

Abstract
Ipomoea L. is the largest genus within the Convolvulaceae and contains 600–700 species. Ipomoea species (morning glories) are economically valuable as horticultural species and scientifically valuable as ecological model plants to investigate mating systems, molecular evolution, and both plant–herbivore and plant–parasite interactions. Furthermore, the dried seeds of I. nil or I. purpurea are used in Korean traditional herbal medicines. In this study, chloroplast (cp) genomes were sequenced from six Ipomoea species, namely, I. nil and I. purpurea and, for the first time, I. triloba, I. lacunosa, I. hederacea, and I. hederacea var. integriuscula. The cp genomes were 161,354–161,750 bp in length and exhibited conserved quadripartite structures. In total, 112 genes were identified, including 78 protein-coding regions, 30 transfer RNA genes, and 4 ribosomal RNA genes. The gene order, content, and orientation of the six Ipomoea cp genomes were highly conserved and were consistent with the general structure of angiosperm cp genomes. Comparison of the six Ipomoea cp genomes revealed locally divergent regions, mainly within intergenic spacer regions (petN-psbM, trnI-CAU-ycf2, ndhH-ndhF, psbC-trnS, and ccsA-ndhD). In addition, the protein-coding genes accD, cemA, and ycf2 exhibited high sequence variability and were under positive selection (Ka/Ks > 1), indicating adaptive evolution to the environment within the Ipomoea genus. Phylogenetic analysis of the six Ipomoea species revealed that these species clustered according to the APG IV system. In particular, I. nil and I. hederacea had monophyletic positions, with I. purpurea as a sister. I. triloba and I. lacunosa in the section Batatas and I. hederacea and I. hederacea var. integriuscula in the section Quamoclit were supported in this study with strong bootstrap values and posterior probabilities. We uncovered high-resolution phylogenetic relationships between Ipomoeeae. Finally, indel markers (IPOTY and IPOYCF) were developed for the discrimination of the important herbal medicine species I. nil and I. purpurea. The cp genomes and analyses in this study provide useful information for taxonomic, phylogenetic, and evolutionary analysis of the Ipomoea genome, and the indel markers will be useful for authentication of herbal medicines.
Keywords: Ipomoeeae, plastid genome, divergent region, phylogenetic relationship, indel marker
Introduction
Chloroplasts (cp) are among the most important organelles in plants, having important roles in photosynthesis and carbon fixation as well as in the biosynthesis of starch, fatty acids, amino acids, and pigments (Jansen and Ruhlman, 2012; Daniell et al., 2016). Cp genomes in higher plants are 120–180 kb and, in general, exhibit a quadripartite structure consisting of two single-copy regions, namely, the large single-copy (LSC) and small single-copy (SSC), as well as two copies of a larger inverted repeat (IR) region. Angiosperm cp genomes generally contain 80 protein-coding genes, 4 ribosomal RNA (rRNA) genes, and 30 transfer RNA (tRNA) genes (Wicke et al., 2011). While the majority of cp genomes exhibit highly conserved structures, some reveal structural variations, IR loss, and gene loss as a result of adaptation to their environments (Delannoy et al., 2011; Wicke et al., 2013). Next-generation technologies have allowed the rapid sequencing of many cp genomes in recent years. These abundant cp genomes have facilitated the verification of evolutionary relationships and allowed detailed phylogenetic classifications to be conducted at group, family, and even genus level in Plantae (Jansen et al., 2007; Parks et al., 2009). Furthermore, cp genomes can be used for species identification through the use of DNA barcodes and molecular markers that allow morphologically similar species to be distinguished (Kim et al., 2015; Park et al., 2017a,b). Thus, cp genomes can be used for practical applications such as species identification as well as for fundamental research into biological processes and evolutionary relationships.
Ipomoea is the largest genus in the Convolvulaceae family, with 600–700 species (Austin and Huáman, 1996; Wilkin, 1999). Ipomoea species are widely distributed across tropical, subtropical, and some temperate regions worldwide (Austin and Huáman, 1996; Wilkin, 1999). I. nil, I. purpurea, I. tricolor, and I. batatas are particularly well-known Ipomoea species. I. nil exhibited spontaneous mutations related to floricultural traits. These mutants have been exploited as ornamental plants in horticulture (Hoshino et al., 2016). Ipomoea is an emerging model system for ecological genomics studies (Baucom et al., 2011; Eserman et al., 2014). Ecological studies of Ipomoea have answered many diverse questions about the Ipomoea mating system, the evolution of floral color pathways, and both plant–herbivore and plant–parasite interactions (Baucom et al., 2011). Visitations by natural pollinators and the selfing rate in various Ipomoea species vary in proportion to the number of offspring derived from self-fertilization. These findings are exemplified by extreme differences in floral color in Ipomoea, which ranges from white or yellow to red or purple (Ennos and Clegg, 1983; Epperson and Clegg, 1992; Baucom et al., 2011). These flower colors (which are associated with anthocyanin pigments) have evolved via parallel evolution due to various factors, such as enzyme-coding genes (F3′H) or regulatory modifications (Des Marais and Rausher, 2008; Streisfeld and Rausher, 2009). Furthermore, I. purpurea and I. hederacea are model plants used to study plant–herbivore interactions based on ecological evolution (Tiffin and Rausher, 1999; Baucom et al., 2011). Several studies show that insects have affected natural selection for plant resistance in Ipomoea and the tradeoff between resistance and tolerance in plant defense responses. In addition, studies of plant–herbivore interactions point to the coevolution between I. purpurea and I. hederacea and their competitors (Rausher and Fry, 1993; Simonsen and Stinchcombe, 2007). Studies examining the evolution of Ipomoea in response to plant pathogens show that quantitative resistance to Colletotrichum dematium is genetically correlated to quantitative resistance to an insect herbivore and that an oomycete exhibits host specialization in Ipomoea (Simms and Rausher, 1993; Sato et al., 2009). Therefore, Ipomoea species represent highly important resources that have contributed strongly to ecological studies.
In Korean traditional medicine, the dried seeds of I. nil or I. purpurea are an important herbal medicine, namely, Pharbitidis Semen, which is used to eliminate toxins or heat, as a diuretic, and as a treatment for constipation relief treatment [Korea Institute of Oriental Medicine (KIOM), 2016]. Pharbitidis Semen is designated as a medicine in Korea, and is regulated by the Ministry of Food and Drug Safety due to its pharmaceutical activity and potential toxicity (Korean Food Standard Codex, 2010). Only seeds of I. nil or I. purpurea are considered to be authentic Pharbitidis Semen. In general, Ipomoea seeds are trigonous and are brown to dark-brown in color (McDonald, 1995), and seeds from different Ipomoea species are morphologically similar and difficult to distinguish with the unaided eye. As a result, Pharbitidis Semen in Korean and Chinese herbal markets often contains a mixture of seeds from I. nil and I. purpurea and seeds from other Ipomoea species. Indiscriminate use of these adulterated Pharbitidis Semen preparations could cause unforeseen side-effects and threaten its use as a safe and reliable medication. Methods are therefore needed to distinguish good quality Pharbitidis Semen preparations from adulterated preparations.
Molecular tools can be used for accurate species identification and authentication of herbal medicine. In particular, the universal DNA barcode markers ITS, matK, and rbcL are widely used for species classification and phylogenetic analysis in Plantae (Semagn et al., 2006; Sucher and Carles, 2008; Chen et al., 2010; Hollingsworth et al., 2011). These barcode markers offer rapid and accurate species identification from short DNA sequences. However, some plants, particularly closely related species, cannot be readily distinguished using these markers. The cp genome has emerged as an alternative to DNA barcoding markers for species identification and phylogenetic studies. Comparison of cp genomes highlighted several variable regions that could be used for the development of markers to allow species discrimination (Kim et al., 2015; Park et al., 2017a). While the cp genome was generally more highly conserved than the nuclear genome, abundant genetic variations such as insertion/deletions (indels) and single nucleotide polymorphisms (SNPs) were identified between species. Several studies developed cp markers for identification of closely related species, including indel and SNP markers for Panax ginseng subspecies (Kim et al., 2015), and indel tandem repeat copy number variation markers for Fagopyrum tataicum and F. esculentum (Cho et al., 2015). In another example, sequence characterized amplified region markers were developed to resolve Aconitum species. Two indel markers derived from large variable regions were used to distinguish three Aconitum species, A. pseudolaeve, A. longecassidatum, and A. barbatum, and a small species-specific 6 bp insertion was used to distinguish A. coreanum (Park et al., 2017a,b). Chenopodium quinoa and C. album were distinguished using indel tandem repeat copy number variation markers (Hong et al., 2017). These examples illustrate the utility of the cp genome for plant species identification and for the authentication and identification of herbal medicines.
Previous phylogenetic analysis of the genus Ipomoea was unclear, with unresolved monophyly at the subgenera level (Manos et al., 2001; Stefanoviæ et al., 2002). Previously, molecular phylogenetic relationships within the Convolvulaceae were evaluated using ITS and four cp loci (Stefanoviæ et al., 2002; Miller et al., 2004), but this analysis identified only monophyletic or weak relationships in tribe Ipomoeeae. A separate analysis of four cp loci divided tribe Ipomoeeae into two clades, Astripomoeinae and Argyreiinae, but morphological features were not considered (Stefanoviæ et al., 2003). Recently, Eserman et al. (2014), described high-resolution phylogenetic relationships in tribe Ipomoeeae and the Astripomoeinae and Argyreiinae clades and identified similar divergence times (23–26 MYA) based on whole cp genomes. Analysis of 32 cp genomes from magnoliids, monocots, and eudicots verified phylogenetic relationships for sweet potato (I. batatas) (Yan et al., 2015). Analysis of the completed nuclear genome identified a whole-genome duplication event in I. nil and showed divergence from Solanaceae at 75.25 MYA (Hoshino et al., 2016). Examination of Ipomoea, which contains hundreds of species, identified a range of useful genomic information, but this was not sufficient for high-resolution determination of phylogenetic relationships in Ipomoeeae. Further research is needed to understand the evolutionary relationships within tribe Ipomoeeae as well as the Ipomoea genus.
Here, samples of herbal medicine species I. nil and I. purpurea and four closely related Ipomoea species with similar seed structures were collected and their cp genomes were compared. This study aimed to (1) characterize six Ipomoea cp genomes and identify genetically variable regions by comparison of their global structures, (2) develop novel molecular markers for use in authentication of herbal medicine species, and (3) understand evolutionary relationships within tribe Ipomoeeae through enhanced phylogenic studies in conjunction with previously reported cp genomes.
Materials and Methods
Plant Materials
Fresh leaves of six Ipomoea species were collected from native habitats in Korea and used for cp genome sequencing. I. nil, I. purpurea, I. hederacea, I. hederacea var. integriuscula, I. lacunosa, and I. triloba were assigned identification numbers, and specimens were registered in the Korean Herbarium of Standard Herbal Resources (Index Herbariorum code KIOM) at the KIOM. The plant samples used for cp genome analysis and indel validation in this study are listed in Supplementary Table S1.
Genome Sequencing and Assembly
DNA was extracted using a DNeasy Plant Maxi Kit (Qiagen, Valencia, CA, United States) according to the manufacturer’s instructions. Illumina short-insert paired-end sequencing libraries were constructed and generated using the NextSeq platform (Illumina, San Diego, CA, United States). De novo assembly was used to construct cp genomes from low-coverage whole-genome sequences. Trimmed paired-end reads (Phred scores ≥20) were assembled using CLC genome assembler (ver. 4.06 beta, CLC Inc., Aarhus, Denmark) with default parameters. SOAP de novo gap closer was used to fill gaps based on alignment of paired-end reads (Luo et al., 2012). Principal contigs representing the cp genome were retrieved from total contigs using Nucmer (Delcher et al., 2003), and aligned contigs were ordered using the cp genome sequence of I. nil (AP017304) as a reference (Hoshino et al., 2016).
Genome Annotation and Comparative Analysis
Gene annotation of the six Ipomoea cp genomes was performed using GeSeq (Tillich et al., 2017), and the annotation results were concatenated using an in-house script pipeline. Protein-coding sequences were manually curated and confirmed using Artemis (Carver et al., 2008), and checked against the NCBI protein database. The tRNAs were confirmed with tRNAscan-SE 1.21 (Lowe and Eddy, 1997). IR region sequences were confirmed using IR finder and RepEx (Warburton et al., 2004; Gurusaran et al., 2013). Circular maps of the six Ipomoea cp genomes were obtained using OGDRAW (Lohse et al., 2007). GC content and relative synonymous codon usages (RSCU) were analyzed using MEGA6 software (Tamura et al., 2013). The mVISTA program in Shuffle-LAGAN mode was used to compare the six Ipomoea cp genomes using the I. nil cp genome as a reference. DnaSP version 5.1 (Librado and Rozas, 2009) was used to calculate nucleotide variability (Pi) among the six Ipomoea cp genomes. Substitution rates Ka and Ks were estimated with PAL2NAL (Suyama et al., 2006). LSC/IR, IR/SSC, SSC/IR, and IR/LSC regions of completed cp genomes were validated using PCR-based sequencing. Primer information and sequence alignment results are listed in Supplementary Tables S2, S3.
Repeat Analysis
SSRs in six Ipomoea cp genomes were detected using MISA (Thiel, 2003) with the minimum number of repeat parameters set to 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. Tandem repeats were ≥20 bp with minimum alignment score and maximum period size of 50 and 500, respectively, and the identity of repeats was set to ≥90% (Benson, 1999).
Phylogenetic Analysis
A total of 38 cp genomes, including 36 from Convolvulaceae, were used for phylogenetic analyses, along with Nicotiana tabacum (GenBank acc. NC_001879.2) and Capsicum annuum var. glabriusculum (GenBank acc. KJ619462.1) as outgroups. Of these, 32 cp genome sequences were downloaded from the NCBI GenBank (Supplementary Table S4). MAFFT (Katoh et al., 2002) was used to construct molecular phylogenetic trees from alignments of 48 conserved protein-coding genes, and the sequences were manually adjusted using Bioedit (Hall, 1999). The best-fitting model of nucleotide substitutions was determined using Akaike Information Criterion in JModeltest V2.1.10 (Darriba et al., 2012). The GTR + I + G model was used in both. Maximum likelihood (ML) analysis was performed using RaxML v 8.0.5 (Stamatakis, 2014) with 1000 bootstrap replicates. Bayesian Inference (BI) analysis was performed using MrBayes 3.2.2 (Ronquist et al., 2012) with two-independent runs and four chains using Markov Chain Monte Carlo run simultaneously for one million generations. Trees were sampled every 5,000,000 generations, with the first 25% discarded as burn-in. Trees were determined from 50% majority-rule consensus trees to estimate posterior probabilities (PP). The reconstructed trees were visualized using Fig tree V.1.4.2 (Rambaut, 2012).
InDel Marker Development and Validation for I. nil and I. purpurea
Indel regions were detected by the alignment of six Ipomoea cp genome sequences and comparison of mVISTA similarities. Primers for indel markers were designed using NCBI Primer-BLAST. Specificity of indel markers was confirmed using PCR amplification with 20 ng of genomic DNA extracted from 23 samples of 6 Ipomoea species in a 20 μl PCR mixture with 10 pmol of IPOTY or IPOYCF indel primers. Amplification of both IPOTY and IPOYCF was conducted on a Pro Flex PCR system (Applied Biosystems, Waltham, MA, United States) with the following amplification parameters: initial denaturation at 95°C for 2 min; 35 cycles at 95°C for 50 s, 62°C for 50 s, and 72°C for 50 s; and final extension at 72°C for 5 min. PCR products were separated on a 2% agarose gel for 40 min at 150V. DNA fragments were extracted from agarose using a Gel Extraction Kit, subcloned into the pGEM-T Easy vector (Promega, WI, United States), and sequenced on a DNA sequence analyzer (ABI 3730, Applied Biosystems Inc., CA, United States). The six Ipomoea species germplasms used are listed in Supplementary Table S1.
Results and Discussion
Chloroplast Genome Organization of Six Ipomoea Species
Illumina sequencing generated 1.3–1.6 Gb of trimmed paired-end reads from six Ipomoea species (Supplementary Table S5). The six species yielded complete circular chloroplast cp genomes of 161,354–161,750 bp, with 384–611× coverage (Supplementary Table S6). As in most land plants, the Ipomoea cp genomes exhibited quadripartite structures consisting of a pair of IRs (61,220–62,122 bp) separated by LSC (87,579–88,134 bp) and SSC (12,039–12,101 bp) regions (Figure 1 and Table 1). The six Ipomoea cp genomes had similar GC contents, with higher GC contents observed in the IR regions (41%) than in the single-copy regions (LSC, 36% and SSC, 32%), consistent with previously reported cp genomes (Eserman et al., 2014; Yan et al., 2015). The gene content, order, and orientation were similar in the six Ipomoea cp genomes. The 112 unique genes consisted of 78 protein-coding genes and 30 tRNAs, with 17 duplicated genes in the 6 genomes (Table 2). Seventeen of the genes contained introns, fourteen with a single intron and two (ycf3 and clpP) with two introns (Supplementary Table S7). The genes psbL and ndhD had the alternative start codon ACG, and rps19 started with GTG. Use of ACG and GTG as start codons is common for several genes in the cp genomes of land plants (Sasaki et al., 2003; Kahlau et al., 2006; Gao et al., 2009; Sanchez-Puerta and Abbona, 2014). The codon usage and anticodon recognition patterns of the six Ipomoea cp genomes are shown in Supplementary Figure S1A. Protein-coding genes comprised 28,280 codons in I. hederacea to 28,434 codons in I. triloba, which was consistent with other plant cp genomes. Codons for leucine, isoleucine, and serine were the most abundant, whereas those for cysteine and tryptophan were found least often (Supplementary Figure S1B). RSCU values revealed synonymous codon usage bias, with a high proportion of synonymous codons having A or T in the third position. As expected, codons for arginine, leucine, and serine had abundant synonymous codons and higher RSCU values due to their importance as components of cp genes related to biosynthetic processes (Wang et al., 2016). The RSCU values of the six Ipomoea cp genomes were consistent with those of other higher plants. This phenomenon is indicative of stable cp evolution, which protects important cp genes against harmful mutations and adaptive selective pressures in important cp genes (Wang et al., 2016; Ivanova et al., 2017; Zuo et al., 2017). The six Ipomoea genomes exhibited typical features of Ipomoea cp genomes and had similar genome structures, gene orders, and gene contents, including introns and base composition, to one another.
FIGURE 1.
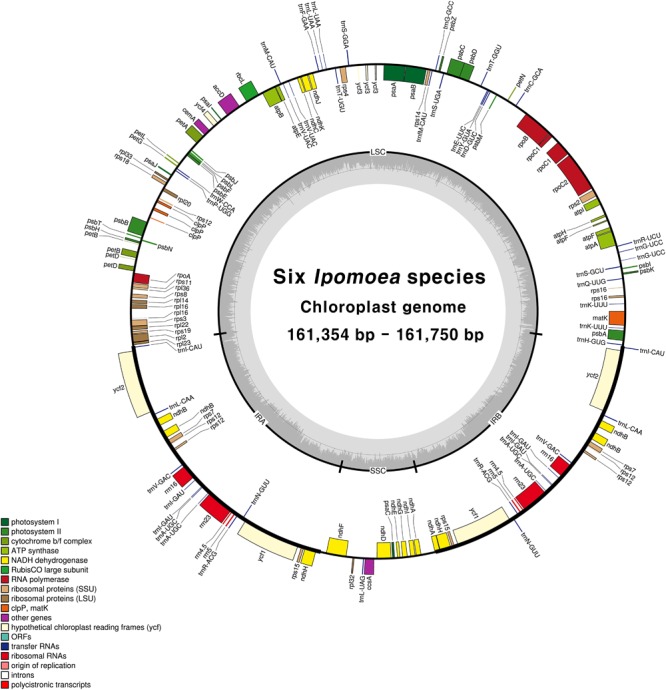
Circular gene map of chloroplast genomes from six Ipomoea species. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. The darker gray in the inner circle represents GC content. The gene map corresponds to I. nil chloroplast genome.
Table 1.
Features of six Ipomoea chloroplast genomes.
| Species | I. nil | I. purpurea | I. hederacea | I. hederacea var. integriuscula | I. lacunosa | I. triloba |
|---|---|---|---|---|---|---|
| Accession number | MG973745 | MG973746 | MG973747 | MG973748 | MG973749 | MG973750 |
| Total cp genome size (bp) | 161,747 | 161,629 | 161,354 | 161,714 | 161,492 | 161,750 |
| Large single-copy (LSC) region (bp) | 88,097 | 88,134 | 88,041 | 88,083 | 87,579 | 87,589 |
| Inverted repeat (IR) region (bp) | 61,564 | 61,394 | 61,220 | 61,538 | 61,862 | 62,122 |
| Small single-copy (SSC) region (bp) | 12,086 | 12,101 | 12,093 | 12,093 | 12,051 | 12,039 |
| Total number of genes (unique) | 112 | 112 | 112 | 112 | 112 | 112 |
| Protein-coding gene (unique) | 78 | 78 | 78 | 78 | 78 | 78 |
| rRNA (unique) | 4 | 4 | 4 | 4 | 4 | 4 |
| tRNA (unique) | 30 | 30 | 30 | 30 | 30 | 30 |
| GC content (%) | 38 | 38 | 38 | 38 | 38 | 38 |
| LSC (%) | 36 | 36 | 36 | 36 | 36 | 36 |
| IR (%) | 41 | 41 | 41 | 41 | 41 | 41 |
| SSC (%) | 32 | 32 | 32 | 32 | 32 | 32 |
Table 2.
Genes in the chloroplast genomes of six Ipomoea species.
| Gene groups | Gene names |
|---|---|
| Photosystem I | psaA, B, C, I, J, ycf32, ycf4 |
| Photosystem II | psbA, B, C, D, E, F, H, I, J, K, L, M, N, T, Z |
| Cytochrome b6/f | petA, B1, D1, G, L, N |
| ATP synthase | atpA, B, E, F1, H, I |
| Rubisco | rbcL |
| NADH oxidoreductase | ndhA1, B1,3, C, D, E, F, G, H3, I, J, K |
| Large subunit ribosomal proteins | rpl2, 14, 161, 20, 22, 23, 32, 33, 36 |
| Small subunit ribosomal proteins | rps2, 3, 4, 73, 8, 11, 12 2-4, 14, 153, 161, 18, 19 |
| RNA polymerase | rpoA, B, C11, C2 |
| Unknown function protein-coding gene | ycf13, 23 |
| Other genes | accD, ccsA, cemA, clpP2, matK |
| Ribosomal RNAs | rrn163, 233, 4.53, 53 |
| Transfer RNAs | trnA-UGC1,3, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC1, trnH-GUG, trnI-CAU3, trnI-GAU1,3, trnK-UUU1, trnL-CAA3, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU3, trnP-UGG, trnQ-UUG, trnR-ACG3, trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UAC, trnW-CCA, trnY-GUA |
1Gene containing a single intron; 2Gene containing two introns; 3Two gene copies in IRs; 4Trans-splicing gene.
SSR and Tandem Repeat Analysis in Six Ipomoea Chloroplast Genomes
SSRs (1–6 nucleotide repeats) were distributed abundantly across the cp genome. SSRs from cp genomes can be used for analysis of phylogenetic relationships and population genetics due to their high polymorphism rates and stable reproducibility (Powell et al., 1995; Dong et al., 2013; Yang et al., 2016). Most SSRs contained A or T units, contributing to the overall AT richness of the cp genome (Qian et al., 2013). Here, MISA software identified 191–202 SSRs in the six Ipomoea cp genomes. Most SSRs were found in single-copy regions (LSC and SSC) and non-coding regions (Figure 2). Mononucleotide motifs were the most abundant repeat type, with dinucleotide motifs, the second most abundant in the six Ipomoea cp genomes (Figure 2C). SSRs were compared between the genomes to identify common and species-specific SSRs. I. purpurea contained seven specific SSRs, whereas no I. hederacea-specific SSRs were identified. The Ipomoea cp SSRs encompassed abundant variation and will be useful genomic resources for marker development and population genetic studies of Ipomoea species. Tandem repeat sequences influence genome structure with respect to genome size, genome rearrangement, and gene duplication (Nie et al., 2012). Here, 27–33 tandem repeats of >20 bp were identified in the six Ipomoea genomes, averaging 56–74 bp in length (Figure 3). Of these, most were located in non-coding LSC and SSC regions (Figure 3A). On average, long tandem repeats (>70 bp) constituted 27% of all tandem repeats in the six Ipomoea cp genomes. I. triloba contained the longest repeat (484 bp). While most of the tandem repeats were conserved, a small number of specific tandem repeats were detected in the six Ipomoea cp genomes (Figure 3D). In particular, indel regions with different repeat copy numbers were identified for I. nil in trnN-ycf1 (68 bp × one copy in I. nil / 68 bp × two copies in other Ipomoea) and I. purpurea in ycf1 (18 bp × two copies in I. purpurea / 18 bp × three copies in other Ipomoea). These characteristics allowed indel markers for distinguishing I. nil and I. purpurea from other Ipomoea species to be developed in this study.
FIGURE 2.
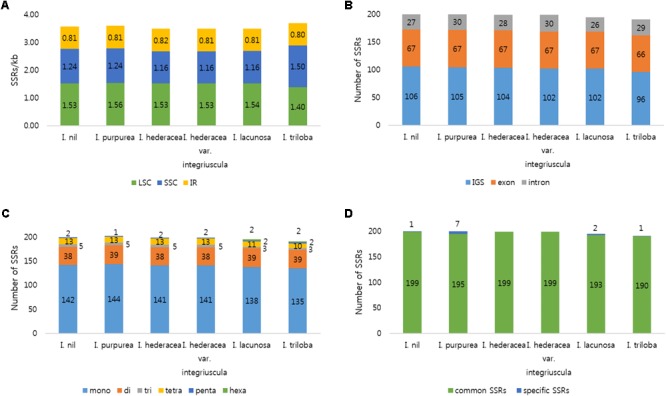
Distribution of SSRs in the six Ipomoea chloroplast genomes. (A) Number of SSRs per unit length in genomic regions. (B) Distribution of SSRs in intergenic spacer (IGS), exon, and intron regions. (C) Distribution of SSR types. (D) Number of common and species-specific SSRs among the six Ipomoea chloroplast genomes.
FIGURE 3.
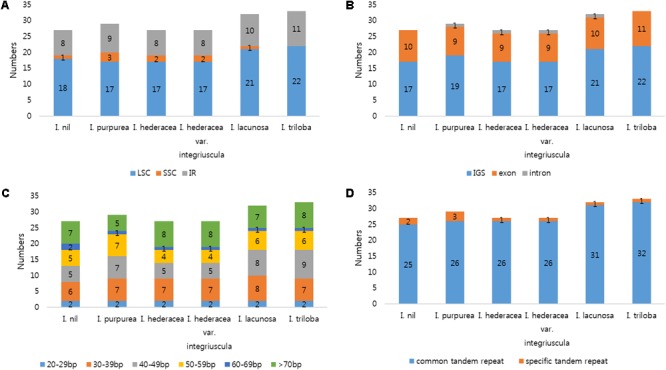
Analysis of tandem repeats in six Ipomoea chloroplast genomes. (A) Distribution of tandem repeats in genomic regions. (B) Distribution of tandem repeats in intergenic spacer (IGS), exon, and intron regions. (C) Distribution and lengths of tandem repeats. (D) Number of common and species-specific tandem repeats among the six Ipomoea chloroplast genomes.
Comparative Analysis of Six Ipomoea Chloroplast Genomes
Overall, the six Ipomoea cp genomes were highly conserved, with 98.5–99.8% similarity, conserved genomic structure, and conserved gene order and orientation (Supplementary Table S8). Pairwise determination of divergent regions was conducted using mVISTA (Figure 4). In general, non-coding regions were more diverged than coding regions. Five non-coding regions, petN-psbM, trnI-CAU-ycf2, ndhH-ndhF, psbC-trnS, and ccsA-ndhD, exhibited high divergence among the six Ipomoea. Coding regions were generally more conserved, with the exception of ycf1, matK, and rbcL, which are commonly used as representative plant DNA barcoding regions (CBOL Plant Working Group, 2009). Previous phylogenetic analysis of divergent non-coding regions allowed identification of potential molecular markers and DNA barcoding analysis (Shaw et al., 2007; Xu et al., 2017). Pi in the six Ipomoea cp genomes was calculated to show divergence at the sequence level (Figure 5). As expected, IR regions were more conserved than the LSC and SSC regions, with average Pi values of 0.003 for IR and 0.006 in SC (for regions other than those with a Pi value = 0). The average Pi value for coding regions was 0.00315 (range, 0.00038–0.00955; accD = 0.00955). The Pi value for intron-containing IGS averaged 0.00752 (range, 0.0005–0.00336; psbC-trnS in LSC = 0.00336). In the SSC, ccsA-ndhD exhibited a Pi value of 0.03, higher by an order of magnitude. Although the six Ipomoea cp genomes were generally highly conserved, the intergenic regions were particularly divergent. This is consistent with previous research with angiosperm cp genome (Ivanova et al., 2017). Similarly, the relatively higher divergence seen in the ycf1, matK, rbcL, and accD genes than in other coding regions was similar to observations in other cp genomes (Yukawa et al., 2006; Nie et al., 2012; Liu et al., 2013; Song et al., 2015).
FIGURE 4.
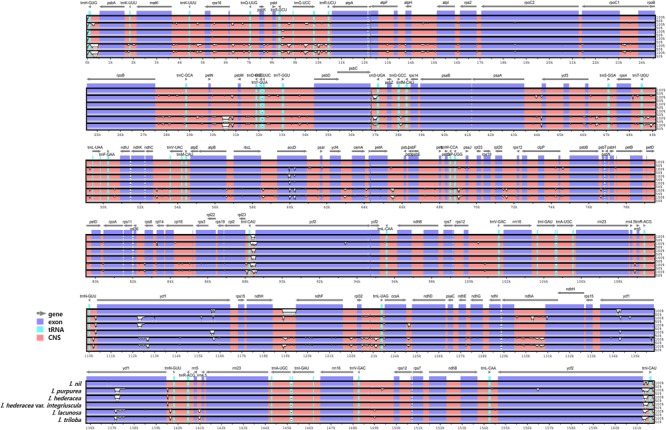
Comparison of six Ipomoea chloroplast genomes using mVISTA. Complete cp genomes of six Ipomoea species were compared, with I. nil as a reference. Blue block: conserved genes, sky-blue block: transfer RNA (tRNA) and ribosomal RNA (rRNA), and red block: conserved non-coding sequences (CNS). White represents regions with sequence variation among the six Ipomoea species.
FIGURE 5.
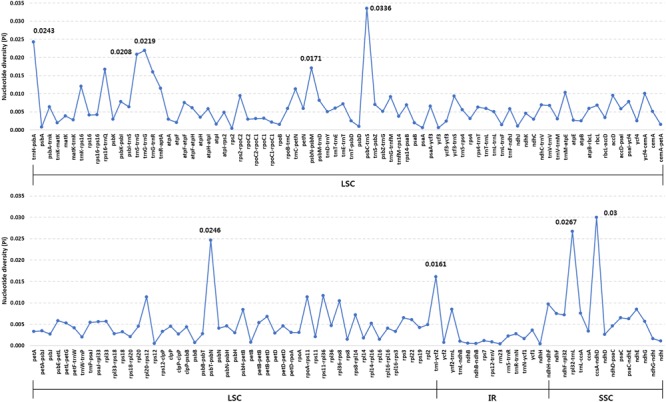
Comparison of nucleotide diversity (Pi) values among the six Ipomoea species.
IR regions were more highly conserved than SC regions due to copy correction by gene conversion in IR regions (Khakhlova and Bock, 2006). However, these events frequently lead to genome length variation through genome contraction and expansion (Raubeson et al., 2007), and IR contraction and expansion was previously used as an evolution criterion when examining cp genomes (Hansen et al., 2007; Huang et al., 2014). Here, we compared border regions and embedded genes among the six Ipomoea cp genomes (Supplementary Figure S2). The rpl23 gene region in the six Ipomoea cp genomes was located in the LSC. All trnH genes were located in the LSC, 62–69 bp away from the IRb/LSC boundary. The ndhH genes were positioned in regions IRa and IRb. In particular, the ndhA gene extended into IRb. The rpl23 gene shifted from the IR to the LSC, and ndhH exhibited gene duplication in IRs when compared with dicotyledon cp genomes (Yan et al., 2015). Although the genome structure of the IR region was highly conserved among the cp sequences of the six Ipomoea species, extreme gene shifting and duplication have occurred in the Ipomoea genus. To determine the selection pressure on protein-coding genes, we examined Ka/Ks (non-synonymous substitution to synonymous substitution) ratios from collinear genes as a marker of evolution (Supplementary Figure S3). Genes under positive selection are considered to be undergoing adaptive evolution in response to their environment (Kimura, 1989; Raman and Park, 2016; Ivanova et al., 2017). The most highly conserved genes exhibited purifying selection (Ka/Ks ratio, 0–0.001). The Ka/Ks ratios for most photosynthetic apparatus genes were close to 0. No significant gene evolution was observed according to regional groupings (i.e., LSC, IR, or SSC). Within the 6 Ipomoea cp genomes, 31 genes had Ka and Ks values >0.001, and the average Ka and Ks values were 0.0042 and 0.0168, respectively. The highest Ks value across the six Ipomoea species was 0.0717 (for ndhE), and the highest Ka/Ks ratio was 2.654 (for accD between I. purpurea and I. lacunosa). Thus, although the six Ipomoea cp genomes exhibited highly conserved organization, positive selection pressure (Ka/Ks > 1) was observed for accD, cemA, and ycf2. Positive selection of these three genes suggests that they are undergoing essential adaptations to their environment despite the weak selection pressures experienced by Ipomoea. Previous studies show that these genes are generally lost from, or are highly divergent in, angiosperms (Wicke et al., 2011; Ivanova et al., 2017). McNeal et al. (2007) showed that photosynthesis-related genes are under strong selection constraint in parasitic plants of the Cuscuta genus. Also, they reported that accD, cemA, and ycf2 genes in both Ipomoea and Cuscuta were under purifying selection pressure. However, in the current study, we found that these genes were under positive selection pressure. Although plants within the Cuscuta genus express markedly diverse genes to adapt to life as parasitic plants, these genes might be rapidly evolving in the Ipomoea genus. Several studies show that nuclear genes in Ipomoea are under positive selection pressure. In particular, genes encoding dihydeoflavonol-4 reductase (DFR) and chalcone synthase (CHS) in Ipomoea are under positive selection pressure (Yang et al., 2004; Des Marais and Rausher, 2008). DFR is an important factor in the anthocyanin biosynthetic pathway. Des Marais and Rausher (2008) demonstrated that escape from adaptive conflict via repeated positive selection occurred after DFR genes duplicated in I. purpurea. Thus, DFR genes exhibit adaptive evolutionary changes. The CHS genes (which function in flavonoid biosynthesis) experienced selective pressure to promote divergence via increasing gene duplication in Ipomoea. Ipomoea has extremely diverse flower colors, a rapid generation time, and various growth forms. We suggest that these ecological characteristics of Ipomoea reflect their remarkable adaptability to various environments due to diverse positive selection pressure on genes in the nucleus or plastid.
Phylogenic Relationships of Six Ipomoea Within Ipomoeeae
Cp genomes are valuable genomic resources for reconstruction of accurate and high-resolution phylogenies, and have been used as such in several studies (Jansen et al., 2007; Moore et al., 2007), for example, in angiosperms (Wu et al., 2010; Nie et al., 2012). To identify the phylogenetic positions of the six Ipomoea species within the Convolvulaceae, we aligned 48 protein-coding sequences shared by 38 cp genomes (Figure 6 and Supplementary Figure S4). The alignment length was 38,229 bp. All except two nodes were supported by a Bayesian PP of 1.0. Ipomoea and Cuscuta had the closest phylogenetic relationship within the Convolvulaceae. Consistent with previous analysis, the tribe Ipomoeeae was divided into 2 major clades, Atstipomoeinae and Atgyreiinae, with 28 Ipomoea species within 7 sections (Stefanoviæ et al., 2003). Most Ipomoea species were within Quamoclit and Batatas. The positions of the six Ipomoea examined in this study were strongly supported with BI and PP values. I. nil and I. hederacea formed a monophyletic cluster as a sister to I. purpurea within Quamoclit I. lacunosa and I. triloba formed a monophyletic cluster in Batatas. Previous analysis of 28 Ipomoea cp genomes clarified the evolutionary relationships within the 2 major clades of Ipomoeeae (Eserman et al., 2014). Quamoclit species were divided into two clades (Miller et al., 2004), whereas our phylogenetic results revealed their monophyly. In this study, I. hederacea and I. hederacea var. integriuscula were clustered with I. nil in a monophyletic relationship, but that I. purpurea was paraphyletic with these species. A previous study indicated that I. nil and I. purpurea share a monophyletic relationship. Here, we obtained more accurate information about the relationship between I. nil and I. hederacea and I. hederacea var. integriuscula and I. purpurea. Furthermore, I. triloba and I. lacunosa were positioned in Batatas as a monophyletic group with I. trifida and I. cordatotriloba. Therefore, we performed high-resolution phylogenetic analysis of the positions of Ipomoea species in Batatas in the phylogenic tree. The reconstructed phylogenic trees were clearly consistent with previous studies according to the APG IV system (Austin, 1978; Eserman et al., 2014; The Angiosperm Phylogeny Group, 2016). The results of this study are strongly supported by those of previous studies; however, we further clarified the phylogenetic relationships within the Ipomoeeae. Based on the phylogenetic positions of the six Ipomoea species determined in the present study, I. nil and I. purpurea (whose seeds are used for Pharbitidis Semen) share closer relationships with I. hederacea and I. hederacea var. integriuscula than with I. triloba and I. lacunosa. Thus, there is a strong possibility for confusion between I. hederacea and I. hederacea var. integriuscula. We suspect that the most frequent adulterations of Pharbitidis Semen are seeds of I. hederacea and I. hederacea var. integriuscula.
FIGURE 6.
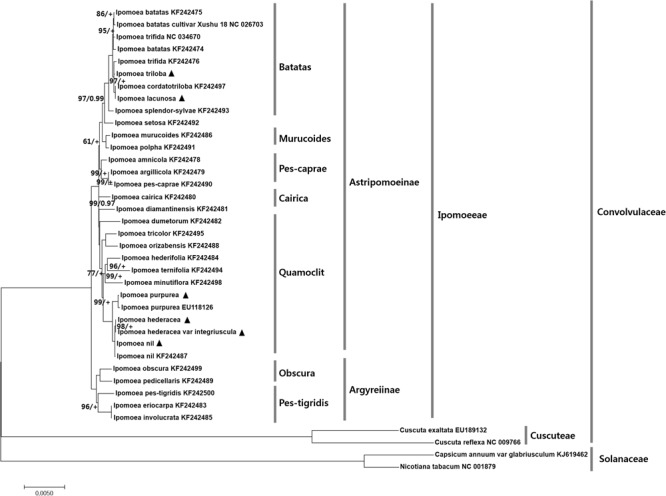
Phylogenetic tree based on 48 protein-coding genes from 6 Ipomoea and 28 other Ipomoea species using maximum likelihood (ML) bootstraps and Bayesian posterior probabilities (PP). ML topology is shown with ML bootstrap support values/ Bayesian PP given at each node. 100% bootstrap and 1.0 PP support are not marked. + Indicated 1.0 PP support values. Black triangles indicate the cp genomes of six Ipomoea species examined in this study.
New Indel Markers for Distinguishing Herbal Medicine Plants
Dried seeds of I. nil and I. purpurea are used in traditional herbal medicine in Korea (Korea Institute of Oriental Medicine [KIOM], 2016). However, seeds from other Ipomoea strongly resemble those of I. nil and I. purpurea and are often inappropriately included in herbal preparations. Although the phylogenetic analysis in this study indicated that I. triloba and I. lacunosa were phylogenetically distant from I. nil and I. purpurea, the highly similar seed shapes present a challenge for identifying authentic herbal medicines, and a molecular approach would be beneficial. Here, DNA barcode analysis was performed for I. nil and I. purpurea and four related Ipomoea species, I. triloba, I. lacunosa, I. hederacea, and I. hederacea var. integriuscula, with similar seed shapes. I. purpurea was distinguished from other Ipomoea species with the ITS2 and matK regions (Supplementary Figure S5). However, the sequence of I. nil at ITS2 was the same as that of I. hederacea, and the sequence of I. nil at matK was the same as that of I. lacunosa and I. triloba, highlighting the limitations of universal DNA barcode sequences for distinguishing species. To resolve this problem, divergent regions within the cp genome were examined with the aim of distinguishing I. nil and I. purpurea. This analysis revealed species-specific divergent regions at trnN-ycf1 and ycf1 for I. nil and I. purpurea, respectively, with respect to copy number variation in tandem repeats. To develop indel markers, specific primers were designed against conserved regions of trnN-ycf1 and ycf1 (Table 3). The primer pairs, respectively named IPOTY and IPOYCF, successfully amplified sequences from I. nil and I. purpurea (Figure 7). The markers were tested with other Ipomoea germplasms (23 samples for IPOTY and 22 samples for IPOYCF), and the five I. nil samples and six I. purpurea samples were clearly distinguishable. Amplified fragments from all of the tested Ipomoea samples were sequenced to identify exact amplicon size. IPOTY primers yielded a 525 bp amplicon with I. nil, and IPOYCF primers yielded a 467 bp fragment with I. purpurea. Predicted deletion or insertion sizes from cp genomes were consistent with those resulting from the Ipomoea germplasms used in this study. Indel markers for variable copy numbers at tandem repeats were also used previously to distinguish closely related Fagopyrum and Chenopodium species, indicating the utility of these markers in species identification (Cho et al., 2015; Hong et al., 2017). Copy number variation at tandem repeats in cp genomes may, therefore, prove broadly useful in distinguishing closely related plant species where universal barcode sequences are non-discriminatory. The IPOTY and IPYCF indel markers developed in this study will be useful for Ipomoea species identification and authentication of herbal medicines.
Table 3.
Primer information for indel markers IPOTY and IPOYCF.
| Primer name | Primer sequence (5′ > 3′) | Position |
|---|---|---|
| IPOTY_F | TAACGGTCAAAGCGAGCCCC | trnN-GUU–ycf1 |
| IPOTY_R | AAGTCCAGCCGCAAGAACTGA | |
| IPOYCF_F | GGTCGCGGTAAATCCCAGCA | ycf1 |
| IPOYCF_R | TCTTCCCAGAATTTGTGCGGC |
FIGURE 7.

Schematic of indel markers IPOTY and IPOYCF for I. nil and I. purpurea. (A) Primers for IPOTY were tested with 23 Ipomoea germplasms. (B) Primers for IPOYCF were tested with 22 Ipomoea germplasms. Germplasm details are provided in Supplementary Table S1. 1, Ipomoea nil; 2, I. purpurea; 3, I. hederacea; 4, I. hederacea var. integriuscula; 5, I. lacunosa; and 6, I. triloba.
Conclusion
Six Ipomoea cp genomes were sequenced in this study. Overall, the cp genomes were highly conserved with respect to gene content, gene orientation, and GC content, but local variations in sequence and structure were observed. Tandem repeats and SSRs were identified with the aim of developing molecular markers for species identification and authentication of herbal medicines. The most divergent regions among the six genomes were found in non-coding regions petN-psbM, trnI-CAU-ycf2, ndhH-ndhF, psbC-trnS, and ccsA-ndhD, and coding regions accD, cemA, and ycf2. The accD, cemA, and ycf2 genes exhibited positive selection. Phylogenetic analysis of cp genome sequences yielded more accurate phylogenetic relationships within the Ipomoea genus than previous studies. Novel indel markers based on copy number variation at tandem repeats were developed for identification of I. nil and I. purpurea. These markers, named IPOTY and IPOYCF, were able to discriminate between authentic I. nil and I. purpurea and other inauthentic Ipomoea species, respectively, and will be useful for authentication of herbal medicines containing these two species. The cp genomes and analyses in this study are valuable for species identification, clarification of taxonomy, and understanding evolutionary history in the Ipomoea genus.
Author Contributions
IP designed the experimental framework and drafted and revised the manuscript. SY and BCM collected and identified plant materials. WJK and PN performed the experiments. HOL carried out sequence analysis. BCM revised the manuscript. All authors contributed to the experiments and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer AZ and the handling Editor declared their shared affiliation.
Acknowledgments
The authors would like to thank the “Classification and Identification Committee of the KIOM” for the identification of plant materials and the Herbarium of Korea Standard Herbal Resources (Index Herbariorum code KIOM) for the provision of plant materials.
Footnotes
Funding. This work was supported by a grant of the Development of Foundational Techniques for the Domestic Production of Authentic Herbal Medicines based on the Establishment of Molecular Authentication Systems (K17403 and K18403) from the Korea Institute of Oriental Medicine (KIOM), South Korea.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00965/full#supplementary-material
References
- Austin D. F. (1978). The Ipomoea batatas complex-I.Taxonomy. Bull. Torrey Bot. Club 105 114–129. 10.2307/2484429 [DOI] [Google Scholar]
- Austin D. F., Huáman Z. (1996). A synopsis of Ipomoea (Convolvulaceae) in the Americas. Taxon 45 3–38. 10.2307/1222581 [DOI] [Google Scholar]
- Baucom R., Chang S., Kniskern J., Rausher M., Stinchcombe J. (2011). Morning glory as a powerful model in ecological genomics: tracing adaptation through both natural and artificial selection. Heredity 107 377–385. 10.1038/hdy.2011.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T., Berriman M., Tivey A., Patel C., Bohme U., Barrell B. G., et al. (2008). Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24 2672–2676. 10.1093/bioinformatics/btn529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CBOL Plant Working Group (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U.S.A. 106 12794–12797. 10.1073/pnas.0905845106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Yao H., Han J., Liu C., Song J., Shi L., et al. (2010). Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5:e8613. 10.1371/journal.pone.0008613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K. S., Yun B. K., Yoon Y. H., Hong S. Y., Mekapogu M., Kim K. H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS One 10:e0125332. 10.1371/journal.pone.0125332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniell H., Lin C. S., Yu M., Chang W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. 10.1186/s13059-016-1004-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delannoy E., Fujii S., des Francs-Small C., Brundrett M., Small I. (2011). Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 28 2077–2086. 10.1093/molbev/msr028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A. L., Salzberg S. L., Phillippy A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. Chapter 10:Unit 10.13. 10.1002/0471250953.bi1003s00 [DOI] [PubMed] [Google Scholar]
- Des Marais D. L., Rausher M. D. (2008). Escape from adaptive conflict after duplication in an anthocyanin pathway gene. Nature 454 762–765. 10.1038/nature07092 [DOI] [PubMed] [Google Scholar]
- Dong W., Xu C., Cheng T., Lin K., Zhou S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5 989–997. 10.1093/gbe/evt063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennos R. A., Clegg M. T. (1983). Flower color variation in the morning glory, Ipomoea purpurea. J. Hered. 74 247–250. 10.1093/oxfordjournals.jhered.a109778 [DOI] [Google Scholar]
- Epperson B., Clegg M. (1992). Unstable white flower color genes and their derivatives in morning glory. J. Hered. 83 405–409. 10.1093/oxfordjournals.jhered.a111242 [DOI] [Google Scholar]
- Eserman L. A., Tiley G. P., Jarret R. L., Leebens-Mack J. H., Miller R. E. (2014). Phylogenetics and diversification of morning glories (tribe Ipomoeeae. Convolvulaceae) based on whole plastome sequences. Am. J. Bot. 101 92–103. 10.3732/ajb.1300207 [DOI] [PubMed] [Google Scholar]
- Gao L., Yi X., Yang Y. X., Su Y. J., Wang T. (2009). Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 9:130. 10.1186/1471-2148-9-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurusaran M., Ravella D., Sekar K. (2013). RepEx: repeat extractor for biological sequences. Genomics 102 403–408. 10.1016/j.ygeno.2013.07.005 [DOI] [PubMed] [Google Scholar]
- Hall T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp. Ser. 41 95–98. [Google Scholar]
- Hansen D. R., Dastidar S. G., Cai Z., Penaflor C., Kuehl J. V., Boore J. L., et al. (2007). Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 45 547–563. 10.1016/j.ympev.2007.06.004 [DOI] [PubMed] [Google Scholar]
- Hollingsworth P. M., Graham S. W., Little D. P. (2011). Choosing and using a plant DNA barcode. PLoS One 6:e19254. 10.1371/journal.pone.0019254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. Y., Cheon K. S., Yoo K. O., Lee H. O., Cho K. S., Suh J. T., et al. (2017). complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front. Plant Sci. 8:1696. 10.3389/fpls.2017.01696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A., Jayakumar V., Nitasaka E., Toyoda A., Noguchi H., Itoh T., et al. (2016). Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat. Commun. 7:13295. 10.1038/ncomms13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Shi C., Liu Y., Mao S. Y., Gao L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. 10.1186/1471-2148-14-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova Z., Sablok G., Daskalova E., Zahmanova G., Apostolova E., Yahubyan G., et al. (2017). Chloroplast genome analysis of resurrection tertiary relict Haberlea rhodopensis highlights genes important for desiccation stress response. Front. Plant Sci. 8:204. 10.3389/fpls.2017.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R. K., Cai Z., Raubeson L. A., Daniell H., Leebens-Mack J., Müller K. F., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A 104 19369–19374. 10.1073/pnas.0709121104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R. K., Ruhlman T. A. (2012). “Plastid genomes of seed plants,” in Genomics of Chloroplasts and Mitochondria: Advances in Photosynthesis and Respiration (Including Bioenergy and Related Processes) Vol. 35 eds Bock R., Knoop V. (Dordrecht: Springer; ). [Google Scholar]
- Kahlau S., Aspinall S., Gray J. C., Bock R. (2006). Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 63 194–207. 10.1007/s00239-005-0254-5 [DOI] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K. I., Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhlova O., Bock R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46 85–94. 10.1111/j.1365-313X.2006.02673.x [DOI] [PubMed] [Google Scholar]
- Kim K., Lee S. C., Lee J., Lee H. O., Joh H. J., Kim N. H., et al. (2015). Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLoS One 10:e0117159. 10.1371/journal.pone.0117159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. (1989). The neutral theory of molecular evolution and the world view of the neutralists. Genome 31 24–31. 10.1139/g89-009 [DOI] [PubMed] [Google Scholar]
- Korea Institute of Oriental Medicine [KIOM] (2016). Defining Dictionary for Medicinal Herbs. Available at: http://boncho.kiom.re.kr/codex/ [Google Scholar]
- Korean Food Standard Codex (2010). Ministry of Food and Drug Safety (Republic of Korea). Available at: http://www.mfds.go.kr/files/upload/eng/9_Foods_Labeling_Standards.pdf [Google Scholar]
- Librado P., Rozas J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Liu Y., Huo N., Dong L., Wang Y., Zhang S., Young H. A., et al. (2013). Complete chloroplast genome sequences of Mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS One 8:e57533. 10.1371/journal.pone.0057533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M., Drechsel O., Bock R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52 267–274. 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- Lowe T. M., Eddy S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 955–964. 10.1093/nar/25.5.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. 10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manos P. S., Miller R. E., Wilkin P. (2001). Phylogenetic analysis of Ipomoea. Argyreia, Stictocardia, and Turbina suggests a generalized model of morphological evolution in morning glories. Syst. Bot. 26 585–602. [Google Scholar]
- McDonald J. A. (1995). Revision of Ipomoea section Leptocallis (Convolvulaceae). Harv. Pap. Bot. 1 97–122. [Google Scholar]
- McNeal J. R., Kuehl J. V., Boore J. L., de Pamphilis C. W. (2007). Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta. BMC Plant Biol. 7:57. 10.1186/1471-2229-7-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. E., McDonald J. A., Manos P. S. (2004). Systematics of Ipomoea subgenus Quamoclit (Convolvulaceae) based on ITS sequence data and a Bayesian phylogenetic analysis. Am. J. Bot. 91 1208–1218. 10.3732/ajb.91.8.1208 [DOI] [PubMed] [Google Scholar]
- Moore M. J., Bell C. D., Soltis P. S., Soltis D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104 19363–19368. 10.1073/pnas.0708072104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie X., Lv S., Zhang Y., Du X., Wang L., Biradar S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS One 7:e36869. 10.1371/journal.pone.0036869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I., Kim W. J., Yang S., Yeo S. M., Li H., Moon B. C. (2017a). The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS One 12:e0184257. 10.1371/journal.pone.0184257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I., Yang S., Choi G., Kim W. J., Moon B. C. (2017b). The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum Subgenus Lycoctonum. Molecules 22:E2012. 10.3390/molecules22112012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks M., Cronn R., Liston A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. 10.1186/1741-7007-7-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell W., Morgante M., McDevitt R., Vendramin G., Rafalski J. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92 7759–7763. 10.1073/pnas.92.17.7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J., Song J., Gao H., Zhu Y., Xu J., Pang X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS One 8:e57607. 10.1371/journal.pone.0057607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman G., Park S. (2016). The complete chloroplast genome sequence of Ampelopsis: gene organization, comparative analysis, and phylogenetic relationships to other angiosperms. Front. Plant Sci. 7:341. 10.3389/fpls.2016.00341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. (2012). FigTree v 1.4. 2: Molecular Evolution, Phylogenetics and Epidemiology. Edinburgh: University of Edinburgh. [Google Scholar]
- Raubeson L. A., Peery R., Chumley T. W., Dziubek C., Fourcade H. M., Boore J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. 10.1186/1471-2164-8-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausher M. D., Fry J. D. (1993). Effects of a locus affecting floral pigmentation in Ipomoea purpurea on female fitness components. Genetics 134 1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Hohna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Puerta M. V., Abbona C. C. (2014). The chloroplast genome of Hyoscyamus niger and a phylogenetic study of the tribe Hyoscyameae (Solanaceae). PLoS One 9:e98353. 10.1371/journal.pone.0098353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Yukawa Y., Miyamoto T., Obokata J., Sugiura M. (2003). Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol. Biol. Evol. 20 1028–1035. 10.1093/molbev/msg098 [DOI] [PubMed] [Google Scholar]
- Sato T., Okamoto J., Degawa Y., Matsunari S., Takahashi K., Tomioka K. (2009). White rust of Ipomoea caused by Albugo ipomoeae-panduratae and A. ipomoeae-hardwickii and their host specificity. J. Gen. Plant Pathol. 75 46–51. 10.1007/s10327-008-0142-0 [DOI] [Google Scholar]
- Semagn K., Bjørnstad Å., Ndjiondjop M. (2006). An overview of molecular marker methods for plants. Afr. J. Biotechnol. 5 2540–2568. [Google Scholar]
- Shaw J., Lickey E. B., Schilling E. E., Small R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94 275–288. 10.3732/ajb.94.3.275 [DOI] [PubMed] [Google Scholar]
- Simms E. L., Rausher M. D. (1993). Patterns of selection on phytophage resistance in ipomoea purpurea. Evolution 47 970–976. 10.1111/j.1558-5646.1993.tb01252.x [DOI] [PubMed] [Google Scholar]
- Simonsen A. K., Stinchcombe J. R. (2007). Induced responses in Ipomoea hederacea: simulated mammalian herbivory induces resistance and susceptibility to insect herbivores. Arthropod Plant Interact. 1 129–136. 10.1007/s11829-007-9009-0 [DOI] [Google Scholar]
- Song Y., Dong W., Liu B., Xu C., Yao X., Gao J., et al. (2015). Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 6:662. 10.3389/fpls.2015.00662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanoviæ S., Austin D. F., Olmstead R. G. (2003). Classification of Convolvulaceae: a phylogenetic approach. Syst. Bot. 28 791–806. [Google Scholar]
- Stefanoviæ S., Krueger L., Olmstead R. G. (2002). Monophyly of the Convolvulaceae and circumscription of their major lineages based on DNA sequences of multiple chloroplast loci. Am. J. Bot. 89 1510–1522. 10.3732/ajb.89.9.1510 [DOI] [PubMed] [Google Scholar]
- Streisfeld M. A., Rausher M. D. (2009). Genetic changes contributing to the parallel evolution of red floral pigmentation among Ipomoea species. New Phytol. 183 751–763. 10.1111/j.1469-8137.2009.02929.x [DOI] [PubMed] [Google Scholar]
- Sucher N. J., Carles M. C. (2008). Genome-based approaches to the authentication of medicinal plants. Planta Med. 74 603–623. 10.1055/s-2008-1074517 [DOI] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34 W609–W612. 10.1093/nar/gkl315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Angiosperm Phylogeny Group (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181 1–20. 10.1111/boj.12385 [DOI] [Google Scholar]
- Thiel T. (2003). MISA—Microsatellite Identification Tool. Available at: http://pgrc.ipk-gatersleben.de/misa/ [Google Scholar]
- Tiffin P., Rausher M. D. (1999). Genetic constraints and selection acting on tolerance to herbivory in the common morning glory Ipomoea purpurea. Am. Nat. 154 700–716. 10.1086/303271 [DOI] [PubMed] [Google Scholar]
- Tillich M., Lehwark P., Pellizzer T., Ulbricht-Jones E. S., Fischer A., Bock R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45 W6–W11. 10.1093/nar/gkx391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhan D.-F., Jia X., Mei W.-L., Dai H.-F., Chen X.-T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the malvales order. Front. Plant Sci. 7:280. 10.3389/fpls.2016.00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton P. E., Giordano J., Cheung F., Gelfand Y., Benson G. (2004). Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res. 14 1861–1869. 10.1101/gr.2542904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Müller K. F., de Pamphilis C. W., Quandt D., Wickett N. J., Zhang Y., et al. (2013). Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the Broomrape family. Plant Cell 25 3711–3725. 10.1105/tpc.113.113373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S., Schneeweiss G. M., dePamphilis C. W., Muller K. F., Quandt D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76 273–297. 10.1007/s11103-011-9762-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin P. (1999). A morphological cladistic analysis of the Ipomoeeae (Convolvulaceae). Kew Bull. 853–876. 10.3732/ajb.89.9.1510 [DOI] [PubMed] [Google Scholar]
- Wu F. H., Chan M. T., Liao D. C., Hsu C. T., Lee Y. W., Daniell H., et al. (2010). Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 10:68. 10.1186/1471-2229-10-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Dong W., Li W., Lu Y., Xie X., Jin X., et al. (2017). Comparative analysis of six Lagerstroemia complete chloroplast genomes. Front. Plant Sci. 8:15. 10.3389/fpls.2017.00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Lai X., Li X., Wei C., Tan X., Zhang Y. (2015). Analyses of the complete genome and gene expression of chloroplast of sweet potato [Ipomoea batata]. PLoS One 10:e0124083. 10.1371/journal.pone.0124083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Gu H., Yang Z. (2004). Likelihood analysis of the chalcone synthase genes suggests the role of positive selection in morning glories (Ipomoea). J. Mol. Evol. 58 54–63. 10.1007/s00239-003-2525-3 [DOI] [PubMed] [Google Scholar]
- Yang Y., Zhou T., Duan D., Yang J., Feng L., Zhao G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7:959. 10.3389/fpls.2016.00959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa M., Tsudzuki T., Sugiura M. (2006). The chloroplast genome of Nicotiana sylvestris and Nicotiana tomentosiformis: complete sequencing confirms that the Nicotiana sylvestris progenitor is the maternal genome donor of Nicotiana tabacum. Mol. Genet. Genomics 275 367–373. 10.1007/s00438-005-0092-6 [DOI] [PubMed] [Google Scholar]
- Zuo L.-H., Shang A.-Q., Zhang S., Yu X.-Y., Ren Y.-C., Yang M.-S., et al. (2017). The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: genome comparative and taxonomic position analysis. PLoS One 12:e0171264. 10.1371/journal.pone.0171264 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.