrolyze ceramides into sphingosine (SPH) which in turn is phosphorylated by sphingosine kinases (SK) 1 and 2 to generate sphingosine-1 phosphate (S1P). Importantly, we have recently shown that inhibition of neutral CDase (nCDase) induces an increase of ceramide in colon cancer cells which decreases cellular growth, increases apoptosis and modulates the WNT/β-catenin pathway. We have also shown that the deletion of nCDase protected mice from the onset and progression of colorectal cancer in the AOM carcinogen model. Here we demonstrate that AKT is a key target for the growth suppressing functions of ceramide. The results show that inhibition of nCDase activates GSK3β through dephosphorylation, and thus is required for the subsequent phosphorylation and degradation of β-catenin. Our findings show that inhibition of nCDase also inhibits the basal activation status of AKT, and we further establish that a constitutively active AKT (AKT T308D, S473D; AKTDD) reverses the effect of nCDase on β-catenin degradation. Functionally, the AKTDD mutant is able to overcome the growth suppressive effects of nCDase inhibition in CRC cells. Moreover, nCDase inhibition induces a growth delay of xenograft tumors from control cells, whereas xenograft tumors from constitutively active AKT cells become resistant to nCDase inhibition. Taken together, these results provide important mechanistic insight into how nCDase regulates cell proliferation. These findings demonstrate a heretofore unappreciated, but critical, role for nCDase in enabling/maintaining basal activation of AKT and also suggest that nCDase is a suitable novel target for colon cancer therapy.
Keywords: Colorectal cancer, Sphingolipids, Ceramide, β-catenin, proliferation, AKT
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide and a leading cause of cancer-related mortality1–3. Five percent of newly diagnosed cases in the United States are due to inherited gene mutations such as Familial adenomatous polyposis or Lynch syndrome and an additional twenty percent of cases occur in people with a family history of the disease. Therefore, approximately seventy-five percent of colon cancer cases are thought to occur sporadically 2. These sporadic carcinomas develop through the accumulation of genetic modifications such as the activation of oncogenes or the inactivation of tumor suppressors 4 and additional epigenetic alterations, leading to activation of oncogenic programs.
Growing evidence established over the last two decades shows that bioactive sphingolipids have a role in colorectal cancer pathogenesis and therapeutics. Among these molecules, sphingosine (SPH), ceramide, and sphingosine-1-phosphate (S1P) have been involved in the regulation of major cellular functions including death, growth, autophagy, angiogenesis, cell adhesion, differentiation, migration, senescence, stress and inflammatory responses5–7.
Ceramides are the central hub in sphingolipid metabolism. Ceramides are produced via the de novo pathway, catabolic pathways and/or salvage pathway 6. Ceramides can be synthesized either de novo or from complex sphingolipids. Conversely, ceramides can be catabolized by CDases into SPH which in turn can be phosphorylated by SK 1 and 2 to generate S1P 8, 9.
Among the five ceramidases 10 identified to date, nCDase in particular is predominantly expressed in the large intestine and is involved in the metabolism of dietary sphingolipids 11. nCDase deficient mice show a modified profile of basal intestinal bioactive sphingolipids with increased levels of C16:0 ceramide as well as less SPH. We have recently shown 12 that inhibition of nCDase induces an increase of ceramide in colon cancer cells, as well as a decrease in growth and an increase in apoptosis. These effects were specific to cancerous intestinal cells. We also demonstrated that nCDase inhibition decreased tumor growth in a cancer xenograft model and that deletion of nCDase prevented the development of tumors in an inducible colon carcinogenesis (AOM) model.
In addition, colon cancer cells proliferation is partially regulated by the Wnt/β-catenin pathway. β-catenin turnover is regulated through a multi-protein complex, termed the β-catenin destruction complex. In the absence of Wnt, this complex composed of: AXIN, adenomatous polyposis coli (APC), casein kinase I-alpha (CK) and GSK3β induces the phosphorylation of β-catenin on serine 33/37 by GSK3β13–15. This is followed by degradation of β-catenin via the 26S proteasome. Although the inhibition of nCDase is associated with an inhibition of the WNT/β-catenin pathway, it remains unclear how nCDase regulates the WNT/β-catenin pathway and what is the role of nCDase in these cells.
Here we show that AKT is a key target for the growth suppressing effects of nCDase inhibition and more importantly that phosphorylation of AKT is sufficient to induce neutral ceramidase dependent activation of WNT/β-catenin. This demonstrates a specific link between AKT and nCDase and the role of AKT in colon cancer biology.
RESULTS
nCDase inhibition induces a decrease of β-catenin level via activation of GSK3β
To investigate the role of nCDase in the growth of colon cancer cells, we used an HCT116 cell line model of colon cancer cells. HCT116 cells are wild type for APC, heterozygous for β-catenin with an in-frame deletion in exon 3 codon 45 16. However, it has been demonstrated that in this cell line β-catenin co-precipitates with APC, E-cadherin, and α-catenin 16. These cells are also wild type for AKT 17. As demonstrated by Garcia-Barros in 2016, we confirm that nCDase inhibition using the specific nCDase inhibitor C6 urea-ceramide (Figure 1A) or using two specific siRNA targeting nCDase (Figure 1B) induces a decrease of β-catenin in the HCT116 colon cancer cell line. Therefore, we evaluated the effects of nCDase inhibition (Figure 1A) or down regulation (Figure 1B) on phosphorylation of GSK3β on serine 9, and the results showed that interfering with nCDase induced a significant decrease in the phosphorylation of GSK3β on serine 9. These effects were time dependent (Supplemental Figure 1).
Figure 1. nCDase inhibition induces a decrease of β-catenin level via activation of GSK3β.
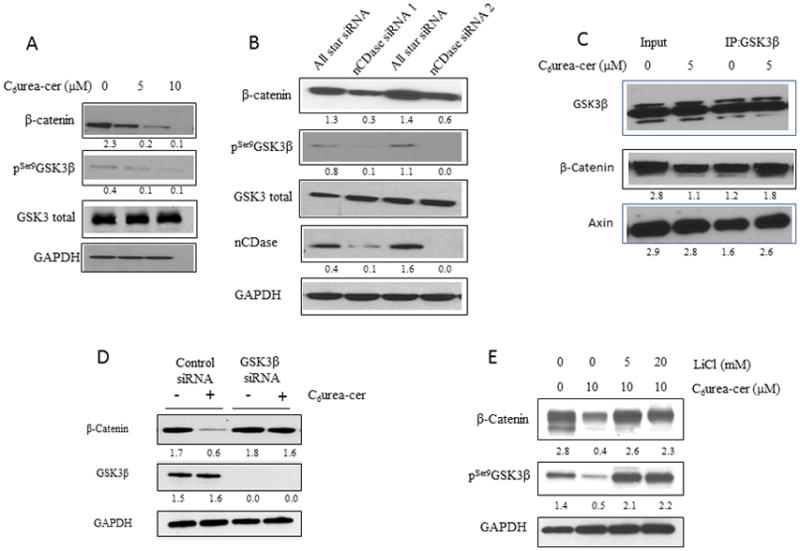
A: HCT116 cells were treated for 24 hours with the indicated concentrations of C6 urea-ceramide or with vehicle. The levels of β-catenin as well as phosphorylation of GSK3β on serine 9 were measured by western blot.
B: Cells were transfected twice for 48 hours with nCDase siRNA or with control siRNA. Efficiency of siRNA downregulation of nCDase was evaluated by western blot as well as levels of β-catenin and phosphorylation of GSK3β on serine 9.
C: HCT116 cells were treated for 24 hours with the indicated concentrations of C6 urea-ceramide or with vehicle. An aliquot of intact cellular extract was set aside. The remainder was incubated with GSK3β antibody overnight and then incubated with protein A-agarose beads and precipitate. Intact cellular extract (Input) as well as the immunoprecipitate (IP:GSK3β) were analyzed by western blot for GSK3β, β-catenin and AXIN.
D: HCT116 cells were transfected for 24 hours with GSK3β or with control siRNA and then treated with (+) or without (−) C6 urea-ceramide (10 μM). Efficiency of siRNA downregulation of GSK3β was evaluated by western blot as well as levels of β-catenin.
E: HCT116 cells were pretreated for 6h with the indicated concentrations of LiCl. Cells were then treated with 10 μM of C6 urea-ceramide for 24h. The level of β-catenin as well as the phosphorylation of GSK3β on serine 9 was measured by western blot
In order to determine if the β-catenin destruction complex was involved in our model, we explored the hypothesis that GSK3β inhibition was the driving event for β-catenin degradation. GSK3β has been well established as a key upstream regulator of β-catenin and, in line with this, our results showed that inhibition of nCDase increased phosphorylation of β-catenin on serine 33/37 12. Next, the association of GSK3β with β-catenin was investigated. The cells were treated with 5 μM of C6 urea-ceramide, and then the immune-precipitate for GSK3β, total cellular extract (input), and immune precipitate (IP:GSK3β) were analyzed by western blot (Figure 1C). The results showed an increase in the association of β-catenin with GSK3β under nCDase inhibition, suggesting that the effects of nCDase inhibition involve the Wnt/β-catenin pathway. Interestingly, under nCDase inhibition, we also observed an increase in the association of GSK3β with Axin.
To further investigate this mechanism, cells were pretreated with a specific siRNA targeting GSK3β (Figure 1D) or with the GSK3β inhibitor lithium chloride (Figure 1E) and then treated with C6 urea-ceramide. The results showed that the decrease of β-catenin level induced by inhibition of nCDase was abolished in response to GSK3β inhibition. These results establish a key role for GSK3β in mediating the effects of nCDase inhibition on β-catenin.
nCDase inhibition induces a decrease of β-catenin level via phosphorylation of AKT upstream of GSK3β
The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway is a central regulatory network involved in many essential pathways for cell signaling. These pathways involve regulation of transcription, cell survival 18, metabolism 19, differentiation 20, growth 21, migration 22, and angiogenesis 23. Dysregulation of this pathway has been observed in several human cancers24–26.
To define the upstream elements of this pathway, we hypothesized that AKT, a known regulator of GSK3β phosphorylation on serine 9, was the upstream element of this pathway. To this end, HCT116 cells were incubated with C6 urea-ceramide (Figure 2A) or with a siRNA targeting nCDase (Figure 2B). AKT phosphorylation was evaluated by western blotting. The results show that nCDase inhibition induced a decrease of AKT phosphorylation on both serine 473 and Thr308. These results connect the nCDase pathway with regulation of AKT.
Figure 2. nCDase inhibition induces a decrease of β-catenin level via phosphorylation of AKT upstream of GSK3β.
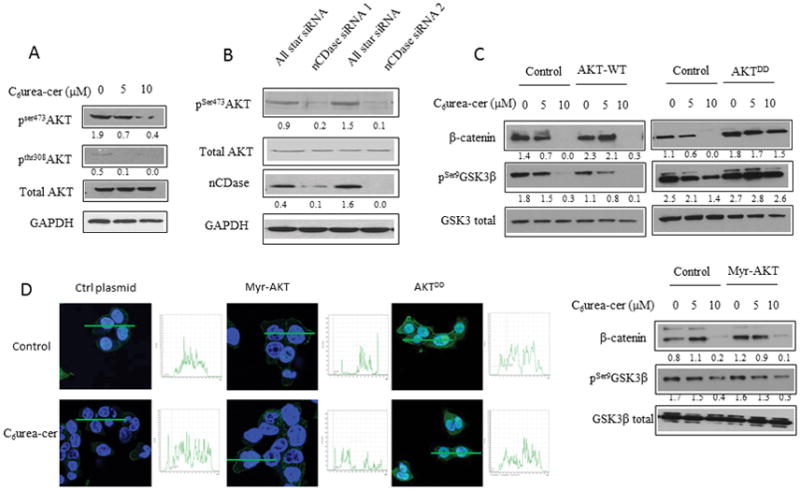
A: HCT116 cells were treated for 24 hours with the indicated concentrations of C6 urea-ceramide or with vehicle. Phosphorylation of AKT on serine 473 and threonine 308 were measured by western blot.
B: Cells were transfected twice for 48 hours with nCDase siRNA or with control siRNA. Phosphorylation of AKT on serine 473 was measured by western blot.
C: HCT116 Cells were transfected with 1μg of a plasmid encoding for a phospho-mimic constitutively active AKT (AKTDD), a wild type AKT (AKT-WT) or a myristoylated AKT (Myr-AKT) for 24 hours. Cells were treated with C6 urea-ceramide for an additional 24 hours (C6Ur-Cer) at the indicated concentrations. The total amount of β-catenin as well as phosphorylation of GSK3-β on serine 9 were measured by western blot.
D: HCT116 Cells were seeded on a glass coverslip and transfected with the indicated plasmid and then treated with C6 urea-ceramide or vehicle at the indicated concentrations for 24 hours. Cells were fixed 20 minutes in formalin, and the total AKT was detected by immunofluorescence (green). Nuclei were stained with DAPI (blue). Quantification is shown in the inset next to each image.
In order to determine if AKT functions upstream of GSK3β and β-catenin in the nCDase pathway, we evaluated the ability of a phospho-mimic mutant of AKT (AKTDD), a myristoylated mutant of AKT (myr-AKT), and the wild type (WT) protein to overcome the effects of inhibition of nCDase. Basally, AKT is localized in the cytoplasm. The AKTDD construct is also mainly localized in the cytoplasm, however myr-AKT is basally present at the plasma membrane (see quantification in the inset of Figure 2D). The results showed that transfection of the cells with the phospho-mimic mutant of AKT (AKTDD) prevented both β-catenin degradation and the decrease in GSK3β phosphorylation (Figure 2C). Interestingly, and unexpectedly, the myristoylated mutant of AKT had no effect on β-catenin degradation nor did it prevent the decrease in phosphorylation of GSK3β under nCDase inhibition.
These latter results prompted us to evaluate AKT localization in presence or absence of C6 urea-ceramide by immunofluorescence (Figure 2D). The results showed no modification of the fluorescence pattern suggesting that nCDase was not able to modulate AKT localization. Taken together, these results define a critical role for nCDase in regulating the activation/phosphorylation of AKT but not its membrane association and that AKT functions as an upstream node in mediating the effects of nCDase inhibition on GSKβ and β-catenin.
Phosphorylation of AKT is sufficient to mediate nCDase downstream effects on growth
As shown previously, inhibition of nCDase induces a decrease of cell proliferation and an increase in apoptosis 27. To determine if the effects of nCDase on AKT are critical in mediating these effects, cells were transfected with the construct expressing the phospho-mimic mutant of AKT (Figure 3). The results showed that C6 urea-ceramide induced a decrease of viable cells as monitored by MTT whereas the phospho-mimic mutant of AKT blocked this effect (Figure 3A). The attached viable cells were also counted after C6 urea-ceramide, and the results showed that the phospho-mimic mutant of AKT blocks the decrease of viable cells under nCDase inhibition (Supplemental Figure 2). These results were further confirmed by demonstrating that the expression of the phopho-mimic of AKT prevented the effects of C6 urea-ceramide on caspase 3 cleavage (Figure 3B) and caspase 3 activity (Figure 3C). As previously demonstrated by Garcia Barros et al, C6 urea-ceramide induced an increase of autophagy measured by LC3I to LC3II conversion 12. Therefore we tested the role of AKT phosphorylation on autophagic flux (Figure 3D), and the results showed an increase of autophagic flux using Bafilomycin A1 with no effect on AKTDD. Interestingly, the myristoylated mutant of AKT did not prevent the decrease of viable cells as monitored by MTT or by cell counting. Thus, these results demonstrate that AKT functions as a key mediator of the effects of nCDase on downstream biology.
Figure 3. Role for AKT in mediating the effects of nCDase on cell growth and apoptosis.
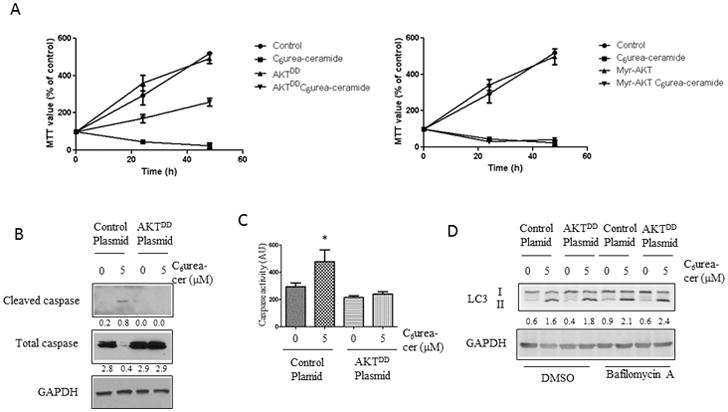
A: HCT116 cells were transfected with 1 μg of AKTDD plasmid, Myr-AKT plasmid or control plasmid. After 24 hours, cells were either treated with 5 μM of C6 urea-ceramide or allowed to remain untreated. After a time course of 0, 24,48h, cells were incubated with 1 mg/ml of MTT for 2 hours and OD was measured at 595 nm. Each measure was done in triplicate, results are presented as mean +/− SEM.
B: HCT116 cells were transfected with 1 μg of AKTDD plasmid or control plasmid. The next day, cells were treated with 5 μM of C6 urea-ceramide or allowed to remain untreated. Caspase 3 cleavage was analyzed by western blot.
C: HCT116 cells were transfected with 1 μg of AKTDD plasmid or control plasmid. The next day, cells were treated with or without 5 μM of C6 urea-ceramide. Caspase-3/7 activity was determined using BioVision™ caspase kit as described in the Materials and Methods and expressed as arbitrary units (AU). The data are a mean ± SEM of 3 independent experiments performed in triplicate. *: p<0.01
D: HCT116 cells were transfected with 1 μg of AKTDD plasmid or control plasmid. The next day, cells were treated with or without 5 μM of C6 urea-ceramide. LC3I to LC3II conversion was analyzed by western blot.
nCDase inhibition does not modulate PI3K activation
To determine if targets upstream of AKT were modulated by nCDase inhibition, we evaluated the ratio of PIP3/PIP2 in HCT116 cells treated with C6 urea-ceramide. Our results showed that the ratio of PIP3/PIP2 was unaffected by nCDase inhibition whereas EGF treatment showed a 25% increase in the PIP3/PIP2 ratio (Figure 4A). To confirm this result, cells were transfected with a plasmid coding for the PH domain of AKT fused to the GFP protein. After 24 hours, the cells were treated for 24 hours with C6 urea-ceramide at the indicated concentrations. As a positive control, we treated transfected cells with EGF for 2 minutes to show conversion of PIP2 to PIP3. The results showed that EGF treatment induced an increase of staining at the plasma membrane (Figure 4B), suggesting a conversion of PIP2 to PIP3. No effect was observed on cells treated with C6 urea-ceramide. These results suggest that nCDase inhibition does not modulate PIP3/PIP2 balance.
Figure 4. Neutral ceramidase inhibition does not modulate PI3K activation.
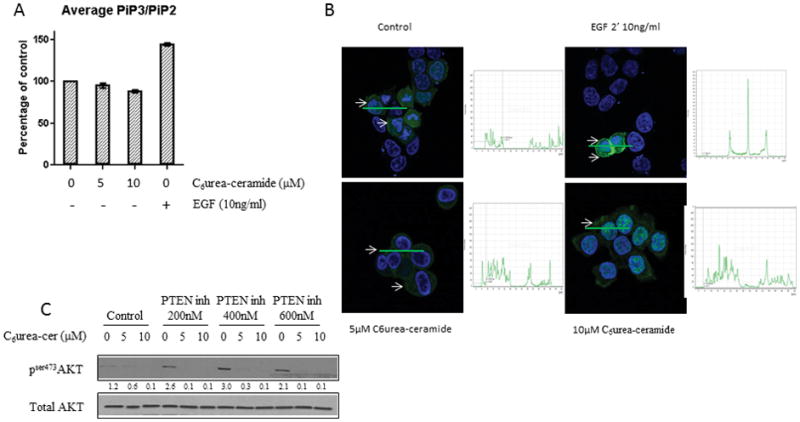
A: HCT116 cells were treated with the indicated concentration of C6 urea-ceramide, cell pellets were frozen, and PIPs were measure by mass spectrometry, results are presented as a mean +/− SEM.
B: HCT116 cells were seeded on a glass coverslip and transfected with a plasmid coding for eGFP fused to the PH domain of AKT and then treated with C6 urea-ceramide or vehicle at the indicated concentration for 24 hours. EGF treatment for 2 minutes at 10ng/ml was used as a positive control of the conversion of PIP2 in PIP3. Cells were fixed for 20 minutes in formalin, and GFP was detected by immunofluorescence (green). Nuclei were stained with DAPI (blue). White arrows: membrane staining. Quantification is shown in the inset next to each image.
C: HCT116 cells were pretreated for 6 hours with the indicated concentrations of VO-OHpic. Cells were then treated with 5 or 10 μM of C6 urea-ceramide for 24 hours. Phosphorylation of AKT on serine 473 was measured by western blot.
AKT activation in the PI3K dependent pathway is also modulated by PTEN. We hypothesized that PTEN was the phosphatase involved in the inhibition of AKT in response to C6 urea-ceramide. Therefore the cells were pretreated with the PTEN inhibitor VO-OHpic at the indicated concentration and then treated with C6 urea-ceramide. The results showed that PTEN inhibition increased basal phosphorylation of AKT, but nCDase inhibition was still able to induce a major decrease of AKT phosphorylation (Figure 4C). These results, taken together, demonstrate that the effects of nCDase on AKT are independent of PI3K.
Decrease of cell growth of CRC cells is strongly associated with effects on β-catenin, GSK3β, and AKT phosphorylation
To investigate whether nCDase-dependent activation of: the wnt/β-catenin pathway, AKT and GSK3β represent a shared mechanism for regulating growth of colon cancer cells, we choose 10 colorectal cancer cell lines that share the most common mutations associated with colorectal tumorigenesis. A summary of these mutations is shown in Supplementary Table 1. We treated 10 different CRC cell lines with C6 urea-ceramide and evaluated cell growth via MTT incorporation, β-catenin level, phosphorylation of GSK3β on serine 9 and phosphorylation of AKT on serine 473. Figure 5A correlates the percentage of β-catenin levels remaining after treatment with the percentage of MTT. This result revealed two groups of cell lines. The first group (HT29, HCT116, T84, HCA-7, SW837 and SW480) demonstrated an approximately 50 percent decrease of β-catenin associated with a greater than 80 percent decrease of the MTT value, similar to what was observed in HCT116. In the other cell lines (RIE-1, SW620 and CaCoBBe), there was little modulation of β-catenin level or MTT incorporation upon nCDase inhibition.
Figure 5. Decrease of cell growth of CRC cells is strongly associated with effects on β-catenin, GSK3β, and AKT phosphorylation.
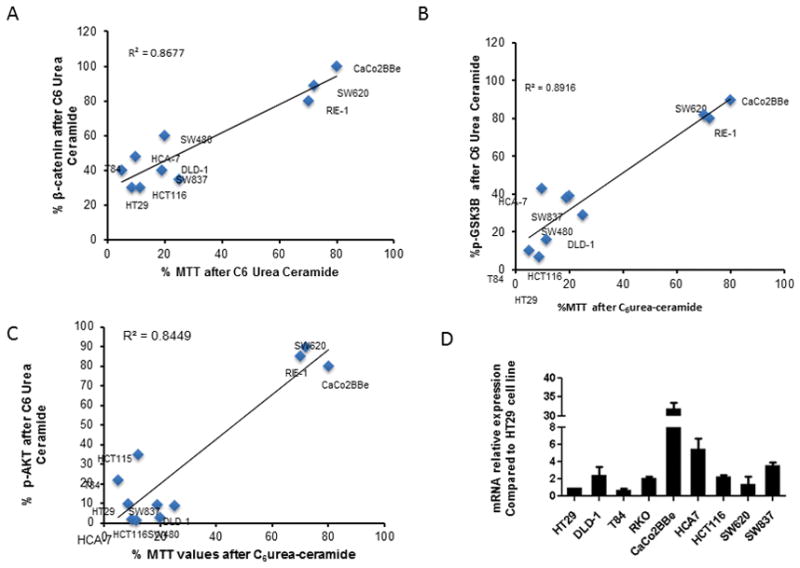
Ten different colon and rectal cancer cell lines were treated with or without 10 μM of C6 urea-ceramide and the effects on MTT were measured. Experiments were performed in triplicate. Treated and control cells were also evaluated for β-catenin levels, as well as total AKT, phosphorylation of AKT on serine 473, total GSK3β and phosphorylation of GSK3β on serine 9.
A: The graph shows for each cell line the percentage of total level of β-catenin after C6 urea-ceramide treatment as a function of the percentage of MTT after C6 urea-ceramide.
B: The graph shows for each cell line the percentage of GSK3β phosphorylation after C6 urea-ceramide treatment as a function of the percentage of MTT after C6 urea-ceramide.
C: Graph shows for each cell line the percentage of AKT phosphorylation after C6 urea-ceramide treatment as a function of the percentage of MTT after C6 urea-ceramide.
D: Graph bars show for each cell line relative mRNA expression of nCDase compared to HT29.
A similar analysis was conducted on the correlation between the decrease of MTT and the percentage of GSK3β phosphorylation in treated cells compared to untreated controls (Figure 5B). Interestingly, the cell lines studied presented the same two groups: a decrease of GSK3β phosphorylation associated with a decrease of MTT in HT29, HCT116, T84, HCA-7, SW837 and SW480 as well as DLD-1 whereas RIE-1, SW620 and CaCoBBe did not modulate GSK3β phosphorylation or MTT in response to inhibition of nCDase.
Nearly identical results were obtained in the analysis of AKT phosphorylation compared to MTT compared to untreated controls (Figure 5C). Given the results from Figure 3 showing that AKT plays a key role in mediating the effects of inhibition of nCDase on growth, these results support a more general role for AKT in various colon cancer cells. Original western blots for this figure are presented in supplemental Figure 3.
We hypothesized that these correlations were associated with a modulation of the expression of nCDase. We therefore evaluated the expression level of nCDase mRNA by RT-QPCR. We were able to detect nCDase mRNA expression in all cell lines. However, the results in Figure 5D showed no link between nCDase expression and MTT value, β-catenin level, AKT phosphorylation or GSK3β phosphorylation.
Phosphorylation of AKT is sufficient to reverse the effects of inhibition of nCDase on HT116 xenografts
Due to the cell studies indicating an important role of AKT in colon cancer cell survival and in mediating the effects of nCDase inhibition, in vivo effects of nCDase inhibition were evaluated next in a colon cancer xenograft animal model. HCT116 cells were transfected with the construct expressing the phospho-mimic mutant of AKT or with a control construct and then were implanted subcutaneously into Nu/J mice. After solid tumors were established (200–250 mm3), animals implanted with the two different cell lines were distributed randomly into 2 different groups each and received daily C6 urea-ceramide treatments (10 mg/kg) or vehicle control (i.p.) for 6 consecutive days. Figure 6A shows that inhibition of nCDase induced growth delay of control xenograft tumor, decreasing tumor volume by 42% at the end of 6 days whereas nCDase inhibition had no effect on the growth of xenograft tumors from cells expressing the phospho-mimic mutant of AKT. Figure 6B shows the over expression of AKT in tumors expressing the phospho-mimic of AKT (right panel) compared to control tumors (left panel). Proliferation was then assessed with 5-Bromo-2-Deoxyuridine (BrdU) immunostaining. Figure 6C shows representative fields of HCT116 control and HCT116 phospho-mimic mutant of AKT xenografts untreated vs. treated with C6 urea-ceramide (positive cells are stained brown). Tumors from HCT116 control cells treated with C6 urea-ceramide had significantly less BrdU-positive cells per mm2 (from 41±4.0 to 19±1.8 positive cells per mm2 per mouse in each group, p<0.0001)) as shown in figure 6D. No changes in animal weight were observed between groups during these studies (data no shown).
Figure 6. Reversal of the effects of nCDase inhibition on HCT116 xenografts by the phospho-mimic mutant of AKT.
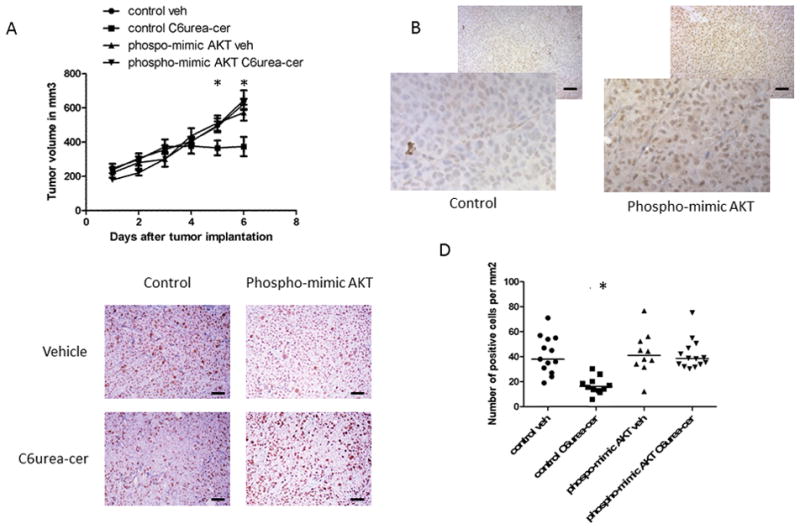
A: Growth pattern of HCT116 xenografts after treatment with C6 urea-ceramide. HCT116 cells (5×106 cell resuspended in PBS) were injected (s.c.) into the nude mice right hind limb and treated with C6 urea-ceramide daily (vehicle, 10 mg/kg, i.p.) for six consecutive days when tumors reached 200 mm3. Tumor volume, measured with calipers, was calculated daily. At least six mice were used in each group, results are presented as the mean +/− SD.
B: Expression of AKT in xenograft tumor implanted in nude mice. Representative 5 μm histologic xenograft sections AKT expression was detected by immunohistochemistry with an antibody specific for AKT.
C: Proliferation of HCT116 xenografts treated with either control vehicle or with C6 urea-ceramide assessed by BrdU staining. Representative 5 μm histologic xenograft sections were subject to BrdU detection by immunohistochemistry. Bar :50μm.
D. Quantification of positive BrDu cells. Positive cells (brown) were quantified manually, and data represent positive cells per mm2 per mouse (5 control; 6 treated mice). At least 20 random fields were evaluated for each mouse. Bar :50μm.
DISCUSSION
The results from this study reveal a key role for nCDase in CRC as well as the key mechanisms by which nCDase regulates growth. Molecular and pharmacologic inhibition of nCDase induces a decrease in β-catenin levels as recently shown by Garcia Barros et al12. Here we demonstrate that this decrease was promoted by a decrease in phosphorylation of GSK3β. In exploring the pathway leading to the degradation of β-catenin, we determined that phosphorylation of AKT was also decreased with inhibition of nCDase. Importantly, the results demonstrate that AKT dephosphorylation was necessary to decrease β-catenin levels and to decrease GSK3β phosphorylation in the setting of nCDase inhibition. Furthermore, the results demonstrate that a phospho-mimic mutant of AKT was able to partially reverse the effects of nCDase inhibition on cell growth, caspase cleavage, and xenograft growth.
Taken together, the results from this study define a specific pathway linking nCDase to AKT to GSK3β and to β-catenin (Figure7). It is known that AKT inactivates GSK3β by inducing its phosphorylation. In this state, phospho GSK3β is unable to phosphorylate β-catenin, thus leading to persistent activation of the latter, which is a major driving force in the pathogenesis of CRC. The current results show that inhibition of nCDase pharmacologically or molecularly reverses these effects, thus placing AKT, GSK3β and β-catenin downstream of the action of nCDase. This is buttressed by demonstrating that a phospho-mimic mutant of AKT is able to overcome the effects of inhibition of nCDase on GSK3β and β-catenin. These results also point to an important, and somewhat unappreciated, role for AKT in the basal operation of the GSK/β-catenin pathway.
Figure 7. Proposed model of regulation of AKT dependent CRC cell growth by nCDase.
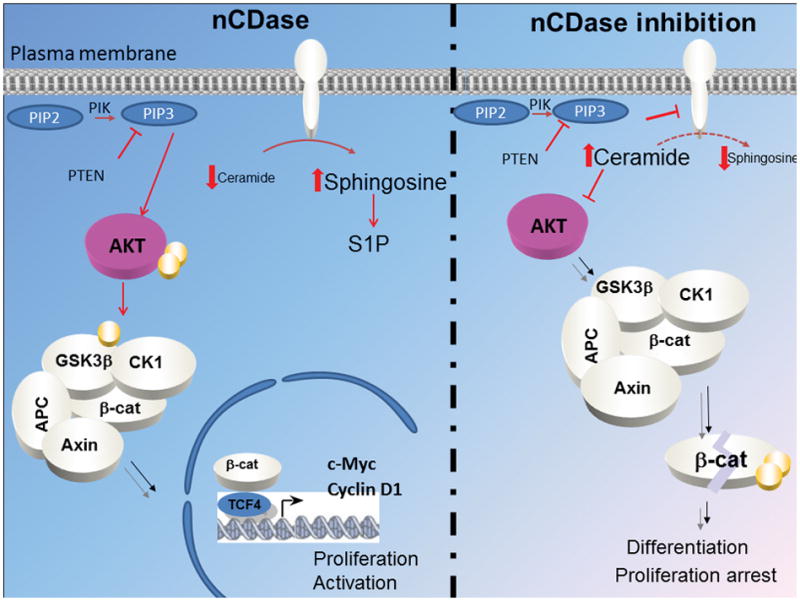
Ceramidase activates the phosphorylation of AKT on serine 473 and threonine 308, through attenuation of the inhibitory action of ceramide. Thus activated, AKT then drives the phosphorylation of GSK3β on serine 9, which inhibits the kinase and prevents it from phosphorylating β-catenin, thus maintaining β-catenin in an active form that that drives cell growth. CK1, casein kinase; APC, Adenomatous polyposis coli; TCF4, Transcription factor 4.
Another major implication from the current results is that the presence of nCDase in a constitutively active form is necessary for optimal function of the AKT/GSK3β/β-catenin pathway. This is a previously unrecognized role for nCDase, and it demonstrates a close coupling between nCDase and AKT that in turn would have implications to our understanding of the repertoire of mechanisms that regulate this enzyme. Clearly, in CRC cells, maintenance of basal activity of AKT requires the presence and activity of nCDase.
Moreover, this conclusion significantly expands the connection of ceramide to the AKT pathway. Previous studies have shown that exogenous ceramides or induction of endogenous ceramide by palmitate supply can regulate AKT, with roles in the metabolic syndrome and obesity 28–31. The current results add a new dimension to the regulation of AKT by ceramide by demonstrating that endogenous ceramide may serve to attenuate activation of AKT and that nCDase has a key function in controlling that function of ceramide.
Mechanistically, it has been proposed that ceramide can inhibit AKT via either dephosphorylation through protein phosphatase 2A activation 32, 33 or by preventing AKT translocation to the plasma membrane via PKC activation 34, 35 The current results demonstrate that endogenously present ceramide can regulate AKT phosphorylation, but not translocation, and that the phospho-mimic, but not the spontaneously translocating AKT, can overcome the ceramide effects.
Multiple studies have suggested a functional role for bioactive sphingolipids in colon cancer. More specifically, sphingolipid based analogues, such as enigmol and sphingadienes, have been proposed to inhibit cancer cell proliferation in a APCmin/+ model decreasing the number of intestinal tumors by half 36, 37. Furthermore, work on human CRC samples also suggests that colon cancer may be associated with decreased ceramide and increased S1P 38. It has also been demonstrated that human colon cancer samples analyzed from a colorectal tissue microarray present 89% less SK1 compared with adjacent normal colon mucosa 39. Interestingly, SK1 has been implicated in inflammation induced CRC 39 whereas nCDase does not share this function40. Thus, these two enzymes appear to target two distinct mechanisms of CRC pathogenesis. In vivo nCDase appears to act on both ceramide in the diet and intracellular ceramide 11. In our model, we suspect the source of ceramide is from activation of sphingomyelinases, but do not yet have proof of this.
From a therapeutic point of view, our results have important implications for CRC treatments by targeting a ceramide-metabolizing enzyme. Current treatment for advanced stage colon cancer focuses on surgical resection often followed by adjuvant chemotherapy with fluorouracil +/− oxiplatinin41, 42. Studies have shown that in patients with stage III colon cancer, there can be as much as a 30 percent reduction in colon cancer recurrence in patients who receive a course of adjuvant chemotherapy as compared to resection alone42. Unfortunately, these therapies can cause significant toxicity to the patient while trying to address the underlying cancer. Moreover, there are only a few limited options for treatment of advanced CRC, mostly with cytotoxic agents and not with mechanism-directed therapies. Multiple trials have evaluated the use of VEGF or EGFR blocking antibodies as targeted adjuvant therapies 43, 44. The long term follow-up data showed no difference in disease free survival in those trials45. Our work creates a new potential pathway for ceramidase inhibition as a novel target for therapy, providing increased chemotherapeutic options for colon cancer, either in adjuvant or therapeutic modality. Recently, the structure of nCDase was solved at 2.6 Å, and the results revealed a catalytic domain, with a narrow, 20 Å deep, hydrophobic pocket with a Zn2+ ion at its base, which suggests a mechanism of action with a transition state and a general acid-base catalysis 46. These evolving insights on the role of nCDase, its mechanism and structure should propel future therapeutic development.
In conclusion, we identify a novel role for nCDase in CRC pathogenesis. This new pathway encourages the development of nCDase as a novel target for colon cancer treatment. Its tissue specificity and unique structure renders nCDase a significant candidate for a targeted approach with little side effects.
MATERIALS AND METHODS
Reagents
Cell culture medium was obtained from Invitrogen (Carlsbad, CA) and fetal bovine serum from Hyclone Thermo (Waltham, MA. #SH300070.03). Antibodies for β-catenin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). LC-3 antibody (NB100-2220) was purchased from Novus biological (Littleton, CO).
GAPDH rabbit mAb (#2118), caspase-3 rabbit mAb (#9665), p-GSK3β (Ser9) rabbit antibody (#5558), GSK3β rabbit antibody (#9315), p-AKT (Thr308) rabbit antibody (#9275), p-AKT (Ser473) rabbit antibody (#4060), AKT rabbit antibody (#9272), Cleaved caspase 3 rabbit antibody (#9664) and caspase 3 rabbit antibody (#9662) were all obtained from Cell Signaling (Beverly, MA).
nCDase antibody (Ab174) was a kind gift from Dr. Rick Proia (Genetics of Development and Disease, NIDDK, Bethesda, MD) 11. The enhanced chemiluminescence kit (ECL) was from ThermoScientific (Rockford, IL). C6 urea-ceramide was provided by the Lipidomics Core facility at MUSC.
Cell culture
Colon cancer cells (HT29, HCT116, T84, SW837, SW480, SW620, DLD-1, HCA-7 and CaCo2BBe) were from ATCC (Manassas, VA) and cultured as recommended. RIE-1 cells were obtained from K.D. Brown (Babraham Institute, Cambridge, UK) and maintained in Dulbecco’s modified Eagle’s supplemented with 10% fetal bovine serum. All cell lines were grown in a 5% CO2 incubator at 37 °C. Mycoplasma tests were performed monthly for each cell line, We include in the supplemental material STR verification of the HCT116 cells.
siRNA transfection
100 000 cells were cultured in 35-mm dishes. After 24 hours, cells were transfected with 20 nM of ASAH2 siRNA (Hs_ASAH2_7 Qiagen, Hilden, Germany and AM16708 Ambion Waltham, MA) or with negative control siRNA (AllStars: Qiagen) using Lipofectamine RMAiMax according to the manufacturer’s instructions (Invitrogen, Carsbad, CA). After 24 hours, cells were trypsinized and seeded at a 1/5 dilution and a day later cells were transfected again with 20 nM of nCDase siRNA or with control siRNA as described previously. Cells were then collected and analyzed as indicated.
Plasmid transfection
Cells were seeded in 35-mm dishes (100 000 cells). After 24 hours, cells were transfected with 1μg of pcDNA3 Myr HA Akt1 (Addgen Cambridge, MA, #9008), Inactive Myr HA Akt1;K179M or pcDNA3-HA: HA-PKB T308D/S473D (Addgen #14751) using Extreme Gene 9-1ml (Sigma, Saint Louis, MO # 6365787001) according to the manufacturer’s instructions. The next day, cells were then treated, collected and analyzed as indicated.
Mass spectrometry measurements of PiPs
Mass spectrometry was used to evaluate PiPs amounts as previously described 47, and as modified by Anderson et al 48. Measurements were done in duplicate per experiment.
Protein extraction and immunoblot analysis
Protein extracts from cells were obtained after harvesting in RIPA buffer (with PMSF, orthovanadate and protease inhibitors) from Santa Cruz Biotechnology. Proteins were sonicated and concentrations was measured using a Bradford Protein Assay. Proteins were separated by SDS-PAGE using the Criterion system (BioRad) and transferred to nitrocellulose membranes. Primary antibodies (1:1 000 dilution) were incubated overnight at 4 °C and horseradish peroxidase-conjugated secondary antibodies (1:5 000 dilution) were incubated for 1 h at room temperature. Immunoreactive proteins were visualized by chemiluminescence via ECL reagent #32106 (Thermo Scientific) on autoradiograph film (LabScientific Highlands, NJ, # XAR ALF 2025).
MTT assay
Methylthiazolyldiphenyl-tetrazolium bromide (MTT) was purchased from Sigma (#M5655). Cells were treated with MTT (1 mg/ml) for 1 h and supernatants were discarded. DMSO was added and absorbance was measured at 560 nm using a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA). Each experimental condition tested was performed in triplicate.
Apoptosis
Caspase-3 activity was measured in HCT116 cells after treatment with C6 urea-ceramide at different doses and times, as specified, using a Caspase-3 Fluormetric Assay Kit from BioVision (#K105-100, Milpitas, CA) according to the manufacturer’s instructions. Results are expressed as fluorescence activity per μg of protein.
Bright field and confocal microscopy
HCT116 cells were treated with nCDase inhibitor at the indicated doses, and immunofluorescent analysis was performed as described previously49. Samples were evaluated using a LSM510 confocal microscope (Carl Zeiss, Inc., Oberkochen, Germany). Photos were obtained and analyzed using Leica software. Bright field microscopy was perform using Carl Zeiss Axio Imager 2 AxioVision and analyzed using axiovision software.
Mice
Six to eight-week-old athymic male nude mice (Nu/J) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were maintained in accordance with the regulations and standard of the U.S. Department of Agriculture and the Department of Health and Human Services. Mice were fed regular chow (PicoLab® Rodent Diet from LabDiet) and had free access to food and water.
Xenograft implantation and C6 urea-ceramide treatment
Stable HCT116 colon cancer cells expressing pcDNA3-HA: HA-PKB T308D/S473D were maintained as indicated, and 5×106 cells were transplanted subcutaneously (s.c.) into right limbs of Nu/J mice referred to as Nude mice. After tumors reached 200–250 mm3, mice were treated with C6 urea-ceramide dissolved in 20% Cremophor EL/80% normal saline at 10mg/kg intraperitoneally (i.p.) over six consecutive days. Tumor volume was measured every day using a caliper and the volume was calculated according Kim et al., 50.
BrdU Staining
Nine mice were injected with BrdU (100 μg/g, i.p.) (Sigma-Aldrich) 2 hours before euthanasia. Immunohistochemistry was performed using an anti-BrdU antibody (Sigma, #B8434) on hydrated sections. Secondary biotinylated anti-mouse antibody and streptavidin-HRP (Sigma) were quantified with a Vectastain ABC kit (Vector Laboratories Inc., Burlingame, CA). Nuclei were visualized with hematoxylin. Data means were compiled from at least 20 random high-resolution fields (10x) from each mouse. Five mice were evaluated per group and the experimenter was blinded during analysis.
Statistical Analysis
Statistical analysis was performed using one-way ANOVA or a Student’s t-test for staining and treatment effects.
Study approval
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Stony Brook University.
Supplementary Material
Acknowledgments
This work was supported by NCI grants CA172517 and CA97132.
We acknowledge the support from the “Research Histology Core Laboratory, Department of Pathology, Stony Brook University”. We would like to thank Ayanna Lewis, MD for proof reading the manuscript.
Sources of support for the work: NCI grants CA172517 and CA97132.
Footnotes
CONFLICT OF INTEREST:
The authors have no conflicts of interest to disclose.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Fakih MG. Metastatic colorectal cancer: current state and future directions. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:1809–1824. doi: 10.1200/JCO.2014.59.7633. [DOI] [PubMed] [Google Scholar]
- 2.Stewart B, Wild C. World Cancer Report 2014. Vol. 1. International Agency for Research on Cancer; Lyon, France: 2014. p. 619. [PubMed] [Google Scholar]
- 3.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2016;25:16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 4.Yeang CH, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22:2605–2622. doi: 10.1096/fj.08-108985. [DOI] [PubMed] [Google Scholar]
- 5.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nature reviews Molecular cell biology. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 6.Hannun YA, Obeid LM. Many ceramides. J Biol Chem. 2011;286:27855–27862. doi: 10.1074/jbc.R111.254359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikolova-Karakashian MN, Rozenova KA. Ceramide in stress response. Advances in experimental medicine and biology. 2010;688:86–108. doi: 10.1007/978-1-4419-6741-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitatani K, Idkowiak-Baldys J, Hannun YA. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cellular signalling. 2008;20:1010–1018. doi: 10.1016/j.cellsig.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO reports. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta. 2008;1781:424–434. doi: 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, et al. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem. 2006;281:7324–7331. doi: 10.1074/jbc.M508382200. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Barros M, Coant N, Kawamori T, Wada M, Snider AJ, Truman JP, et al. Role of neutral ceramidase in colon cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2016 doi: 10.1096/fj.201600611R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends in biochemical sciences. 2010;35:161–168. doi: 10.1016/j.tibs.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Developmental cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 16.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg KCG, Eide PW, Eilertsen IA, Johannessen B, Bruun J, Danielsen SA, et al. Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Molecular cancer. 2017;16:116. doi: 10.1186/s12943-017-0691-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 19.Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends in endocrinology and metabolism: TEM. 2010;21:268–276. doi: 10.1016/j.tem.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Muller EJ, Williamson L, Kolly C, Suter MM. Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol. 2008;128:501–516. doi: 10.1038/sj.jid.5701248. [DOI] [PubMed] [Google Scholar]
- 21.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes & development. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki AT, Firtel RA. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. European journal of cell biology. 2006;85:873–895. doi: 10.1016/j.ejcb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Current cancer drug targets. 2008;8:19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 24.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer metastasis reviews. 2009;28:305–316. doi: 10.1007/s10555-009-9198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (review) International journal of oncology. 2012;40:639–644. doi: 10.3892/ijo.2011.1312. [DOI] [PubMed] [Google Scholar]
- 27.Wu BX, Zeidan YH, Hannun YA. Downregulation of neutral ceramidase by gemcitabine: Implications for cell cycle regulation. Biochim Biophys Acta. 2009;1791:730–739. doi: 10.1016/j.bbalip.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- 29.Rahman A, Thayyullathil F, Pallichankandy S, Galadari S. Hydrogen peroxide/ceramide/Akt signaling axis play a critical role in the antileukemic potential of sanguinarine. Free radical biology & medicine. 2016;96:273–289. doi: 10.1016/j.freeradbiomed.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. The Journal of clinical investigation. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoica BA, Movsesyan VA, Lea PMt, Faden AI. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Molecular and cellular neurosciences. 2003;22:365–382. doi: 10.1016/s1044-7431(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 32.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–15530. [PubMed] [Google Scholar]
- 33.Oaks J, Ogretmen B. Regulation of PP2A by Sphingolipid Metabolism and Signaling. Frontiers in oncology. 2014;4:388. doi: 10.3389/fonc.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halder K, Banerjee S, Bose A, Majumder S, Majumdar S. Overexpressed PKCdelta downregulates the expression of PKCalpha in B16F10 melanoma: induction of apoptosis by PKCdelta via ceramide generation. PloS one. 2014;9:e91656. doi: 10.1371/journal.pone.0091656. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Powell DJ, Hajduch E, Kular G, Hundal HS. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Molecular and cellular biology. 2003;23:7794–7808. doi: 10.1128/MCB.23.21.7794-7808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Symolon H, Bushnev A, Peng Q, Ramaraju H, Mays SG, Allegood JC, et al. Enigmol: a novel sphingolipid analogue with anticancer activity against cancer cell lines and in vivo models for intestinal and prostate cancer. Molecular cancer therapeutics. 2011;10:648–657. doi: 10.1158/1535-7163.MCT-10-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar A, Pandurangan AK, Lu F, Fyrst H, Zhang M, Byun HS, et al. Chemopreventive sphingadienes downregulate Wnt signaling via a PP2A/Akt/GSK3beta pathway in colon cancer. Carcinogenesis. 2012;33:1726–1735. doi: 10.1093/carcin/bgs174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Barros M, Coant N, Truman JP, Snider AJ, Hannun YA. Sphingolipids in colon cancer. Biochim Biophys Acta. 2014;1841:773–782. doi: 10.1016/j.bbalip.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, et al. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23:405–414. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snider AJ, Wu BX, Jenkins RW, Sticca JA, Kawamori T, Hannun YA, et al. Loss of neutral ceramidase increases inflammation in a mouse model of inflammatory bowel disease. Prostaglandins & other lipid mediators. 2012;99:124–130. doi: 10.1016/j.prostaglandins.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andre T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. The New England journal of medicine. 2004;350:2343–2351. doi: 10.1056/NEJMoa032709. [DOI] [PubMed] [Google Scholar]
- 42.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, et al. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3109–3116. doi: 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 43.Carrato A. Adjuvant treatment of colorectal cancer. Gastrointestinal cancer research : GCR. 2008;2:S42–46. [PMC free article] [PubMed] [Google Scholar]
- 44.Verdaguer H, Tabernero J, Macarulla T. Ramucirumab in metastatic colorectal cancer: evidence to date and place in therapy. Therapeutic advances in medical oncology. 2016;8:230–242. doi: 10.1177/1758834016635888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson VM, Benson AB., 3rd Status of targeted therapies in the adjuvant treatment of colon cancer. Journal of gastrointestinal oncology. 2013;4:245–252. doi: 10.3978/j.issn.2078-6891.2013.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Airola MV, Allen WJ, Pulkoski-Gross MJ, Obeid LM, Rizzo RC, Hannun YA. Structural Basis for Ceramide Recognition and Hydrolysis by Human Neutral Ceramidase. Structure. 2015;23:1482–1491. doi: 10.1016/j.str.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nature methods. 2011;8:267–272. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson KE, Juvin V, Clark J, Stephens LR, Hawkins PT. Investigating the effect of arachidonate supplementation on the phosphoinositide content of MCF10a breast epithelial cells. Advances in biological regulation. 2016;62:18–24. doi: 10.1016/j.jbior.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gandy KA, Canals D, Adada M, Wada M, Roddy P, Snider AJ, et al. Sphingosine 1-phosphate induces filopodia formation through S1PR2 activation of ERM proteins. The Biochemical journal. 2013;449:661–672. doi: 10.1042/BJ20120213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JH, Alfieri AA, Kim SH, Young CW. Potentiation of radiation effects on two murine tumors by lonidamine. Cancer research. 1986;46:1120–1123. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.