

Summary
Although the “universal” genetic code is now known not to be universal, and stop codons can have multiple meanings, one regularity remains, namely that for a given sense codon there is a unique translation. Examining CUG usage in yeasts that have transferred CUG away from leucine, we here report the first example of dual coding: Ascoidea asiatica stochastically encodes CUG as both serine and leucine in approximately equal proportions. This is deleterious, as evidenced by CUG codons being rare, never at conserved serine or leucine residues, and predominantly in lowly expressed genes. Related yeasts solve the problem by loss of function of one of the two tRNAs. This dual coding is consistent with the tRNA-loss-driven codon reassignment hypothesis, and provides a unique example of a proteome that cannot be deterministically predicted.
Video Abstract
Keywords: genetic code, codon reassignment, stochastic decoding, competing tRNAs, CUG codon, Ascoidea asiatica, Saccharomycopsis, yeast evolution, proteomics
Graphical Abstract
Highlights
-
•
Ascoidea asiatica stochastically encodes CUG as leucine and serine
-
•
It is the only known example of a proteome with non-deterministic features
-
•
Stochastic encoding is caused by competing tRNALeu(CAG) and tRNASer(CAG)
-
•
A. asiatica copes with stochastic encoding by avoiding CUG at key positions
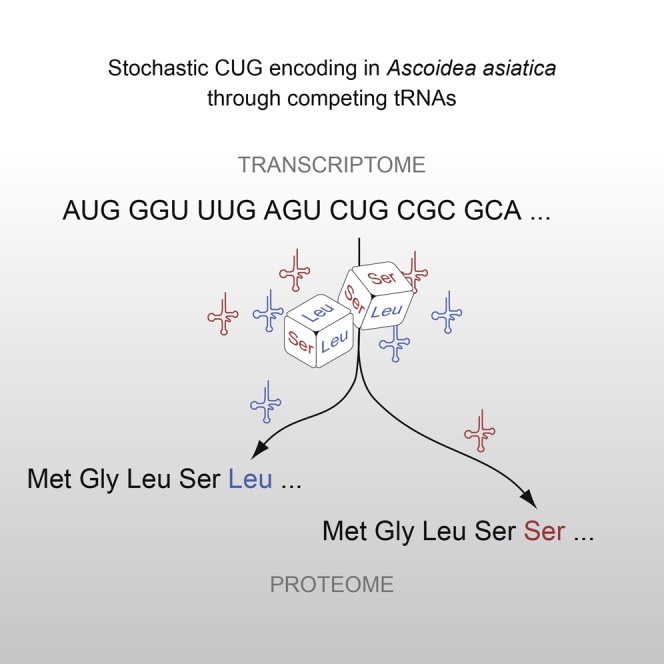
Mühlhausen et al. discover that Ascoidea asiatica stochastically encodes CUG as both serine and leucine, which is most likely caused by two competing tRNAs. This is the first example where the non-ambiguity rule of the genetic code is broken. To minimize its effect, A. asiatica uses CUG only rarely and never at conserved sequence positions.
Introduction
Genetic information, as stored in genomic DNA, is translated into proteins by ribosomes. This process needs tight control and accuracy so that the same functional protein is obtained from the same gene [1, 2, 3]. To preserve accuracy, ribosomes select for cognate aminoacyl-tRNAs matching nucleotide triplets (codons) of the mRNA and discriminate against non- and near-cognate aminoacyl-tRNAs. The correct tRNA charging is secured by highly specific aminoacyl-tRNA synthetases (aaRSs). Assuming there to be selection for “one mRNA-one protein,” it is not surprising that the genetic code is near universal, with there being only a few minor alterations. One such modification is the alternative decoding of the UGA stop codon by selenocysteine, although this affects only a few proteins [4, 5]. Genome-wide changes to the meaning of codons happened in the comparatively tiny organellar genomes of many species, but are extremely rare in nuclear genomes [6, 7]. Except for yeasts, only stop codons are affected by nuclear codon reassignments. In addition to complete reassignments, several ciliates and a trypanosomatid have been discovered in which one or all stop codons have dual or, in case of the UGA stop codon, even threefold meanings [8, 9, 10, 11]. The decoding by standard amino acid, selenocysteine, or stop codon is always context specific and never ambiguous.
Yeasts from the clade comprising the Debaryomycetaceae and Metschnikowiaceae (abbreviated as “DM clade” from here on) and Pachysolen tannophilus are currently the only known species where a sense codon has been reassigned in nuclear genomes. They translate CUG as serine and alanine, respectively [12, 13, 14, 15], rather than as the “universal” leucine. Recently, four genomes from another major yeast clade comprising the Ascoidea and Saccharomycopsis species (named “Ascoidea clade” from here on), have been sequenced [15, 16]. These were proposed to form a monophyletic clade according to a multigene analysis [17] and include Saccharomycopsis fibuligera, the major amylolytic yeast for food fermentation using rice and cassava [18]. In contrast to the suggested CUG decoding by leucine in Ascoidea rubescens [15], the Bagheera webserver for predicting yeast CUG codon translation [19] does not reveal any tRNACAG identity (CAG being the anticodon to CUG). This and lack of CUG codons at conserved sequence positions suggest a novel genetic code. To understand this better, we sought to investigate the evolutionary history of this recoding.
Results
Translation of CUG Is Stochastic in Ascoidea asiatica
To determine the CUG codon translation in Ascoidea clade yeasts, we performed unbiased liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses generating approximately 5.34 million high-quality mass spectra of the following seven yeast proteomes: the four Ascoidea clade yeasts A. asiatica, A. rubescens, S. fibuligera, and Saccharomycopsis malanga; Babjeviella inositovora and Clavispora lusitaniae from the DM clade and Nakazawaea peltata, which is the closest relative to P. tannophilus with a sequenced genome (Table 1; Figure S1; Data S1). To obtain peptide spectrum matches (PSMs) free of CUG-translation bias, 20 replicates for each genome annotation were generated with the CUG codon translated into a different amino acid in each replicate. Spectra searching against these databases resulted in 2.96 million PSMs (394,755 non-redundant peptide matches) with a median mass measurement error of about 408 parts per billion. We identified 31%–67% of the predicted proteins with median protein sequence coverage of 19%–27% and CUG codon recovery of 8%–23% (Table 1; Data S1). To control the quality of the CUG-containing peptide identifications, we considered only those with b- and/or y-type fragment ions around CUG codon positions as fully supported. Unless otherwise stated, all numbers given below refer to fully supported CUG positions.
Table 1.
Mass Spectrometry Data Analysis
| Yeast Species | Aoas (1) | Acr | Safi (1) | Sama | Bai | Cll | Nape |
|---|---|---|---|---|---|---|---|
| Mass spectra | 733,673 | 637,651 | 329,041 | 570,823 | 597,939 | 290,859 | 588,199 |
| PSMs | 246,644 | 411,915 | 332,698 | 298,770 | 223,556 | 125,927 | 263,450 |
| Non-redundant peptides | 31,189 | 41,064 | 43,503 | 49,168 | 33,974 | 34,172 | 41,028 |
| Identified proteins | 2,763 | 3,507 | 3,202 | 3,831 | 3,439 | 3,571 | 3,752 |
| Identified proteins (%) | 35.94 | 52.60 | 51.74 | 61.00 | 54.39 | 60.16 | 66.67 |
| Identified proteins with CUG | 778 | 1,451 | 1,928 | 2,331 | 2,122 | 2,843 | 3,533 |
| Identified proteins with CUG (%) | 33.52 | 46.05 | 49.03 | 56.56 | 47.47 | 56.95 | 66.62 |
| Covered CUG positions | 135 | 449 | 730 | 1,292 | 1,211 | 2,360 | 8,756 |
| PSMs covering CUGs | 1,185 | 2,973 | 3,462 | 5,500 | 4,937 | 5,803 | 51,737 |
| Non-redundant peptides covering CUGs | 210 | 494 | 836 | 1,438 | 1,325 | 2,560 | 10,797 |
| Supported CUG positions | 110 | 361 | 541 | 1,033 | 930 | 1,835 | 7,801 |
| Supported CUG positions (%) | 81.48 | 80.40 | 74.11 | 79.95 | 76.80 | 77.75 | 89.09 |
| Unambiguously translated, supported CUG positions (%)a | 99.09 | 98.89 | 97.78 | 98.74 | 99.03 | 99.24 | 96.45 |
| PSMs with supported CUG = Ser | 418 | 2,038 | 2,192 | 2,984 | 2,635 | 3,031 | 72 |
| PSMs with supported CUG = Leu | 501 | 31 | 36 | 26 | 22 | 14 | 47 |
| PSMs with CUG = Ser at positions also covered by PSMs with CUG = Leu | 357 | 40 | 180 | 102 | 0 | 1 | 2 |
| PSMs with CUG = Leu at positions also covered by PSMs with CUG = Ser | 394 | 18 | 26 | 13 | 0 | 1 | 1 |
| PSMs with supported CUG = Ala | 0 | 3 | 1 | 2 | 5 | 6 | 31,945 |
| PSMs with CUG = Ala at positions also covered by PSMs with CUG = Ser/Leu | 0 | 0 | 0 | 1 | 0 | 1 | 361 |
Aoas (1), A. asiatica sample 1; Acr, A. rubescens; Safi (1), S. fibuligera sample 1; Sama, S. malanga; Bai, B. inositovora; Cll, C. lusitaniae; Nape, N. peltata. See also Data S1 and S2.
“Unambiguously translated” refers to all CUG positions for which only peptides with one translation were found. These not only include CUG positions translated by the expected, cognate tRNA-decoded amino acid but also CUG positions translated by other amino acids that might result from genome sequencing ambiguities and differences between sequenced and analyzed strains. For A. asiatica, we regard translation by both serine and leucine cognate tRNACAG as “unambiguous.”
The A. asiatica coding sequences contain remarkably few CUG codons (4,936 codons as opposed to, for example, 27,696 in the DM clade yeast C. lusitaniae and 53,966 in N. peltata; Data S1). For 110 of the A. asiatica CUGs, we were able to resolve their translation with confidence (Table 1 and Data S1, sample 1). Remarkably, from the 929 PSMs covering those 110 CUG codon positions, 919 PSMs divide into almost equal parts to leucine (501 PSMs, 53.9%; 82 CUG positions) and serine (418 PSMs, 45.0%; 65 CUG positions; Figures 1A, S1B, and S1C; Table 1; Data S1). In contrast, we find that S. fibuligera and S. malanga both primarily translate CUG as serine, as evident in the 2,192 (93.0%; S. fibuligera sample 1) and 2,984 (96.8%; S. malanga) PSMs covering 513 and 997 CUG positions translated as serine, respectively (Figure 1A; Table 1; Data S1). A. rubescens contains similarly low numbers of CUG codons as A. asiatica (7,359 codons), but translates them unambiguously as serine (Figure 1A). Of the 2,119 PSMs covering 361 CUG codon positions, 2,038 (96.2%; 333 CUG positions) contain CUG codons translated as serine (Table 1; Data S1). Observed percentages of “only” about 95% PSMs covering correctly translated CUG codons compare to those observed in the DM clade yeasts B. inositovora and C. lusitaniae that both unambiguously translate CUG as serine: 95.3% (2,635 PSMs; 881 CUG positions) and 95.9% (3,031 PSMs; 771 CUG positions; Table 1; Data S1) of PSMs are translated as serine in B. inositovora and C. lusitaniae, respectively. Rather, the majority of the PSMs with other translations represent differences between sequenced and analyzed yeast strains or base-calling and coverage-dependent genomic ambiguities, because in general about 99% of the CUG positions are unambiguous (Table 1; Data S1). This ratio is neither restricted to translation as serine nor to low codon recovery, as evident in 96.5% (31,945 PSMs; 7,662 CUG positions; Table 1; Data S1) of PSMs translated as alanine in N. peltata (CUG codon recovery of 23.5%). Percentages of CUG codon positions supported by b-/y-type fragment ions are similar in all samples.
Figure 1.

Stochastic Translation of the A. asiatica CUG Codon
(A) Translations found in PSMs at b- and/or y-type fragment ion-supported CUG codon positions. A. asiatica is found to translate CUG stochastically into both leucine and serine, whereas A. rubescens, S. fibuligera, and S. malanga translate CUG codons into serine with a level comparable to that found for B. inositovora and C. lusitaniae, which both branch inside the DM clade. N. peltata is the second species found to use the Pachysolen genetic code. The size of the squares corresponds to the percentage of PSMs with supported CUG with the respective translation (see also Table 1, Figure S1, and Data S1 and S2).
(B) Number of PSMs covering CUG codon positions translated as leucine or serine in A. asiatica: for each CUG position; the number of PSMs with a leucine (blue bars; upper) and serine (orange bars; lower) at the CUG codon is given. Gray bars denote the number of all PSMs, including those where the respective leucine or serine is not supported by b-/y-type ions. Those CUG positions that are exclusively translated into leucine or serine in this sample but also found with the respective other translation (considering positions with b-/y-type ion support only) in another sample are highlighted by dots (see also Figure S2).
(C) The proportion of CUG codons translated as serine and leucine was determined for every CUG codon position for which PSMs with both translations were available, and the number of CUGs is plotted in bins of 10%: e.g., the first bin contains all CUGs with proportions from 0% to 10%.
(D) For every CUG codon position, we separately determined the proportion of PSMs with leucine and the proportion of PSMs with serine, and piled the number of PSMs for the respective proportion in bins of 10%. By far, most PSMs are found at positions with balanced translation, whereas only few PSMs are found at the CUG positions with unbalanced leucine-serine translation.
(E) The boxplot contrasts the number of PSMs found at CUG positions with stochastic translation with the number of PSMs found at CUG positions with only leucine or serine translation.
The unparalleled equal distribution of leucine and serine in A. asiatica could be caused by an endogenous, stochastic CUG codon translation or, as with stops recoded for selenocysteine, by flanking motifs determining that certain CUGs are always leucine and certain others are always serine. To test between these two possibilities, we considered what happens at any given position. At 44 CUG codon positions (40%), we found PSMs with both translations, and these positions are covered in total with almost as many PSMs with leucine (394 PSMs) as PSMs with serine (357; Figures 1B and S1C; Table 1; binomial test, p = 0.19). Most importantly, the distribution of PSMs with leucine and with serine is very similar for every single position (Figures 1C and 1D). At another 59 sites with fully supported CUG positions, we only recovered PSMs with either leucine (107 PSMs at 38 positions; mean of 2.8 per site) or serine (61 PSMs at 21 positions; mean of 2.9 per site) (Figure 1E). Because we observe unique translation into either leucine or serine only at CUG positions with low coverage, it seems plausible that deeper proteomic coverage would lead to observation of stochastic translation at these sites, too.
To exclude bias from sample preparation, we generated proteomics datasets from further, independent samples grown in different media (Data S1). Analysis of these data showed similar stochastic CUG translation in all samples (Figure S2A), considerable overlap of the covered CUG positions (Figure S2B), and, most notably, recovery of some of the CUG positions translated into only serine or leucine in sample 1 with the respective other amino acid (Figure 1B). We conclude that exceptionally, A. asiatica has stochastic translation of CUG to two possible fates. Analysis of the other 11 leucine and serine codons, of which CUC, AGC, and UCG have similarly low codon frequency as CUG, showed these to be translated unambiguously (Figure 1A; Data S2). This indicates that cognate tRNAs are functional and unambiguous, and that the stochastic CUG translation is indeed not an artifact caused by the low CUG codon coverage in the proteomics data.
Stochastic Encoding of CUG Is Best Explained by Competing tRNAs
The observed stochastic CUG translation in A. asiatica could either result from competing and or from misaminoacylation of one species of tRNACAG. The fact that the translation to leucine occurs at approximately the same rate as to serine is more compatible with the competing tRNA model, as prior examples of misaminoacylation give only very weak skews. In particular, misaminoacylation has been reported for Candida zeylanoides and Candida albicans, where their might be leucylated by the LeuRS to about only 3% [20, 21]. We are aware of no example where misaminoacylation occurs at 50:50 rates. By contrast, the high rates are potentially easily explained by the presence of two competing species of functional active tRNAs. Moreover, there is precedent for two different types of tRNA for the same codon in eukaryotes, albeit only in the context of deterministic translation, i.e., the UGA stop codon, where selenocysteine translation is extremely rare and highly specified by the selenocysteine insertion sequence (SECIS) element [8, 9]. Similarly, a few bacteria were also suggested to use sense codons for decoding selenocysteine, but in every case selenocysteine incorporation is specified by the SECIS element [22].
In silico and natural knockout analysis strongly support the viability of the competing tRNA model. The competing tRNA model predicts the presence of at least two distinct species of tRNACAG in A. asiatica, and this is indeed consistent with in silico evidence. To resolve the identities of the Ascoidea yeast tRNACAG, we predicted tRNAs in 137 sequenced yeast species and performed phylogenetic analyses of tRNACAG together with representatives from all isoacceptor Leu-, Ser-, and Ala-tRNAs (Figure 2A). Notably, A. asiatica, S. fibuligera, and S. malanga are predicted to each contain both a and a (A. asiatica contains two copies of ; Figure 2B). A. rubescens, by contrast, has only a gene. All four species encode , a tRNA that is capable of decoding CUG through wobble base pairing and has, incidentally, been lost in DM clade species.
Figure 2.

Yeast tRNACAG Phylogeny and Sequences
(A) An unrooted phylogenetic network of Leu-, Ser-, and Ala-tRNAs was generated with SplitsTree. Selections of Val-, Met-, Thr-, Arg-, Asn-, and Phe-tRNAs were added as outgroups. Leu-, Ser-, and Ala-tRNAs are colored by isoacceptor. CUG-encoding tRNAs of P. tannophilus (Pta), N. peltata (Nape), A. rubescens (Acr), A. asiatica (Aoas), S. fibuligera (Safi), S. malanga (Sama), B. inositovora (Bai), and C. lusitaniae (Cll) are highlighted for better orientation. Identical tRNA identity assignments are obtained in phylogenetic analyses using maximum-likelihood and Bayesian approaches.
(B) Sequences of the tRNACAG of the analyzed species. Some nucleotides are highlighted for faster orientation. The Saccharomycopsis differ most strikingly from the A. asiatica at the important position 20a, at which the presence of purine nucleotides has been shown to dramatically reduce leucylation efficiency. Guanine nucleotides 5′ and 3′ of the anticodon have been shown to cause low-level misleucylation in vitro, but the whole-cell proteomics data of C. lusitaniae show unambiguous CUG translation as serine in vivo. The two A. asiatica differ from the A. rubescens by only 1 and 2 nt and both substitutions are at variable positions.
Three species thus appear to have two species of tRNACAG, tempting the question: what is happening in the other two species, S. fibuligera and S. malanga? Here we see no evidence for 50:50 encoding. In these two, only 1.48% and 0.39% CUG positions (of 541 and 1,033 CUG positions covered in total) show dual translation, respectively. In addition, for those eight (S. fibuligera) and four (S. malanga) CUG positions with serine-leucine ambiguity, there are 6.9 and 7.8 times more PSMs with serine than PSMs with leucine, respectively, indicating extremely low usage or efficiency of (Table 1). Thus A. asiatica is exceptional. Importantly, this exceptionalism is reflected in the structure of its . In contrast to their , the from A. asiatica and the two Saccharomycopsis are distinct: they group differently in the phylogenetic trees and most likely have different origins (Figure 2). Consistent with the competing tRNA model, the Ascoidea clade contains all elements shown to be important for leucylation specificity and accuracy, such as a methylated G37, extended variable loop, and discriminator base A73 [23, 24, 25, 26]. The Saccharomycopsis , by contrast, differs from the A. asiatica and the Leu-tRNA consensus pattern by pyrimidine nucleotides at position 20a (Figure 2B). We also identified a in N. peltata that only shares the Ala-tRNA consensus nucleotides with the P. tannophilus , including the invariable G3-U70 base pair and the A73 discriminator base identity elements (Figure 2) [27, 28, 29]. Similar to the Ascoidea clade , these two have most likely been derived from different ancestors. Thus, the proteomics data evidence that the Saccharomycopsis yeasts and A. rubescens have switched CUG translation from the universal leucine to serine but that A. asiatica has been left with two functional tRNAs in the process.
Might it be possible that A. asiatica functions as an ambiguous tRNA? The presence of a unique in the close relative A. rubescens suggests not. This species translates CUG as serine, in accord with its unique tRNA. Importantly, the two A. asiatica are identical to the A. rubescens except for only 1 and 2 nt, respectively, and differ only in the variable loop from the Saccharomycopsis (Figure 2B). Importantly, all Ascoidea clade contain the conserved Ser-tRNA identity elements, the presence of a variable loop, and the discriminator base G73 [30, 31]. A37 has also been shown to be an antidiscriminant against the LeuRS [20]. Thus, the presence of a near-identical and unambiguously translated in A. rubescens provides a near-perfect natural knockout study looking at the effect of not having the . Because the two A. asiatica sequences are near identical to the A. rubescens sequence, both tRNAs can also be considered functional and unambiguously serylated. Assuming as much, leucines at CUG codons in A. asiatica must result from , which accordingly must be functional as well.
Although the evidence suggests the of A. asiatica must be functional (being such a strong resemblance to the functional species in A. rubescens), might the be misserylated? This seems highly unlikely, because the sequence contains all Leu-tRNA identity elements and is consistent with the Leu-tRNA consensus pattern, and the SerRS is highly specific for Ser-tRNAs, as evident from the unambiguous decoding of the five leucine codons (Data S2). Regardless, the A. asiatica must be a competitive decoding adaptor, because we found slightly more leucine than serine at CUG positions in all samples, although the ratio of to is 1 to 2.
Incidentally, from the Ascoidea clade yeasts are not related to from the DM clade. They belong to the tRNAGCU family (for AGY codons), whereas the monophyletic are from the DM clade group within the HGA isoacceptors (Figure 2A). The AGA, CGA, and UGA isoacceptors (for UCU, UCG, and UCA codons, respectively) do not form monophyletic groups. Thus, the DM clade could have originated from any of these isoacceptors and not necessarily from a tRNACGA ancestor, as suggested by the few tRNA sequences available 20 years ago [13, 32].
Overall, the situation in A. asiatica rather resembles an experiment in C. albicans, where expression of a heterologous in wild-type background resulted in increased leucine incorporation at CUG sites in a reporter protein to 28% [21]. Similar to misaminoacylation, RNA-editing processes can also not explain the observed stochastic translation into both leucine and serine, even more so because it would require the editing of at least 2 nt to switch a CUG into a serine codon. Decoding of CUG by the isoacceptor through wobble base pairing could be responsible for some ambiguity (as seen in A. rubescens [0.83%] and the Saccharomycopsis species [0.38% and 1.48%]) but not for 50:50 stochasticity. Thus, all evidence suggests that CUG translation in A. asiatica is in fact the result of the presence of competing and . Definitive evidence would require detailed biochemistry of tRNA-amino acid association, but this is currently not tractable in this non-model species.
A. asiatica Copes with Stochastic Coding by Avoiding CUG in Key Locations
Ambiguous decoding is expected to be a very unstable intermediate state and to be resolved by loss of one of the tRNAs. To determine how A. asiatica copes with such a sub-optimal condition, we analyzed the positions of CUG codons in alignments of 26 proteins from 137 sequenced Saccharomycotina yeasts and 11 fungal outgroup species. First, Ascoidea species have considerably fewer CUG codons at conserved protein alignment positions than other yeasts with reassigned CUG: whereas both B. inositovora and C. lusitaniae have discriminatory CUG codons at highly conserved serine positions, as has N. peltata at highly conserved alanine positions, all four Ascoidea clade yeasts lack CUG codons at highly conserved protein alignment positions (Figures 3 and S3). In A. asiatica in particular, none of the CUG codons fall at even moderately conserved alignment positions. This is not an effect of low codon usage, because all other leucine and serine codons show similar distributions on highly conserved alignment positions (Figures 3 and S3). Instead, this is likely to be the result of the stochastic codon translation selecting against CUG at positions of any importance (Figures 4A and 4B).
Figure 3.

Conservation of the CUG Codon in Comparison to CUN and AGY Box Codons
The plot on top denotes the total number of CUN box and AGY box codons in the concatenated cytoskeletal and motor protein sequence alignment, whereas the plots below show the percentage of each codon present at alignment positions with a certain conservation score. On the left, codons found at alignment positions enriched in the expected amino acid are shown, contrasted by codons found at alignment positions enriched in an unexpected amino acid on the right. For the remaining leucine and serine codons, see also Figure S3.
Figure 4.

Amino Acid Composition at CUG Positions and Codon Usage in A. asiatica
(A) Amino acid composition at the 30 positions with A. asiatica CUG codons in the alignment of the cytoskeletal and motor proteins. The upper plot shows the amino acid frequency at each CUG codon position. For comparison, the number of sequences at each position contributing to the frequency plot is shown in the lower plot. The number of sequences never reaches 148 (100%) because some yeasts always miss one or the other gene. 14 of the 30 positions are at alignment positions present in the majority of the 148 species, whereas the other 16 positions are in loop regions of variable length or species- or clade-specific protein extensions. At these positions, only some yeast sequences are present.
(B) Global amino acid frequency over all 30 positions with CUG codons in A. asiatica.
(C) Codon usage in the genome versus proteome. The scatterplot presents the fraction of each codon per family box according to its usage in the genome, determined by analysis of the gene prediction dataset, versus its usage in the proteome, determined by analysis of the MS/MS data. Serine and leucine family box codons and the CUG codon are highlighted by red, blue, and purple color, respectively. The CUG codon is the leucine (and serine, respectively) codon least used. Serine and leucine codons preferably used in the proteome are indicated for orientation. See also Figure S4.
(D) Percentage difference between theoretical and actual codon usage. Proteins actually captured by LC-MS/MS show a preference/rejection of certain codons compared to their average usage in the genome. The CUG codon is among those found less often than expected, meaning that it is used more often in lowly than in highly expressed genes. Leucine and serine codons are highlighted the same as in (C).
In contrast to the above, a low level of leucine (mis)incorporation at CUG positions does not select against CUG at conserved serine positions. DM clade species have similar numbers of CUG at conserved serine positions independent of having a potentially slightly misleucylated or having showing 100% serine identity due to the A37 antideterminant against LeuRS [33]. Unambiguous translation of CUG as serine both in B. inositovora and in C. lusitaniae (Figure 1A; Table 1; Data S1) also suggests that the m1G37 nucleotide, which was shown to cause minor-level misleucylation in vitro [20], might not have any effect on correct serylation in vivo because the C. lusitaniae contains m1G37 (Figure 2B). Also, there is no correlation of the number of CUGs at conserved serine positions with a free-living or pathogenic lifestyle of the Candida species [33]. Thus, it is considerably more likely that stochastic decoding can reduce or remove CUG from conserved positions whereas low-level mistranslation cannot.
Second, CUG codons are avoided in A. asiatica in general (Data S1) and, if used, used only in genes with very low to low expression levels (Figures 4C and 4D), both reducing the effective costs of stochastic encoding. In Ascoidea clade yeasts, CUG codons are genome-wide among the codons with lowest to third-lowest frequency. Accordingly, CUG is by far the least used codon of the serine codon box, with the lowest level in A. asiatica (1.2%; 1.3% when considered part of the leucine codon box) and slightly higher levels in the other Ascoidea clade yeasts (2.4%–4.9%; Figure S4). In contrast, CUG codons are well established in B. inositovora (7.4%) and C. lusitaniae (10.6%), and the CUG codon in N. peltata is, with 27.5%, the second-most used alanine codon (Figure S4). In addition to this genome-wide reduction, effective CUG usage is further decreased by maintaining CUG codons in lowly expressed genes only as evidenced by the codon usage found in the proteomes. In the A. asiatica proteome, 0.4% of serine codons (0.2% with respect to leucine codons) are CUG codons, as are 0.6%–1.8% of the serine codons of the other Ascoidea clade yeasts (Figure S4). This suggests that A. asiatica has in part solved the problem of stochastic CUG translation by avoidance of the problem.
CUG Stochasticity Was Probably Resolved by Loss of Function of the Gene in Other Species
How did A. asiatica’s closest relatives resolve codon ambiguity? To determine the most likely position and timing of the divergence of the Ascoidea and Saccharomycopsis yeasts, we combined concatenation of multiple genes with deep taxonomic sampling (Figures 5 and S5). The resulting phylogenies strongly support monophyly of the Ascoidea clade yeasts and their branching before the split of the branch containing the DM clade and Pichiaceae species and the branch containing the Phaffomycetaceae, Saccharomycetaceae, and Saccharomycodaceae. Mapping the tRNA data onto the tree shows that the origin of the Ascoidea dates back 190–230 Mya to the common origin of Ascoidea and Saccharomycopsis, whereas the are divergent in Ascoidea and Saccharomycopsis and presumably appeared only after the split of these two branches (Figure S6). The S. fibuligera and S. malanga are very similar, denoting a common origin in the ancient Saccharomycopsis. Given that these species predominantly translate CUG as serine, the ancient was either non-functional in the first place already, or became non-functional after a period of codon ambiguity. If the ancient was never functional, there would have been no constraint on reintroducing CUG codons at serine positions early. In this scenario, one would expect a considerable number of CUG codon positions to be shared between the two Saccharomycopsis, similar to the CUG position conservation seen in DM clade species (Figures 6 and S7) [33]. Such position conservation is, however, not found between the Saccharomycopsis, which in turn suggests that the ancestor of the Saccharomycopsis indeed experienced some time of codon ambiguity before its became non-functional. Notably, the Saccharomycopsis have purine nucleotides at position 20a in the D loop instead of the usual pyrimidine found in all yeast Leu-tRNAs including the A. asiatica (Figure 2B). Such purine nucleotides have been shown to reduce leucylation efficiency in human tRNALeu by a factor of 25 while not changing their tRNA identity [24]. These data suggest that even if the Saccharomycopsis are expressed at competitive levels, only a minor fraction is likely to be leucylated and functional. This is supported by analysis of RNA sequencing expression data for S. fibuligera under low and high glucose and sulfur limitation [16] showing the presence of the unprocessed (intron-containing) and in all conditions. For unknown reasons, these non-functional tRNAs were not disbanded already and are instead still kept in the genomes. In contrast, A. rubescens does not have a and therefore has either never experienced codon ambiguity or has resolved it by a more recent loss of its . The absence of any CUG codons at highly conserved serine positions and the very low total number of CUG codons strongly support the second scenario. Future sequencing efforts redeeming the present undersampling might well reveal Saccharomycopsis species without or Ascoidea relatives still containing a non-functional .
Figure 5.

Yeast Phylogeny
RAxML-generated phylogeny of 137 yeast species and 11 fungal outgroup species. Support values for major branches are given as bootstrapping replicates (RAxML-generated tree, numbers above branches) and posterior probabilities (MrBayes-generated tree, numbers below branches). For display purposes, species of major lineages have been collapsed into groups, with the numbers in parentheses denoting the number of species and the corners of the triangles representing the shortest and longest distances within the groups. Species and groups employing alternative genetic codes are indicated by color: “CTG clade” (DM clade) species A. rubescens, S. malanga, and S. fibuligera encode CUG as serine (red), N. peltata and P. tannophilus as alanine (green), and A. asiatica encodes CUG as both leucine and serine (purple). The grouping of the Ascoidea yeasts is consistent with a recent phylogenomics study [15]. Discrepancies with other published trees, which placed Ascoidea sister to the Phaffomycetaceae/Saccharomycetaceae/Saccharomycodaceae [34] or the Pichiaceae [17], are best explained by the deeper taxonomic sampling of early-branching yeasts (24 versus 10) in our study and the considerably increased sequence data (26 proteins versus 5), respectively. For each yeast, the presence (colored dots) and absence (white dots) of tRNA isoacceptors encoding the leucine (blue colors) and serine (red colors) codons are depicted at the side of the tree. For lineages collapsed in the tree, the tRNA repertoire of representative species is given. See also Figures S5 and S6.
Figure 6.

CUG Codon Position Conservation across Yeasts
The proportion of CUG codon positions in the concatenated cytoskeletal and motor protein sequence alignment shared between each two species or species groups. Proportions are calculated in relation to the number of CUG positions in the species or group with fewer CUG positions. Boxes on the diagonal represent the number of distinct CUG positions per species or species group. Given that CUG positions are present every 3.6th alignment position on average, there is remarkable clustering of the DM clade species and of species with CUG translated as leucine. In contrast, the Ascoidea yeasts share CUG positions only at background level, the level they share CUG positions with other yeasts. Notably, P. tannophilus and N. peltata do not share more CUG codon positions with each other than with any other yeast. This is consistent with their divergent , suggesting independent capture of the CUG codon. See also Figure S7.
The findings in A. asiatica’s relatives render it most parsimonious that they experienced a phase of CUG stochasticity that was in turn resolved by loss of function of the gene. Given the rate of introduction of CUGs at important positions in the DM clade yeasts, the finding that only few CUGs are found at highly conserved serine positions in Saccharomycopsis, and none in A. rubescens, is most parsimonious, with the possibility that resolving codon ambiguity was a rather recent event in these species. Interestingly, both A. rubescens and the two Saccharomycopsis independently opted for the same tRNA, the one coding for serine. This is even more surprising, as it should be favorable to reestablish the complete leucine codon box and subsequently profit from simpler codon mutating schemes and decoding redundancy. A reason might be that in the case of 2-fold codon capture, the tRNA charged with the less deleterious amino acid (i.e., less important for protein stability) will be selected for.
Although it is suggestive that the solution seen in A. asiatica is an unstable solution and is generally deleterious and that A. asiatica is expected to also evolve to a position where it loses one of the two tRNAs in its evolutionary future, A. asiatica seems to have been living with stochastic translation for already 100 million years (Figure S6). Stochastic translation might have been present in the ancestor of Ascoidea for an additional 100 million years before the split of A. rubescens. Thus, dramatically reducing the frequency of a certain codon and only using this codon in lowly expressed genes seems to be sufficient for a species to retain long-time viability. The growth rate of A. asiatica was similar to that of the other yeasts, indicating that the endogenous stochastic translation is not detrimental to A. asiatica’s fitness in rich medium. Although the evidence suggests that stochastic encoding is simply tolerated, whether there might be unusual circumstances where stochastic translation is beneficial is worthy of consideration.
Discussion
Here we have shown that A. asiatica has an exceptional system in which the codon CUG is translated as either leucine or serine at high relative rates in a stochastic manner. A consequence of this is that the proteome is not deterministically predictable from the genome. This is tolerated by selection against CUG generally, and especially at key locations and in highly expressed genes. The most parsimonious model to explain this supposes that the species has two functional tRNA species for the translation of CUG.
Are there any precedents? It has been reported that several nematodes encode leucine-type tRNAs with anticodons matching among others mainly glycine or isoleucine codons (together termed “nev-tRNAs”) [35], and that bacteria from the Clostridia, Proteobacteria, and Acidobacteria phyla contain novel types of tRNAs (termed “allo-tRNAs”), which are structurally similar to Sec-tRNA and have identity elements of Ser-tRNAs but contain anticodons corresponding to 35 distinct codons [22, 36]. In vitro aminoacylation experiments demonstrated that the nev-tRNAs are leucylated and that these tRNAs are able to decode GGG and AUA codons in translation assays [35]. However, whole-cell proteome analyses of Caenorhabditis elegans did not reveal detectable levels of leucines at GGG glycine codons, indicating that these nev-tRNAs are not used in vivo [37]. Similarly, multiple allo-tRNAs have been shown to be aminoacylated in vitro and to be used in translation in E. coli, but usage in their host organism has not been demonstrated yet [36]. Furthermore, although these allo-tRNAs suggest altered genetic codes in the respective hosts, genomic data demonstrating the presence or absence of standard cognate tRNAs are missing. Thus, these bacteria could have the genetic code strictly maintained or could employ alternative codes, and some might even show stochastic translation of one of the respective codons.
Our finding that stochastic translation is in general selected against but may still survive hundreds of millions of years in rare cases such as in Ascoidea clade species suggests that similar codon ambiguity might be present in other species as well although not yet detected. Bacteria with allo-tRNAs might be the best candidates to look for and investigate potential further cases of stochastic translation. Other principally deleterious codon reassignments, such as the dual decoding of stop codons, have also been found in independent species [9, 10, 11].
Do the new data fit into existing models of codon reassignment? At first glance, the situation found in A. asiatica seems to represent a prime example of the ambiguous intermediate hypothesis, according to which a new mutant tRNA appears and competes with the original cognate tRNA [38, 39]. This competition is thought to cause gradual codon frequency reduction and codon identity change followed by loss of the former cognate tRNA, and finally results in codon reassignment. One of the main ideas behind this scenario is that there should be faster evolutionary processes, such as selection, than genome-wide mutation and drift in codon frequency, which are the main causes for codon reassignment according to the codon capture hypothesis [40, 41]. However, considering the new findings about genetic codes in the Ascoidea clade, a global scenario for the entire yeast clade based on ambiguous intermediate states with competing tRNAs seems highly unlikely. First, at least six independent CUG capture events by completely different types of tRNAs with a combined probability of at most (1/64)6 would have to be considered (different types of in the DM clade and the Ascoidea clade, divergent in Pachysolen and Nakazawaea, and divergent in Ascoidea and Saccharomycopsis branches, plus divergent in Saccharomycetales). Even if the and Ascoidea clade had been of common ancestry, there would have been still three independent ambiguous intermediate events (combined probability of (1/64)3). Second, the ambiguous intermediate scenario fails to explain the polyphyly of in Saccharomycetales and offers no apparent explanation for the complete absence of cognate in Saccharomycodaceae and many Saccharomycetaceae [14]. Third, codon reassignments do not necessarily happen by fast, selection-driven processes, as evidenced by 100 million years of codon ambiguity in A. asiatica and up to 100 million years in the ancestors of the Ascoidea and the Saccharomycopsis. All these findings can, however, be well explained by the recently proposed tRNA-loss-driven codon reassignment hypothesis [14]. Indeed, both the further reassignments in independent yeast branches and the CUG capture by GCU-type Ser-tRNAs are predictions of this theory. According to this theory, the reassignments in yeasts originated from a single event, the loss of the original cognate before the split of the Ascoidea clade. The free codon could have subsequently been captured by any , , or (being the only tRNA species where the anticodon is not part of the aaRS recognition site). Although not considered when the theory was originally proposed, the tRNA-loss-driven codon reassignment scenario also allows for capture by two different tRNAs, as found in the Ascoidea clade. The Saccharomycopsis can thus be regarded as silenced cases of dual-codon capture, whereas A. asiatica is a frozen accident of dual-codon capture trapped in ambiguity for about 200 million years.
Previous examples of codons with dual and triple meanings were stop codons with the respective translation highly regulated and specified by codon context. Our finding of endogenous stochastic decoding by competing tRNAs provides the first example of a living species where the proteome cannot be deterministically predicted from the genome.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Ascoidea asiatica NRRL Y-17576 genome assembly | NCBI | BCKQ01000001-BCKQ01000071 |
| Ascoidea rubescens DSM 1968 genome assembly | NCBI [15] | LYBR01000001-LYBR01000326 |
| Babjeviella inositovora NRRL Y-12698 genome assembly | NCBI [15] | LWKQ01000001-LWKQ01000211 |
| Clavispora lusitaniae ATCC 42720 genome assembly | NCBI [42] | AAFT01000001-AAFT01000088 |
| Saccharomycopsis fibuligera KPH12 genome assembly | NCBI [16] | CP012823-CP012829 |
| Saccharomycopsis malanga NRRL Y-7175 genome assembly | NCBI | BCGJ01000001-BCGJ01000044 |
| Ascoidea asiatica NRRL Y-17576 genome annotation | NBRP | N/A |
| Ascoidea asiatica NRRL Y-17576 | This paper | N/A |
| Ascoidea rubescens DSM 1968 genome annotation | Ensembl Fungi [43] | N/A |
| Babjeviella inositovora NRRL Y-12698 genome annotation | Ensembl Fungi [43] | N/A |
| Clavispora lusitaniae ATCC 42720 genome annotation | Ensembl Fungi [43] | N/A |
| Nakazawaea peltata NRRL Y-6888 | NBRP | N/A |
| Saccharomycopsis fibuligera KPH12 | This paper | N/A |
| Saccharomycopsis malanga NRRL Y-7175 | NBRP | N/A |
| tRNA identification | [14]; This paper | N/A |
| Sequence data | Figshare [33]; This paper | https://doi.org/10.6084/m9.figshare.6086639 |
| Phylogenetic trees | Figshare; This paper | https://doi.org/10.6084/m9.figshare.6086639 |
| Mass spectrometry data | ProteomeXchange via PRIDE [44] | PXD009494 |
| Experimental Models: Organisms/Strains | ||
| Ascoidea asiatica | NRRL | Y-17576 |
| Ascoidea rubescens | DSMZ | 1968 |
| Babjeviella inositovora | NRRL | Y-12698 |
| Clavispora lusitaniae | NRRL | Y-11827 |
| Nakazawaea peltata | NRRL | Y-6888 |
| Saccharomycopsis fibuligera | NRRL | Y-2388 |
| Saccharomycopsis malanga | NRRL | Y-7175 |
| Software and Algorithms | ||
| Custom scripts for data generation and parsing | [14]; This paper | N/A |
| Gene prediction | AUGUSTUS [45] | http://bioinf.uni-greifswald.de/augustus/binaries/ |
| Mass spectrometry analysis and search | MaxQuant [46] | http://www.coxdocs.org/doku.php?id=maxquant:common:download_and_installation |
| tRNA identification | tRNAscan [47] | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| Alignment redundancy reduction | CD-HIT [48] | http://weizhongli-lab.org/cd-hit/ |
| Maximum likelihood tree calculation | RAxML v8.2.10 [49] | https://github.com/stamatak/standard-RAxML |
| Maximum likelihood tree calculation | FastTree v2.1.9 [50] | http://www.microbesonline.org/fasttree/#Install |
| Maximum likelihood tree calculation | IQ-TREE v1.63b [51] | https://github.com/Cibiv/IQ-TREE |
| Scoring of substitution models for (tRNA) ML-tree generation | jModelTest v2.1.10 [52] | https://github.com/ddarriba/jmodeltest2 |
| Scoring of substitution models for (protein) ML-tree generation | ProtTest v3.4.2 [53] | https://github.com/ddarriba/prottest3 |
| Bayesian tree calculation (tRNA) | Phase v3.0 [54] | https://github.com/james-monkeyshines/rna-phase-3 |
| Bayesian tree calculation (protein) | MrBayes v3.2.6 [55] | http://mrbayes.sourceforge.net/download.php |
| Phylogenetic network calculation | SplitsTree v4.14.4 [56] | http://ab.inf.uni-tuebingen.de/data/software/splitstree4/download/welcome.html |
| Alignment position reduction | Gblocks v0.91b [57] | http://molevol.cmima.csic.es/castresana/Gblocks.html |
| Divergence time estimation | treePL [58] | https://github.com/blackrim/treePL |
| Tree visualization | FigTree v1.4.3 (Rambaut and Drummond) | http://tree.bio.ed.ac.uk/software/figtree/ |
| Gene structure reconstruction | WebScipio [59] | http://www.webscipio.org/ |
| Alignment position conservation calculation | conservation code toolbox [44] | http://compbio.cs.princeton.edu/conservation/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Martin Kollmar (mako@nmr.mpibpc.mpg.de).
Method Details
Growth and lysis of yeast species
Babjeviella inositovora NRRL Y-12698, Clavispora lusitaniae NRRL Y-11827 (CBS 6936), Nakazawaea peltata NRRL Y-6888, Saccharomycopsis fibuligera NRRL Y-2388 (ATCC 36309) and Saccharomycopsis malanga NRRL Y-7175 were obtained from the Agricultural Research Service (ARS) Culture Collection Database (NRRL - Northern Regional Research Laboratory). C. lusitaniae was grown in YEPD medium (containing [% w/v]: bacto peptone 2.0; yeast extract 1.0; glucose 2.0) at 25°C. B. inositovora, N. peltata and S. malanga were grown in YM medium (NRRL Medium No. 6, containing [% w/v]: yeast extract 0.3; malt extract 0.3; peptone 0.5; glucose 1.0) at 25°C. S. fibuligera samples were grown in YM medium (sample [1]) and malt extract medium (sample [2]; ATCC Medium 325 [Blakeslee’s formula; % w/v]: malt extract 2.0; glucose 2.0; peptone 1.0) at 25°C. Cells were harvested by centrifugation (5′ at 4,400 x g), and washed with water. Aliquots of cells were lysed in 2 M NaOH and 5% mercaptoethanol, and proteins precipitated with 10% trichloroacetic acid (TCA) (both steps with 10 min incubation on ice). For neutralizing, the pellet was rinsed once with 1.5 M TRIS-base and proteins were resuspended in SDS sample buffer. Proteins were resolved on 4%–12% SDS-PAGE.
Growth and lysis of Ascoidea rubescens
Ascoidea rubescens DSM 1968 ( = NRRL Y-17699) was obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ - Deutsche Sammlung von Mikroorganismen und Zellkulturen). Cells were grown in malt-soya peptone medium (containing [% w/v]: malt extract 3.0, soya peptone 0.3) at 22°C. Clusters of A. rubescens cells were recovered using a loop. After washing with water cells were ground in liquid nitrogen. Sample buffer was added to the extract and the suspension was collected and fractionated by SDS-PAGE.
Growth and lysis of Ascoidea asiatica
Ascoidea asiatica NRRL Y-17576 was obtained from the Agricultural Research Service (ARS) Culture Collection Database (NRRL - Northern Regional Research Laboratory) and grown in malt-soya peptone medium (sample [1]), malt extract medium (samples [2] and [4]), and YM (sample [3]) at 22°C. A. asiatica cells from sample [1] were collected by centrifugation and washed. After washing with water cells were ground in liquid nitrogen. Cells from samples [2] to [4] were harvested by centrifugation (5′ at 4,400 x g), and washed with water. Aliquots of cells were lysed in 2 M NaOH and 5% mercaptoethanol, and proteins precipitated with 10% trichloroacetic acid (TCA) (both steps with 10 min incubation on ice). For neutralizing, the pellet was rinsed once with 1.5 M TRIS-base. Sample buffer was added to the extracts and the suspensions were collected and fractionated by SDS-PAGE.
Genome assemblies and annotation
All genome assemblies were obtained from NCBI with the following GenBank accessions: Ascoidea asiatica NRRL Y-17576: BCKQ01000001-BCKQ01000071; Ascoidea rubescens DSM 1968: LYBR01000001-LYBR01000326 [15], Babjeviella inositovora NRRL Y-12698: LWKQ01000001-LWKQ01000211 [15]; Clavispora lusitaniae ATCC 42720: AAFT01000001-AAFT01000088 [42]; Saccharomycopsis fibuligera KPH12: CP012823-CP012829 [16]; and Saccharomycopsis malanga NRRL Y-7175: BCGJ01000001-BCGJ01000044. Genome annotations for Ascoidea rubescens DSM 1968 [15], Babjeviella inositovora NRRL Y-12698 [15] and Clavispora lusitaniae ATCC 42720 [42] were obtained from Ensembl Fungi [43]. The genome annotations for Ascoidea asiatica NRRL Y-17576, Nakazawaea peltata NRRL Y-6888 and Saccharomycopsis malanga NRRL Y-7175 were obtained from the National BioResource Project (NBRP) program web page (http://www.jcm.riken.jp/cgi-bin/nbrp/nbrp_list.cgi). Ascoidea asiatica NRRL Y-17576 and Saccharomycopsis fibuligera KPH12 genes were predicted with AUGUSTUS [45] using the parameter “genemodel=complete,” the gene feature set of Candida albicans, and the standard codon translation table.
Mass spectrometry sequencing
SDS-PAGE-separated protein samples were processed as described by Shevchenko et al. [60]. The resuspended peptides in sample loading buffer (2% acetonitrile and 0.05% trifluoroacetic acid) were separated and analyzed by an UltiMate 3000 RSLCnano HPLC system (Thermo Fisher Scientific) coupled online to a Q Exactive HF or a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). First, the peptides were desalted on a reverse phase C18 pre-column (Dionex 5 mm long, 0.3 mm inner diameter) for 3 min. After 3 min the precolumn was switched online with the analytical column (30 cm long, 75 μm inner diameter) prepared in-house using ReproSil-Pur C18 AQ 1.9 μm reversed phase resin (Dr. Maisch GmbH). The peptides were separated with a linear gradient of 5%–35% buffer (80% acetonitrile and 0.1% formic acid) at a flow rate of 300 nl/min (with back pressure 500 bars) over 88 min gradient time. The pre-column and the column temperature were maintained at 50°C. In the Q Exactive Plus the MS data were acquired by scanning the precursors in mass range from 350 to 1600 m/z at a resolution of 70,000 at m/z 200. Top 20 precursor ions were chosen for MS2 by using data-dependent acquisition (DDA) mode at a resolution of 17,500 at m/z 200 with maximum IT of 50 ms. In the Q Exactive HF the MS data were acquired by scanning the precursors in mass range from 350 to 1600 m/z at a resolution of 60,000 at m/z 200. Top 30 precursor ions were chosen for MS2 by DDA mode at a resolution of 15,000 at m/z 200 with maximum IT of 50 ms. Data for Ascoidea asiata, Saccharomyces fibuligera and Ascoidea rubescens where measured on Q Exactive Plus instrument. All other Data where measured on Q Exactive HF instrument.
Mass spectrometry analysis
Data analysis and search were performed using MaxQuant v.1.5.2.8 [46] as search engine with 1% FDR. To obtain peptide mappings free of CUG-translation bias, 20 replicates for each genome annotation were generated with the CUG codon translated as different amino acid in each replicate. To reduce database size and redundancy, predicted proteins were split at lysine and arginine residues into peptides resembling trypsin proteolysis. Peptides containing CUG codons were fused together with the two subsequent peptides so that CUG-containing fragments can be detected with up to two missed cleavages. The remaining peptides were fused back together as long as they formed consecutive blocks. By this process we could reduce database size and redundancy by 31%–89% depending on CUG-usage in the respective coding sequences. Search parameters for searching the precursor and fragment ion masses against the databases were as described in Oellerich et al. [61] except that all peptides shorter than seven amino acids were excluded. The datasets were searched with the gene prediction dataset for the respective species, except for the second sample [2] of A. asiatica that was searched with both the gene prediction dataset from NBRP [ = 2A] and the newly generated AUGUSTUS gene prediction dataset [ = 2B]. To claim CUG codon translations with high confidence, we determined CUG positions with b- and y-type fragment ions at both sides that allow for determining the amino acids’ mass. Only those positions were regarded as fully supported by the data. In addition, we regard the first two amino acids as combinedly fully supported if a b- and/or y-type fragment ion exists for the C-terminal site of this di-peptide and the combined mass of the two amino acids is unambiguous.
tRNA gene identification and alignment
tRNA genes from 60 Saccharomycetes and four Schizosaccharomycetes were taken from a previous analysis [14]. tRNA genes for additional 77 Saccharomycetes sequenced since then were identified with tRNAscan [47] using standard parameters. The tRNAs from the 60 Saccharomycetes and four Schizosaccharomycetes were sorted by anticodon. From the newly sequenced yeasts, only the tRNAs with CAG anticodon were extracted from the predictions and added to the other tRNACAG. The tRNAs from each anticodon group were aligned and mitochondrial tRNAs, fragmented tRNAs and obviously unusual tRNAs were removed manually. To generate a dataset with a broad and unbiased sampling of as many tRNA types as possible, redundancy for all anticodon groups but CAG was reduced to 90% sequence identity by applying the CD-HIT suite [48]. The CAG anticodon group was first split into leucine-, serine, and alanine-encoding tRNAs and then reduced to 95% sequence identity.
To prepare a representative tRNA dataset for tRNA-type determination, all tRNACAG from the reduced alignments, the first six tRNAs from each leucine, serine, alanine, valine, phenylalanine, asparagine and methionine anticodon alignment, and the first six tRNAs from tRNACAG the AGU threonine anticodon alignment were combined.
tRNA phylogeny
tRNA phylogenies were inferred using maximum likelihood, Bayesian and split networks methods. 1) Maximum likelihood trees were computed with RAxML v8.2.10 [49], FastTree v2.1.9 [50], and IQ-TREE v1.63b [51]. First, a substitution model was selected using jModelTest v2.1.10 [52]. jModelTest found the GTR +G +I model to be the best under the AICc framework followed by GTR +G as second best model. RAxML was run with substitution model GTR +G +I and 1,000 bootstrap replicates. FastTree does not allow to control for proportion of invariable sites, and was therefore started with the second best substitution model, GTR +G. IQ-Tree was run with the model selected by its build-in ModelSelector according to BIC (Ala-tRNA alignment: TIMe+G4; Ser-tRNA alignment: TVMe+I+G4; Leu-tRNA alignment: TPM2u+I+G4; alignment of representative tRNAs: TVM+R5). To assess branch support, the analyses were performed with 1,000 bootstrap replicates. 2) Bayesian trees were inferred using Phase v3.0 [54] and MrBayes v3.2.6 [55]. Phase was started with a mixed model consisting of REV +G for loops and RNA7D +G for stem regions as suggested by the developers in their example control files. 750,000 burn-in cycles and 1,500,000 sampling cycles with a sampling period of 150 cycles have been performed. Met-tRNAs were defined to form a monophyletic cluster. MrBayes was started with the 4by4 option, two independent runs with 1,000,000 generations, four chains, and a random starting tree. Trees were sampled every 1,000th generation and the first 25% of the trees were discarded as “burn-in” before generating a consensus tree. A separate run was performed with structural information and a partitioned model with option 4by4 for loop regions and doublet for stem regions. 3) An unrooted phylogenetic network was computed using SplitsTree v4.14.4 [56] with the neighbor-net method and 1,000 bootstrap replicates.
Generating the protein sequence alignment
The protein sequences of the actin and actin-related, CapZ, dynein heavy chain, kinesin, myosin and tubulin proteins of 81 yeasts, four Pezizomycotina and three Basidiomycota were added to the already existing multiple sequence alignments from 60 yeast species following the previously described approach [33]. A 148-taxa, 26-protein supermatrix was then constructed for further analysis, resulting in an alignment of 35,202 columns. A reduced alignment was generated using Gblocks v0.91b [57] with parameters allowing less stringent block selection (smaller final blocks, gap positions within the final blocks, less strict flanking positions). Gblocks reduced the alignment to 7,942 amino acid positions in 385 blocks.
Inferring species phylogeny
Phylogenetic trees were generated on both the full and the gblocks-reduced alignments using two different methods: 1) Bayesian trees were inferred using MrBayes v3.2.6 [55] with the mixed amino acid option, two independent runs with 1,000,000 generations, four chains, and a random starting tree. 2) Maximum likelihood trees were inferred with RAxML v8.2.10 [49] and IQ-TREE v1.63b [51]. RAxML was run with substitution model LG +G +I, which was the best-fitting model according to the Bayesian information criterion (BIC) determined by ProtTest v3.4.2 [53], and 1,000 bootstrap replicates. IQ-Tree was run with the model selected by its build-in ModelSelector according to BIC, LG +F +R12 for the full alignment and LG +F +R11 for the gblocks-reduced alignment. To assess branch support, the analyses were performed with 1,000 bootstrap replicates. Both ML methods gave effectively identical results, as did gblocks-reduced and full alignments, indicating that the results are not software specific. The divergence times of species were estimated with the penalized-likelihood approach as implemented in treePL [58] based on the RAxML-generated tree of the full alignment. The splits between Saccharomyces cerevisiae and Candida albicans, and C. albicans and Neurospora crassa [62] were constrained simultaneously. All phylogenetic trees were visualized using FigTree v1.4.3 [63].
Calculating CUG position conservation
Gene structures of the assembled protein sequences were reconstructed with WebScipio [59], and the structures of all “complete” genes (e.g., genes that do not contain a sequence shift) were mapped onto the concatenated protein sequence alignment allowing any kind of codon-based comparisons. Overall, the mapped genes contain 34,517 CTG codons that distribute to 9,857 alignment positions.
Conservation of leucine, serine and alanine alignment positions
Conservation scores were calculated for all alignment positions containing leucine, serine or alanine with the conservation code toolbox [44], a window size of 3 and the property entropy as conservation estimation method. Alignment blocks of 15 positions before and after the respective position of interest were generated to reduce any further influence of the rest of the alignment on the scoring process. Sequences with CUG codons in the block have been retained. Any stop codons present in the concatenated alignment have been replaced by ‘X’ for calculating scores.
Quantification and Statistical Analysis
Binomial test was implemented using R function binom.test, with p = 0.5 and employing a two sided test.
Data and Software Availability
The mass spectrometry data from this study have been submitted to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE [64] partner repository with the dataset identifier PXD009494. Sequence data and phylogentic trees are available from Figshare (https://doi.org/10.6084/m9.figshare.6086639).
Acknowledgments
The authors would like to thank Rikiya Endoh, PhD, and the National BioResource Project (NBRP) program for generating the yeast genome assemblies and annotations and for permitting us to use the data prior to publication. In particular, we thank Rikiya Endoh for his comments on and careful reading of the manuscript. M.K. would like to thank Prof. Dr. Christian Griesinger for his continuous generous support. This work was supported by the European Research Council (advanced grant ERC-2014-ADG 669207 to L.D.H.) and the Medical Research Council (MR/L007215/1 to L.D.H.).
Author Contributions
M.K. conceived the study. S.M. generated genome annotations and performed MS/MS data and phylogenetic analyses. H.D.S. prepared experimental samples. K.-T.P. and U.P. performed MS/MS experiments. H.U. supervised MS/MS analyses. L.D.H. was involved in data interpretation and manuscript writing. M.K. assembled and analyzed protein and tRNA sequences. S.M. and M.K. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: June 14, 2018
Footnotes
Supplemental Information includes seven figures and two data files and can be found with this article online at https://doi.org/10.1016/j.cub.2018.04.085.
A video abstract is available at https://doi.org/10.1016/j.cub.2018.04.085#mmc5.
Supplemental Information
Mass spectrometry analysis of yeast proteomes.
Mass spectrometry analysis of the A. asiatica (1) proteome, leucine and serine codons.
References
- 1.Zaher H.S., Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wohlgemuth I., Pohl C., Mittelstaet J., Konevega A.L., Rodnina M.V. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:2979–2986. doi: 10.1098/rstb.2011.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohler K., Ibba M. Translational fidelity and mistranslation in the cellular response to stress. Nat. Microbiol. 2017;2:17117. doi: 10.1038/nmicrobiol.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leinfelder W., Zehelein E., Mandrand-Berthelot M.A., Böck A. Gene for a novel tRNA species that accepts L-serine and cotranslationally inserts selenocysteine. Nature. 1988;331:723–725. doi: 10.1038/331723a0. [DOI] [PubMed] [Google Scholar]
- 5.Metanis N., Hilvert D. Natural and synthetic selenoproteins. Curr. Opin. Chem. Biol. 2014;22:27–34. doi: 10.1016/j.cbpa.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Keeling P.J. Genomics: evolution of the genetic code. Curr. Biol. 2016;26:R851–R853. doi: 10.1016/j.cub.2016.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Kollmar M., Mühlhausen S. Nuclear codon reassignments in the genomics era and mechanisms behind their evolution. BioEssays. 2017 doi: 10.1002/bies.201600221. Published online March 20, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Turanov A.A., Lobanov A.V., Fomenko D.E., Morrison H.G., Sogin M.L., Klobutcher L.A., Hatfield D.L., Gladyshev V.N. Genetic code supports targeted insertion of two amino acids by one codon. Science. 2009;323:259–261. doi: 10.1126/science.1164748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swart E.C., Serra V., Petroni G., Nowacki M. Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell. 2016;166:691–702. doi: 10.1016/j.cell.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heaphy S.M., Mariotti M., Gladyshev V.N., Atkins J.F., Baranov P.V. Novel ciliate genetic code variants including the reassignment of all three stop codons to sense codons in Condylostoma magnum. Mol. Biol. Evol. 2016;33:2885–2889. doi: 10.1093/molbev/msw166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Záhonová K., Kostygov A.Y., Ševčíková T., Yurchenko V., Eliáš M. An unprecedented non-canonical nuclear genetic code with all three termination codons reassigned as sense codons. Curr. Biol. 2016;26:2364–2369. doi: 10.1016/j.cub.2016.06.064. [DOI] [PubMed] [Google Scholar]
- 12.Kawaguchi Y., Honda H., Taniguchi-Morimura J., Iwasaki S. The codon CUG is read as serine in an asporogenic yeast Candida cylindracea. Nature. 1989;341:164–166. doi: 10.1038/341164a0. [DOI] [PubMed] [Google Scholar]
- 13.Miranda I., Silva R., Santos M.A.S. Evolution of the genetic code in yeasts. Yeast. 2006;23:203–213. doi: 10.1002/yea.1350. [DOI] [PubMed] [Google Scholar]
- 14.Mühlhausen S., Findeisen P., Plessmann U., Urlaub H., Kollmar M. A novel nuclear genetic code alteration in yeasts and the evolution of codon reassignment in eukaryotes. Genome Res. 2016;26:945–955. doi: 10.1101/gr.200931.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riley R., Haridas S., Wolfe K.H., Lopes M.R., Hittinger C.T., Göker M., Salamov A.A., Wisecaver J.H., Long T.M., Calvey C.H. Comparative genomics of biotechnologically important yeasts. Proc. Natl. Acad. Sci. USA. 2016;113:9882–9887. doi: 10.1073/pnas.1603941113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choo J.H., Hong C.P., Lim J.Y., Seo J.-A., Kim Y.-S., Lee D.W., Park S.-G., Lee G.W., Carroll E., Lee Y.-W., Kang H.A. Whole-genome de novo sequencing, combined with RNA-seq analysis, reveals unique genome and physiological features of the amylolytic yeast Saccharomycopsis fibuligera and its interspecies hybrid. Biotechnol. Biofuels. 2016;9:246. doi: 10.1186/s13068-016-0653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurtzman C.P., Robnett C.J. Relationships among genera of the Saccharomycotina (Ascomycota) from multigene phylogenetic analysis of type species. FEMS Yeast Res. 2013;13:23–33. doi: 10.1111/1567-1364.12006. [DOI] [PubMed] [Google Scholar]
- 18.Chi Z., Chi Z., Liu G., Wang F., Ju L., Zhang T. Saccharomycopsis fibuligera and its applications in biotechnology. Biotechnol. Adv. 2009;27:423–431. doi: 10.1016/j.biotechadv.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Mühlhausen S., Kollmar M. Predicting the fungal CUG codon translation with Bagheera. BMC Genomics. 2014;15:411. doi: 10.1186/1471-2164-15-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki T., Ueda T., Watanabe K. The ‘polysemous’ codon—a codon with multiple amino acid assignment caused by dual specificity of tRNA identity. EMBO J. 1997;16:1122–1134. doi: 10.1093/emboj/16.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes A.C., Miranda I., Silva R.M., Moura G.R., Thomas B., Akoulitchev A., Santos M.A.S. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007;8:R206. doi: 10.1186/gb-2007-8-10-r206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukai T., Englert M., Tripp H.J., Miller C., Ivanova N.N., Rubin E.M., Kyrpides N.C., Söll D. Facile recoding of selenocysteine in nature. Angew. Chem. Int. Ed. Engl. 2016;55:5337–5341. doi: 10.1002/anie.201511657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breitschopf K., Gross H.J. The exchange of the discriminator base A73 for G is alone sufficient to convert human tRNA(Leu) into a serine-acceptor in vitro. EMBO J. 1994;13:3166–3169. doi: 10.1002/j.1460-2075.1994.tb06615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breitschopf K., Achsel T., Busch K., Gross H.J. Identity elements of human tRNA(Leu): structural requirements for converting human tRNA(Ser) into a leucine acceptor in vitro. Nucleic Acids Res. 1995;23:3633–3637. doi: 10.1093/nar/23.18.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soma A., Kumagai R., Nishikawa K., Himeno H. The anticodon loop is a major identity determinant of Saccharomyces cerevisiae tRNA(Leu) J. Mol. Biol. 1996;263:707–714. doi: 10.1006/jmbi.1996.0610. [DOI] [PubMed] [Google Scholar]
- 26.Yao P., Zhu B., Jaeger S., Eriani G., Wang E.-D. Recognition of tRNALeu by Aquifex aeolicus leucyl-tRNA synthetase during the aminoacylation and editing steps. Nucleic Acids Res. 2008;36:2728–2738. doi: 10.1093/nar/gkn028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou Y.M., Schimmel P. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988;333:140–145. doi: 10.1038/333140a0. [DOI] [PubMed] [Google Scholar]
- 28.Hou Y.M., Schimmel P. Evidence that a major determinant for the identity of a transfer RNA is conserved in evolution. Biochemistry. 1989;28:6800–6804. doi: 10.1021/bi00443a003. [DOI] [PubMed] [Google Scholar]
- 29.Shi J.P., Francklyn C., Hill K., Schimmel P. A nucleotide that enhances the charging of RNA minihelix sequence variants with alanine. Biochemistry. 1990;29:3621–3626. doi: 10.1021/bi00467a005. [DOI] [PubMed] [Google Scholar]
- 30.Achsel T., Gross H.J. Identity determinants of human tRNA(Ser): sequence elements necessary for serylation and maturation of a tRNA with a long extra arm. EMBO J. 1993;12:3333–3338. doi: 10.1002/j.1460-2075.1993.tb06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Normanly J., Ollick T., Abelson J. Eight base changes are sufficient to convert a leucine-inserting tRNA into a serine-inserting tRNA. Proc. Natl. Acad. Sci. USA. 1992;89:5680–5684. doi: 10.1073/pnas.89.12.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueda T., Suzuki T., Yokogawa T., Nishikawa K., Watanabe K. Unique structure of new serine tRNAs responsible for decoding leucine codon CUG in various Candida species and their putative ancestral tRNA genes. Biochimie. 1994;76:1217–1222. doi: 10.1016/0300-9084(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 33.Mühlhausen S., Kollmar M. Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage. Genome Biol. Evol. 2014;6:3222–3237. doi: 10.1093/gbe/evu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen X.-X., Zhou X., Kominek J., Kurtzman C.P., Hittinger C.T., Rokas A. Reconstructing the backbone of the Saccharomycotina yeast phylogeny using genome-scale data. G3 (Bethesda) 2016;6:3927–3939. doi: 10.1534/g3.116.034744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamashima K., Fujishima K., Masuda T., Sugahara J., Tomita M., Kanai A. Nematode-specific tRNAs that decode an alternative genetic code for leucine. Nucleic Acids Res. 2012;40:3653–3662. doi: 10.1093/nar/gkr1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukai T., Vargas-Rodriguez O., Englert M., Tripp H.J., Ivanova N.N., Rubin E.M., Kyrpides N.C., Söll D. Transfer RNAs with novel cloverleaf structures. Nucleic Acids Res. 2017;45:2776–2785. doi: 10.1093/nar/gkw898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamashima K., Mori M., Andachi Y., Tomita M., Kohara Y., Kanai A. Analysis of genetic code ambiguity arising from nematode-specific misacylated tRNAs. PLoS ONE. 2015;10:e0116981. doi: 10.1371/journal.pone.0116981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultz D.W., Yarus M. Transfer RNA mutation and the malleability of the genetic code. J. Mol. Biol. 1994;235:1377–1380. doi: 10.1006/jmbi.1994.1094. [DOI] [PubMed] [Google Scholar]
- 39.Schultz D.W., Yarus M. On malleability in the genetic code. J. Mol. Evol. 1996;42:597–601. doi: 10.1007/BF02352290. [DOI] [PubMed] [Google Scholar]
- 40.Osawa S., Jukes T.H. Codon reassignment (codon capture) in evolution. J. Mol. Evol. 1989;28:271–278. doi: 10.1007/BF02103422. [DOI] [PubMed] [Google Scholar]
- 41.Osawa S., Jukes T.H., Watanabe K., Muto A. Recent evidence for evolution of the genetic code. Microbiol. Rev. 1992;56:229–264. doi: 10.1128/mr.56.1.229-264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler G., Rasmussen M.D., Lin M.F., Santos M.A.S., Sakthikumar S., Munro C.A., Rheinbay E., Grabherr M., Forche A., Reedy J.L. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kersey P.J., Allen J.E., Armean I., Boddu S., Bolt B.J., Carvalho-Silva D., Christensen M., Davis P., Falin L.J., Grabmueller C. Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res. 2016;44(D1):D574–D580. doi: 10.1093/nar/gkv1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Capra J.A., Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- 45.Stanke M., Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 46.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 47.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 49.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price M.N., Dehal P.S., Arkin A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darriba D., Taboada G.L., Doallo R., Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darriba D., Taboada G.L., Doallo R., Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jow H., Hudelot C., Rattray M., Higgs P.G. Bayesian phylogenetics using an RNA substitution model applied to early mammalian evolution. Mol. Biol. Evol. 2002;19:1591–1601. doi: 10.1093/oxfordjournals.molbev.a004221. [DOI] [PubMed] [Google Scholar]
- 55.Ronquist F., Teslenko M., van der Mark P., Ayres D.L., Darling A., Höhna S., Larget B., Liu L., Suchard M.A., Huelsenbeck J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huson D.H., Bryant D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 57.Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 58.Smith S.A., O’Meara B.C. treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics. 2012;28:2689–2690. doi: 10.1093/bioinformatics/bts492. [DOI] [PubMed] [Google Scholar]
- 59.Hatje K., Hammesfahr B., Kollmar M. WebScipio: reconstructing alternative splice variants of eukaryotic proteins. Nucleic Acids Res. 2013;41:W504–W509. doi: 10.1093/nar/gkt398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shevchenko A., Wilm M., Vorm O., Jensen O.N., Podtelejnikov A.V., Neubauer G., Shevchenko A., Mortensen P., Mann M. A strategy for identifying gel-separated proteins in sequence databases by MS alone. Biochem. Soc. Trans. 1996;24:893–896. doi: 10.1042/bst0240893. [DOI] [PubMed] [Google Scholar]
- 61.Oellerich T., Bremes V., Neumann K., Bohnenberger H., Dittmann K., Hsiao H.-H., Engelke M., Schnyder T., Batista F.D., Urlaub H., Wienands J. The B-cell antigen receptor signals through a preformed transducer module of SLP65 and CIN85. EMBO J. 2011;30:3620–3634. doi: 10.1038/emboj.2011.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beimforde C., Feldberg K., Nylinder S., Rikkinen J., Tuovila H., Dörfelt H., Gube M., Jackson D.J., Reitner J., Seyfullah L.J., Schmidt A.R. Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol. Phylogenet. Evol. 2014;78:386–398. doi: 10.1016/j.ympev.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 63.Rambaut, A., and Drummond, A. (2016). FigTree v1.4.3. http://tree.bio.ed.ac.uk/software/figtree/.
- 64.Vizcaíno J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44(D1):D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mass spectrometry analysis of yeast proteomes.
Mass spectrometry analysis of the A. asiatica (1) proteome, leucine and serine codons.