

Abstract
Self-folding deep cavitands with variably functionalized upper rims are able to selectively immobilize proteins at a biomimetic supported lipid bilayer surface. The immobilization process takes advantage of the dual-mode binding capabilities of the hosts, combining a defined binding pocket with upper rim charged/H-bonding groups. A variety of proteins can be selectively immobilized at the bilayer interface, either via complementary charge/H-bonding interactions, cavity-based molecular recognition, or a combination of both. The immobilization process can be used to bind unmodified native proteins, epitopes for bioadhesion, or proteins covalently modified with suitable RNMe3+ binding “handles” and charged groups that can either match or mismatch with the cavitand rim. The immobilization process can be monitored in real time using surface plasmon resonance (SPR) spectroscopy, and applied to the construction of cavitand:lipid arrays using the hosts and trehalose vitrified phospholipid vesicles. The selective, dual-mode protein recognition is maintained in the arrays, and can be visualized using SPR imaging.
Introduction
Molecular recognition of proteins and enzymes at extracellular membrane surfaces is vital for controlling cellular function and response.1,2 The complexity of the cellular environment has led to the application of biomimetic lipid bilayers3–6 for the study of protein and enzyme recognition at membrane interfaces.7,8 A wide variety of biological, often macromolecular targets can be bound at synthetic bilayer interfaces, including proteins,9 nucleotides,10,11 glycopeptides,12,13 and glycopolymers.14–16 The most common technique for membrane display is lipidation, whereby a steroid or long lipid chain is attached to an antibody or epitope to allow target recognition,17–20 or covalently attached to the protein target via chemical modification, allowing membrane incorporation.21 This is Nature’s solution: lipidated proteins such as Ras are a vital component of intracellular function.22,23
Of course, the hallmark of molecular recognition at biological membranes is target selectivity, and most importantly the selective discrimination between multiple targets in a single membrane environment. The challenge with lipidation is that there is no selectivity for membrane incorporation: almost all lipids can incorporate into a bilayer. To improve selectivity in target binding at membrane bilayers, and to allow the construction of complex, multicomponent cell surface mimics, we have focused on the use of synthetic receptors for biomimetic molecular recognition. Instead of applying known biological recognition motifs at bilayer interfaces to replicate cellular processes,9–16 the application of synthetic receptors broadens the scope of molecular recognition that can be attempted. We have previously described the use of water-soluble deep cavitands such as 124 as hosts for biomacromolecules in supported lipid bilayers (SLBs).25–28 This allows the controlled recognition of both small and large species at biomimetic interfaces. Cavitand 1 is lipophilic, and smoothly self-incorporates into supported lipid bilayers, allowing real-time measurement of target binding by surface plasmon resonance spectroscopy (SPR). This system is highly effective for target sensing, as the surface construction merely involves sequential injections of lipids and host, with no challenging synthesis or surface modification processes necessary.29
The cavitand is capable of biomacromolecule recognition via two orthogonal, distinct mechanisms (Fig. 1b and c).26,27 It has an open-ended, defined cavity that is capable of selective recognition of substituted trimethylammonium (R-NMe3+) salts such as acetylcholine, driven by favorable shape-filling interactions between host and guest, as well as via cation–π interactions between the softcation and the electron rich aromatic walls of the cavity.30 The open-ended nature of 1 allows the substrates to protrude into the exterior milieu, thus allowing variation in target, and different proteins and small molecules25 can be immobilized via the use of the R-NMe3+ binding “anchor” (Fig. 1c).
Fig. 1.
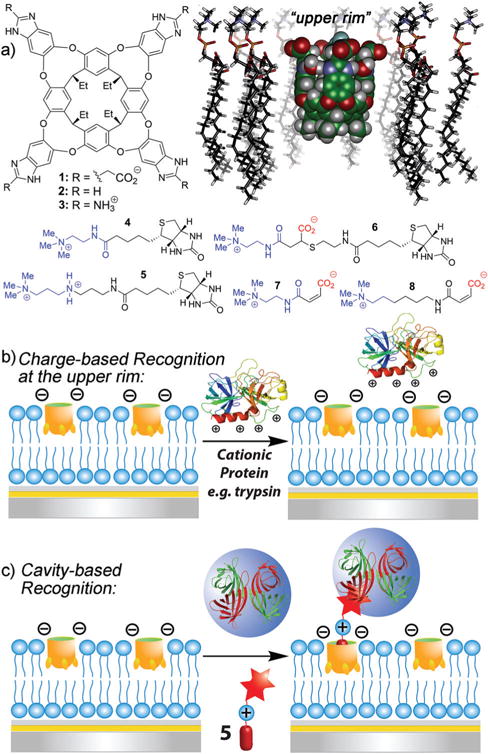
(a) Membrane-incorporating cavitand hosts 1–3, guests 4–8 and a minimized model of cavitand 1 embedded in a POPC bilayer (SPARTAN). Representation of the two target recognition modes exhibited by cavitand 1: (b) multipoint charge-based recognition via the upper rim groups and (c) cavity-based recognition via shape-filling interactions with substituted R-NMe3+ groups.
In addition to cavity-based recognition, a second recognition mechanism is possible (Fig. 1b). Unmodified cationic proteins can be adhered to the negatively charged cavitand 1 rim (not the cavity) by favorable H-bonding/charge interactions.27 These upper rim interactions are relatively weak in aqueous solution, and serve as an additional discriminating factor for selective R-NMe3+ substrate sensing.31 In a bilayer environment, the effect is magnified, enhancing salt bridge interactions between anionic 1 and cationic macromolecules such as trypsin.27 We attribute this difference to the deformation of the bilayer upon cavitand incorporation providing a small hydrophobic “pocket” above the cavitand rim, although the exact cause of the enhanced interactions is not completely clear. This effect is specific to proteins with high isoelectric points (pI) such as trypsin or cytochrome c.27
These two binding methods have only been employed separately so far, but dual, orthogonal recognition motifs in a single receptor scaffold could achieve more complex target discrimination via pairing an “anchor” recognition motif with secondary upper rim effects. The R-NMe3+ (i.e. cavity based) recognition process in bilayers is relatively insensitive to the medium (pH, ionic strength): this is not the case for the solely charge/H-bonding recognition process, which can be completely abrogated in high ionic strength solution. The two binding modes can be individually favored by altering the solution contents. The outcome of this is that charged cavitands show dual-mode binding: as well as the usual shape-fitting interactions with the cavity, they can exploit additional complementary charge and H-bonding interactions at the upper rim. We sought to use this phenomenon in complex membrane environments with our suite of cavitands, to allow tailored guest recognition. Here, we describe the use of a suite of complementary host molecules with varying upper rim functions to investigate the effect of combining different recognition modes to enhance target selectivity. In addition, we exploit lipid array technology to visualize this selectivity in a microarray format. Microarray technology32 is essential for rapid analysis of complex biological samples. Lipid microarrays suitable for SPR analysis33,34 can be formed via spontaneous adsorption and fusion of unilamellar phospholipid vesicles on a solid substrate. We have recently demonstrated a new strategy of generating air-stable membrane arrays by use of trehalose35 vitrified phospholipid vesicles, allowing each element a unique composition, for the label-free and high throughput analysis of biomolecular interactions.36 The resulting SLBs exhibit high lateral mobility, characteristic of fluidic cellular lipid membranes, allowing real time visualization by SPR imaging (SPRi).
Experimental
General information
1-Palmitoyl-2-oleoyl-snglycero-3-phosphocholine (POPC) was purchased from Avanti Polar Lipids. All other materials were obtained from Aldrich Chemical Company (St. Louis, MO), Fisher Scientific (Fairlawn, NJ), or TCI (Tokyo, Japan) and were used as received. Cavitands 1,24 237 and 331,38 were synthesized according to literature procedures. For synthesis and characterization of guests 4–8, see ESI.† Tryptophan synthase was purified via a literature procedure.39 Surface Plasmon Resonance spectroscopic measurements were performed with a dual-channel SPR spectrometer, NanoSPR6-321 (NanoSPR, Chicago, IL), with a GaAs semiconductor laser light source (λ = 650 nm). The device was equipped with a manufacturer-supplied high-refractive index prism (n = 1.61) and a 30 mL flow cell. Surface interactions at the gold interface were monitored using the resonance angle tracking mode. Molecular modeling (semi-empirical calculations) was performed using the AM1 force field using SPARTAN.
Covalent protein modification
5 mg of Bovine serum albumin (BSA) was dissolved in 100 mM PBS buffer in a glass vial equipped with a stir bar. A 152 mM solution of 7/8 was prepared in 100 μL nanopure water, and 10 μL of this solution was added to the BSA solution and gently stirred at room temperature for 16 h. The reaction mixture was transferred to a 50k centrifugal filter and centrifuged in order to filter any unattached 7/8. The product was washed and centrifuged three times with 10 mM PBS buffer. The resulting solid was then dissolved in 1 mL 10 mM PBS buffer and a Bradford assay was performed in order to determine the concentration of the solution. The solution was diluted to a concentration of 1 mg mL−1 using 10 mM PBS buffer and the resulting solution was used in the SPR experiments.
SPR spectroscopic procedure
Calcinated chip preparation
SPR chips were fabricated using BK-7 glass microscope slides. BK-7 substrates were cleaned using boiling piranha solution (3: 1 H2SO4/30% H2O2, CAUTION) for 30 min, followed by rinsing with deionized water and drying under compressed air. A 2 nm thick chromium adhesion layer was attached, followed by the deposition of a 46 nm thick gold layer via e-beam evaporation. The chips were immersed in 10 mM 3-mercaptopropionic acid (MPA) ethanol solution overnight to form a self-assembled monolayer. After extensive rinsing with ethanol and nanopure water and drying with N2 gas, the chips were alternately dipped into poly(allylamine hydrochloride) (MW ~ 17 500) solution (2 mg mL−1, adjusted to pH 8.0) and sodium silicate solution (22 mg mL−1, adjusted to pH 9.5) for 1 min each, to form a layer-by-layer assembly structure, with ultrapure water rinsing between layers. This dipping process was repeated five times to build up a multilayer silicated gold chip, followed by calcination in a furnace by heating to 450 °C at a rate of 17 °C per min and allowing cooling to room temperature 4 h later.
Vesicle preparation
POPC stock solution in chloroform (5 mg mL−1) was dried with N2 gas, and resuspended in 10 mM PBS to a lipid concentration of 1.0 mg mL−1. The suspension was extruded (11 passes) through a polycarbonate membrane of pore size 100 nm to ensure formation of small unilamellar vesicles. The solution was incubated at 4 °C for at least 1 h before use. For the preparation of vesicles preincorporated with cavitand 2, POPC lipid stock solution in chloroform was combined with cavitand 2 stock solution to a final POPC−2 ratio of 98: 2 and SUVs formed as above.
Fabrication of cavitand receptor layer and protein binding SPR measurement
The calcinated gold substrate was rinsed with ethanol and nanopure water, dried with a gentle stream of N2 gas, then clamped on an optical stage containing a 30 μL flow cell. The substrate was put in contact with the high-refractive index prism (n = 1.61) using refractive index matching fluid. POPC vesicles (1 mg mL−1) in 10 mM PBS (150 mM NaCl, pH 7.4) were injected through a flow-injection system (5 mL h−1) and incubated for 15–20 min to allow vesicle fusion on the hydrophilic calcinated gold surface, forming a smooth bilayer membrane. After 5–10 min of rinsing to remove excess lipids, 2 mg mL−1 cavitand 1 in 10% DMSO solution was injected and incubated for 20 min. The surface was extensively rinsed with nanopure water, followed by incubation with 15 μM protein in 10 mM PBS for 20 min. For streptavidin immobilization, 100 μL of a 1 mg mL−1 aqueous solution of biotinylated guest (4–6) was injected before protein, followed by 20 min incubation. Excess proteins were rinsed with water. Control experiments were performed under identical conditions in the absence of cavitand, or by the injection of POPC vesicles pre-incorporated with cavitand 2. The uncertainties in resonance angle changes were determined by applying the standard deviations of variations over multiple (at least 3) repeated runs.
Saturation binding mode25 was applied here to determine the equilibrium dissociation constant (Kd) value for the interaction between cavitand 1 and guests 7/8−BSA. Increasing concentrations of guests 7/8−BSA (0.01–15 μM) were injected over the cavitand 3:membrane complex, and the minimum angle shift was recorded:
where ABeq is the average of response signal at equilibrium and ABmax is the maximum response that can be obtained for guests 7/8·BSA binding and [A] is the concentration of 7/8·BSA injection. ABmax/ABeq was plotted against 1/[A], and the slope is equal to Kd value.
SPR imaging procedure
Arrayed chip preparation
Patterned well SPRi chips were fabricated using BK-7 glass microscope slides. BK-7 substrates were cleaned using boiling piranha solution (3: 1 H2SO4/30% H2O2, Caution) for 30 min, followed by rinsing with deionized water and drying under compressed air. A 2 nm thick chromium adhesion layer was attached, followed by the deposition of a 51 nm thick gold layer via e-beam evaporation. Subsequently, photoresist AZ5214E was spin coated on the gold at 4000 rpm, and the surface was patterned into mesas representing the final array spots using standard photolithography methods. After a second electron beam evaporation of 100 nm of gold, the photoresist was lifted off with acetone, leaving an elevated gold grid behind, defining the array elements (800 × 800 μm). The surface was rendered hydrophilic with ca. 4 nm of SiO2 deposited by plasma enhanced chemical vapor deposition (PECVD).
Fabrication of cavitand receptor array and protein binding SPRi
POPC SUVs (formed as above) were diluted to a final concentration of 1 mg mL−1 in 50 mM trehalose using a trehalose/10 mM PBS mixture. The solution was incubated at 4 °C for at least 1 h before use. 200 nL of this solution was deposited in the array wells, and dried overnight in a vacuum desiccator. The arrayed gold chips were mounted on an optical stage containing a 300 μL flow cell. Each array was put in contact with an equilateral SF2 prism (n = 1.616) using refractive index matching fluid (n = 1.616, Cargille Laboratories, Cedar Grove, NJ). The optical stage was fixed on a goniometer that allows manual selection of the incident light angle. An incoherent light source (LED, λ = 648 nm) was used for SPR excitation, and the reflected images were captured by a cooled 12-bit CCD camera, Retiga 2000R (QImaging, Surrey, BC, Canada) with a resolution of 1600 × 1200 pixels, and 7.4 μm × 7.4 μm pixel size. The dehydrated vesicles were rehydrated under constant flow in the flow cell for 20 min before cavitand injection (either 1 or 3 depending on the system being formed). The cavitand was incubated for 20 min, then rinsed for 5 to 10 min to remove any unincorporated cavitand. Next, either cyt c, biotinylated guest 5 followed by streptavidin, or 7·BSA were injected and incubated for 20 min before washing away any unincorporated sample for 5 to 10 min. Injections of sample solutions into the flow cell were monitored in real time by recording changes in the reflectance every 300 ms inside the gold array wells and for reference purpose on the surroundings. Sensorgrams were obtained by averaging reflected light intensity over each array element using a home-built LabView program (see ESI†).
Results and discussion
The three cavitands used are shown in Fig. 1. We have previously used cavitands 1 and 2 for membrane-based recognition studies,25,26 and they provide negative and neutral charges at the membrane:water interface, respectively. To access cationic upper rim functional groups on the cavitand, we exploited the recently described aminobenzimidazole cavitand scaffold 3.31,38 This host is formed by condensation of cyanogen bromide with the standard octamine cavitand precursor,40 and displays millimolar solubility in water. Cavitand 3 displays an identical cavity to 1 and 2, varying only in the upper rim functionality. We initially tested the ability of 3 to incorporate into POPC SLBs, and compared its efficacy in charge-based immobilization of proteins at the bilayer interface with that of anionic 1 (Fig. 2).
Fig. 2.
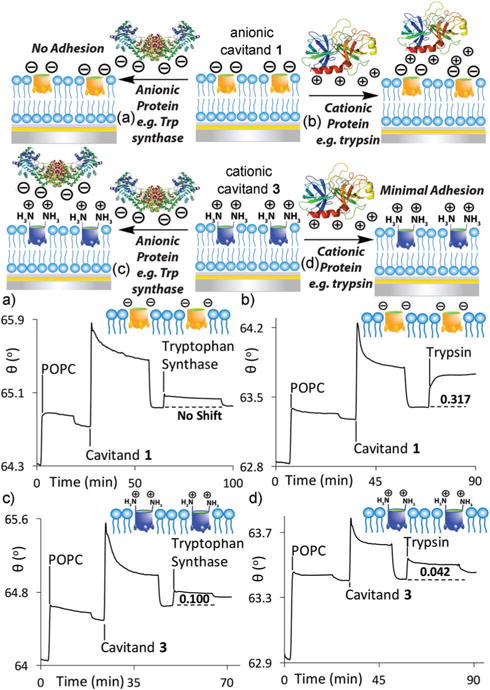
Selective charge-based unmodified protein recognition. SPR sensorgrams of the interactions of (a) anionic tryptophan synthase and (b) cationic trypsin with the POPC·anionic cavitand 1 interface. SPR sensorgrams of the interactions of (c) anionic tryptophan synthase and (d) cationic trypsin with the POPC cationic cavitand 3 interface. Protein injection medium: 10 mM PBS buffer (pH 7.4).
The cavitand-impregnated supported lipid bilayer was generated via established methods, by injecting POPC vesicles atop a cleaned calcinated gold chip in a flowcell apparatus for real time SPR analysis, followed by injection of the cavitand.25 SPR sensorgrams (shown in Fig. 2c and d) illustrate that cavitand 3 can smoothly self-incorporate in POPC SLBs. Upon injection of a 1.8 mM solution of 3 into a preformed POPC bilayer and washing away the unincorporated excess, a 0.1° 0.011 increase in resonance angle was observed. All hosts were injected to the flowcell in a 10% DMSO solution to maximize solubility. Incorporation of cavitand 3 in the bilayer is less efficient than seen with anionic 1, as shown by the lower Δθ value obtained after washing, but still occurs effectively.
The native protein adhesion properties of cationic 3 were compared with those of anionic 1 and neutral 2 (see ESI† for additional sensorgrams). Whereas anionic 1 is capable of immobilizing cationic proteins (i.e. those with a high pI such as trypsin or cytochrome c), we expected that 3 would show charge/H-bond matching with anionic proteins, and show minimal affinity for those of high pI. Table 1 shows the SPR response upon introduction of different proteins with varying pI and size to the two different POPC·cavitand surfaces. The injections were performed in triplicate, and the standard deviations were small, indicating consistent recognition and adhesion. None of the proteins showed any adhesion when exposed to either pristine POPC SLBs or a POPC·cavitand 2 surface,27 as is to be expected due to their lack of charge/ H-bonding interactions at the interface.
Table 1.
Charge/H-bonding-based immobilization of unmodified proteins at the cavitand:POPC interface
| Protein | MW (kDa) | pI | Δθcav 1a(°) | Δθcav 3a(°) |
|---|---|---|---|---|
| Cyt c | 12.4 | 10.5 | 0.13 ± 0.020 | 0.03 ± 0.002 |
| Trypsin | 23.3 | 10.5 | 0.32 ± 0.004 | 0.04 ± 0.004 |
| Avidin | 69 | 10.5 | 0.55 ± 0.022 | 0.12 ± 0.006 |
| Streptavidin | 53 | 5.0 | 0.03 ± 0.002 | 0.12 ± 0.001 |
| Tryptophan synthase | 143 | 5.06 | 0.00 ± 0.003 | 0.10 ± 0.003 |
| BSA | 66.4 | 4.8 | 0.00 ± 0.000 | 0.00 ± 0.000 |
Δθcav (°) = resonance angle change upon target binding in the presence of cavitand. Protein injection medium: 10 mM PBS buffer (pH 7.4), injected [1/3] = 1.6 mM; [protein] = 15 μM.
The proteins were injected in 10 mM PBS buffer solution at pH 7.4, as low ionic strength media is essential for effective charge/H-bonding-based adhesion. As expected, the surface showed selectivity for anionic proteins (Table 1 and Fig. 2). Cationic trypsin (pI = 10.5) gave only a very slight response upon injection (Δθ = 0.04° ± 0.004, Fig. 2d), indicating minimal immobilization. As trypsin is cationic under these experimental conditions, positively charged 3 repels the protein from the interface, preventing adhesion. In contrast, trypsin is strongly immobilized by the POPC·1 interface, causing an SPR resonance angle shift Δθ = 0.32° 0.004 (Fig. 2b).27 Other cationic proteins such as cytochrome c or avidin showed the same selectivity profile, adhering only to the POPC·1 surface. The Δθ values observed in the response varied, and were consistent with the size of the adhered protein: larger species cause an increased change in resonance angle. The sharp signal response from the SPR indicates strong binding affinities between all the proteins and the POPC·host interface. The complex, multivalent interaction precludes simple Kd determination via SPR saturation mode analysis, but CE analysis of a vesicle-based system showed the Kd (POPC·1·cyt c) = 7.6 μM.27 The similar adhesion profile of the other protein:host complexes shown here is consistent with micromolar affinities.
When anionic proteins were tested, inverted selectivity was observed. Tryptophan synthase has a pI of 5.06, and is overall negative under the injection conditions. No affinity was observed for the POPC·1 surface (Fig. 2a), but Trp synthase was successfully immobilized by cationic cavitand 3, showing an observed resonance angle change Δθ = 0.10° 0.003 upon injection (Fig. 2c). The smaller amount of incorporated 3 (when compared to 1) leads to a smaller observed Δθ upon adhesion. The adhesion was persistent, and the Trp synthase remained adhered at the surface after washing with nanopure water. As the adhesion process is a surface-based effect, pI is not the only indicator of binding efficiency: for example, the lipophilic protein bovine serum albumin (BSA) was poorly bound by all cavitands under these conditions.
The tunable, selective, multifunctional protein recognition of proteins by our suite of cavitands in POPC bilayers can be applied to array-based detection. Microarrays allow the construction of individually separated corrals containing membrane bilayer environments. These systems can be applied to high throughput analyte sensing, but reproducible construction of bilayer arrays is challenging.36 The introduction of small molecule hosts in membrane bilayer arrays has, to date, not been attempted. To visualize the adhesion process, we formed an array of cavitand impregnated POPC bilayers, and analyzed the target binding by Surface Plasmon Resonance Imaging (SPRi). Templates were fabricated with individual corrals (800 × 800 μm) on calcinated silicate nanochips by a conventional cleanroom lithographic process, and the SLB arrays were formed with a high precision nanoliter delivery system so that the individual wells are properly separated from each other. After the array chip was spotted with trehalose-vitrified POPC vesicles and dried (see Experimental section), it was placed on the SPRi flowcell and areas on each array spot were selected using the SPRi software to obtain SPRi sensorgrams of those areas. The vesicles were then rehydrated before injecting the designated cavitand (either 1 or 3). After incubation and washing of excess cavitand, the appropriate protein target was injected, incubated and washed. Each chip contains arrays with a single type of cavitand (either 1 or 3) for clarity. Any binding event can be visually detected by the lightening of the array spots in the SPRi, while no change in shade indicated no binding. Difference images (displaying a black background) were also taken to maximize visibility.
Fig. 3 shows the process for the charge: H-bonding recognition of cationic proteins. Cytochrome c, which binds strongly to the POPC·1 interface, was injected to arrays containing POPC lipids and either cavitand 1 or 3. The immobilization of cyt c by the POPC·1 is clearly visible via SPRi (Fig. 3b). After injection of cyt c and washing away any unbound excess, a lightening of the array spots occurs, corresponding to an increase in resonance angle of the SPR image in the individual corrals, indicating that the immobilization process is successful. The difference image also shows the difference between the two array images. In contrast, no change in the SPRi image is seen when cyt c is injected atop the POPC·3 interface. The cationic host 3 cannot immobilize the cationic protein, and no change in reflectivity is seen. This process was repeated for trypsin, another protein that strongly binds to POPC·1 and similar results were seen (see ESI†).
Fig. 3.
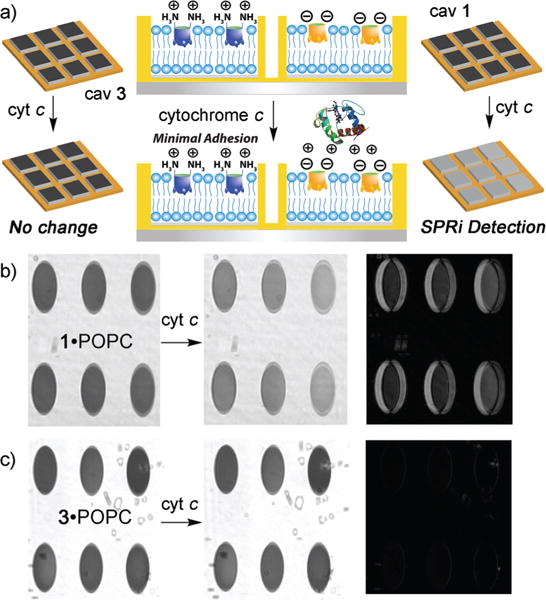
(a) Selective charge-based unmodified protein recognition in SLB arrays. SPRi images (distance vs. gray scale) and difference images of the interactions of (b) cytochrome c·POPC·1; (c) cytochrome c·POPC·3. Protein injection medium: 10 mM PBS. Corral size: 800 × 800 μm.
The upper rim functions can be combined with the cavity-based recognition element to confer dual-mode protein recognition on the system. To this end, we designed guest molecules (4–8, Fig. 1) that can both fit into the pocket and interact with the charged rim of the cavitand. The ability of cavitands 1–3 to bind substituted R-NMe3+ guests is wide-ranging, and most species containing Me3N+ CH2CH2– tails will show affinity for 1, no matter the nature of the external group.24,25,31 There are two known exceptions, however: carnitine24 and phosphocholines.41 In these cases, the guest contains a negatively charged group that is positioned in close proximity at the carboxylates of 1, and this anion–anion charge mismatch abrogates any affinity conferred by the R-NMe3+ group. However, these species are bound by neutral 2, as there is no longer a charge mismatch: the upper rim functions also dictate binding affinity, in addition to the cavity.
The various guest substrates allow different strategies for protein adhesion. Biotinylated guests 4–6 are epitopes for avidin proteins, allowing the POPC cavitand guest construct to immobilize avidin at the interface. The biotin:avidin Ka is extremely high, so the avidin adhesion process is controlled solely by the guest:cavitand interaction. As the avidin size and hydrophobicity is a constant, the only variations in SPR response stem from the host:guest affinity. Guests 4–6 each contain the core Me3N+ CH2CH2– binding handle, a biotin group and variations in their internal structure to allow charge matching and mismatching with cavitands 1 and 3 (Fig. 4a). Guest 425 is a control: the Me3N+ CH2CH2– binding handle is present, but no other matching/mismatching groups are included. Guest 5, synthesized in two steps from commercially available N,N-dimethylpropylenediamine, positions an amine function three atoms from the NMe3+ handle. In neutral aqueous solution, this group will mainly exist as an ammonium ion, and as can be seen in Fig. 4c (by molecular modeling), will confer an extra favorable charge interaction and hydrogen bonding interaction upon binding in 1, as the NH2+ group is positioned directly at the cavitand rim. Similarly, this guest will be disfavored in cationic 3 due to a charge mismatch at the rim. Guest 6 should provide inverse selectivity, as it positions a carboxylate group at the upper rim, which should match with 3 and mismatch with 1 (Fig. 4b). Introduction of negative groups is possible via anionic guest 6, which was easily synthesized by Michael addition of a thiolated biotin derivative with NMe3+-containing maleamic acid 7.
Fig. 4.
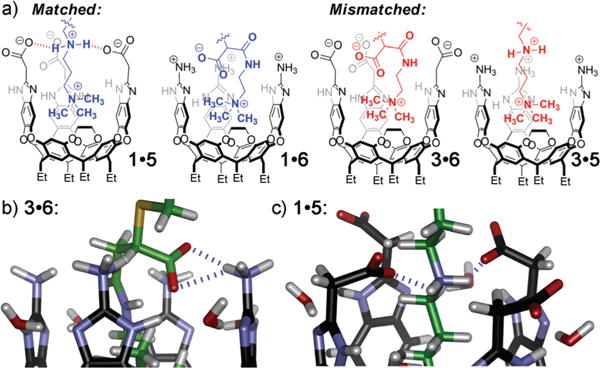
(a) Representations of the dual-mode recognition system, illustrating matched and mismatched secondary interactions between guests and the cavitands’ upper rims; (b and c) close-up views of the upper rim portion of minimized structures of “matched” 3.6 and 1.5, respectively (SPARTAN, AM1 forcefield).
For accurate analysis of the dual-mode binding process shown in Fig. 4, the background adhesion of the protein with the POPC·cavitand surface via charge/H-bonding interactions must be minimized. While avidin was unsuitable due to strong background adhesion even in high ionic strength conditions, streptavidin did not show any background affinity for cavitand 1, and only showed minimal affinity for 3, so was a suitable affinity marker for the guest matching experiments. The selective, dual mode binding effect is shown in Fig. 5. 100 μL of a 1 mg mL−1 solution of 4, 5 or 6 in nanopure water was injected atop either a POPC·1 or POPC·3 surface, and after a short incubation time and washing away the unincorporated excess, streptavidin (15 μM) was injected to the system. The SPR sensorgrams are shown in Fig. 5 and the ESI.†
Fig. 5.
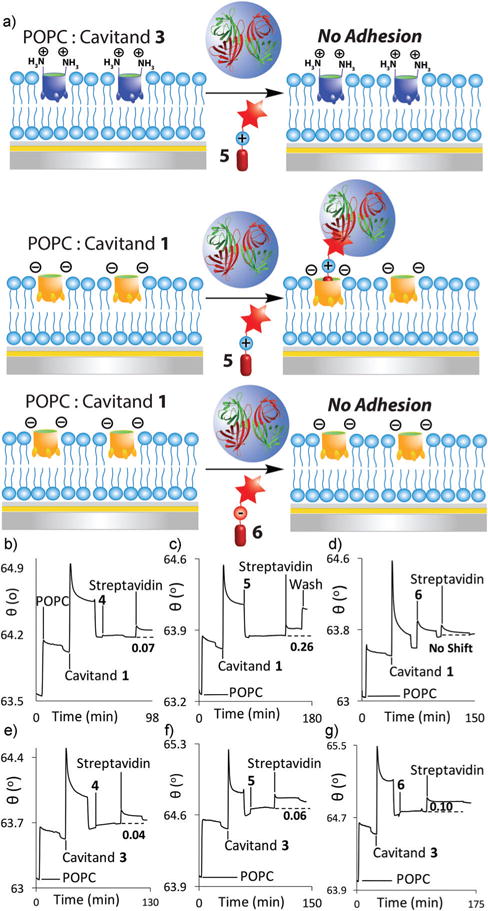
(a) Selective streptavidin immobilization via dual-mode recognition of biotinylated guests. SPR sensorgrams of the variable interactions of streptavidin at the POPC interface with anionic cavitand 1 and (b) control guest 4; (c) matched guest 5; (d) mismatched guest 6, or with cationic cavitand 3, and (e) control guest 4; (f) mismatched guest 5; (g) matched guest 6. Protein injection medium: 100 mM PBS buffer.
The dual-mode binding selectivity is illustrated well with the cationic amine-based guest 5. This substrate is “matched” with anionic 1 and shows good adhesion of streptavidin once encapsulated in 1 (observed Δθ = 0.26° ± 0.004, Fig. 5c). When applied to the cationic POPC·3 interface, however, minimal adhesion occurs (observed Δθ = 0.06° ± 0.007, Fig. 5f). The cationic amine group is repelled from the upper rim of 3, and even though guest 5 contains the requisite NMe3+ binding handle, it is not bound in the host, preventing streptavidin binding.
The recognition selectivity was also tested with guest 6, which contains a carboxylate group that can interact with the rim functions of cavitands 1 or 3 when bound. SPR experiments using this tag showed no immobilization of streptavidin at all at the POPC·1 surface (Fig. 5d), as was expected due to charge–charge repulsion between the carboxylate groups in the host and the guest. Cavitand 3 did show an inverted selectivity profile, and was able to bind the 6−streptavidin guest at the interface (Δθ = 0.10° ± 0.007, Fig. 5g). The amount of target bound was less than for the 5·streptavidin complex with cavitand 1, however. The binding of guest 4 is consistent with this: this species has no matched/mismatched charge interaction with either 1 or 3, and can theoretically bind in both hosts. Anionic 1 is a far more effective host than 3 (Fig. 5b and e), giving an observed Δθ = 0.22° ± 0.004 for 4·streptavidin. Even though there is no mismatch between cavitand 3 and the amide group in guest 4, far less streptavidin adhesion is seen (observed Δθ = 0.04° ± 0.003). This short guest positions the biotin group quite close to the water–bilayer interface, and streptavidin binding, while successful, suffers somewhat from steric clashes with the POPC bilayer, leading to the relatively small observed Δθ value.
The SPR sensorgrams observed for the successful adhesion of avidin proteins with guests 4/5 at the POPC·1 showed an unusual trait, as an unexpected increase in signal occurred during washing after the incubation of streptavidin in the presence of 1 (most notable in Fig. 5c). It is not clear why this effect is so prevalent for the streptavidin:biotin recognition, however, especially as it is not seen with other strongly binding proteins described here and in our other publications.26–28 Different experimental conditions were explored to attain a single binding event, such as increasing the injection time, changing the running solution from water to 10 mM PBS buffer as well as altering the concentrations of protein injected into the system (see ESI†), but the effect was persistent. The qualitative selectivity is not affected by this effect, however: guest 5 matches with cavitand 1, and mismatches with 3, allowing selective streptavidin adhesion.
This process was also visualized by SPRi (see ESI†). Two visualization modes are possible for the SPRi: as well as the simple visual depiction shown in Fig. 3, multiple individual SPR spectra can be recorded at each corral, then averaged to generate an SPR trace. This method is less visually striking than the SPRi image, but allows greater sensitivity in detection. When streptavidin was injected to the “matched” POPC·1·5 surface, a distinct change in reflectivity was seen (see ESI†), indicating that the process was amenable to SPRi, although less efficiently than for the unmodified protein recognition. No change was observed when streptavidin was injected to the “mismatched” POPC·3·5 surface process, as expected.
As well as using cavitand:biotin:avidin interactions to recognize proteins, covalent derivatization of macromolecules to introduce a target R-NMe3+ anchor is an effective strategy, and allows a wider scope of target recognition via binary complex formation. The simple synthesis of guest 6 from 7 via thiolate Michael reaction suggested that maleamic acid-containing guests could be used as a labeling agent for cysteine residues on a protein target, a common strategy for protein modification. BSA was chosen as a suitable protein target, as it contains only one external cysteine group, and shows no charge-based affinity for cavitands 1–3 in its unmodified form (see Table 1). Two maleamic acids of varying length (7 and 8) were tested. BSA was exposed to a buffered aqueous solution of 7 or 8, incubated for 2 h to allow complete reaction of the exposed cysteine with the maleamic acid function in 7/8, and the resultant 7/8·BSA conjugate was injected atop POPC SLBs containing cavitands 1–3, as shown in Fig. 6.
Fig. 6.
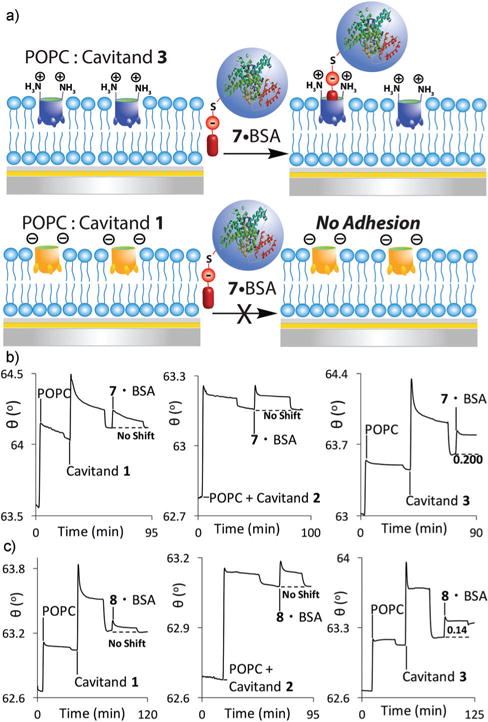
(a) Selective dual mode recognition of covalently labeled BSA. SPR sensorgrams of the variable interactions of: (b) the 7·BSA conjugate; (c) the 8·BSA conjugate at the POPC interface with cavitands 1–3. Protein injection medium: 10 mM PBS buffer.
This recognition method was highly selective: the 7·BSA conjugate showed no affinity at all for negative cavitand 1 in the POPC bilayer (Fig. 6b), but was strongly immobilized (Δθ = 0.20° ± 0.023) by the positive 3. Interestingly, no immobilization of 7·BSA was observed at the POPC 2 interface. Since 2 has no charge at its rim, there is no charge mismatch with 7, and some immobilization would be expected. Cavitand 2 has been shown to bind proteins labeled with longer oligoethyleneglycol-containing R-NMe3+ handles,26 but guest 7 shows no affinity. Only in the “matched” case with cavitand 3 is any protein binding observed. Guest 8 positions the carboxylate group further away from the cavity and lowered response was observed (Δθ = 0.14° ± 0.020) with 3. No immobilization was observed for the covalently modified 8·BSA conjugate at bilayers with either 1 or 2 embedded.
The simple 1: 1 binding complex formed between cavitand 3 and the covalently derivatized 7/8·BSA systems allows determination of binding affinity under saturation mode analysis, and illustrates the effect of charge matching on the system. Increasing concentrations of either 7·BSA or 8·BSA (0.1–15 μM) were injected over the POPC·3 surface and the angle shift was recorded. SPR signal for 7/8·BSA was observed at as little as 0.1 μM protein, and saturation occurred at micromolar concentrations. Plotting the affinities against concentration (see Experimental section) gave dissociation constants Kd (8·BSA−3) = 1.1 ± 0.01 μM, and Kd (7·BSA·3) = 0.29 0.01 μM. This illustrates that the qualitative changes in Δθ are consistent with changes in binding affinity. These affinities are broadly similar to the affinities of other types of R-NMe3+-derivatized macromolecules for cavitand 1, which are on the order of micromolar.26
This system illustrates the effectiveness of small variations in guest structure on target binding. Whereas 7 positions the carboxylate group in close proximity to the cavitand rim, allowing H-bonding/charge interactions with the upper rim groups, guest 8 positions the hydrophobic hexyl group in close proximity with the upper rim functions. Guest 8 is flexible, so some aminobenzimidazole-carboxylate interactions are possible, but they are less efficient than between 3 and 7, where the units are positioned in close proximity. The “matched” CO2−–NH3+ interactions confer a 3-fold increase in association when compared to a hydrocarbon: NH3+ interaction. The R-NMe3+ binding handle allows recognition, but the charge matching interactions confer an extra layer of selectivity.
Conclusions
We have shown that deep cavitands can act as dual-mode recognition elements for protein immobilization at supported lipid membrane bilayer interfaces. Both the defined cavity and the upper rim functions of the hosts can be exploited to allow selective, tailored molecular recognition by multiple different mechanisms. Native protein adhesion is possible in low salt conditions via matched charges and H-bonding interactions in shielded hydrophobic pockets at the bilayer interface, and complementary interactions allow recognition of differently charged proteins using differently charged hosts. By building two recognition components into a small molecule guest, the selectivity can be further enhanced: either by exploiting biotin:avidin interactions and matched ammonium:carboxylate salt bridges in the host, or by covalent derivatization of BSA with tailored maleamic acid guests. Each mechanism of interaction is selective, and the mismatched upper rim interactions can outcompete the highly favorable cavity–NMe3+ interactions. Whereas our previous work had shown that cavitands were pan-specific for substituted R-NMe3+ species, we have now introduced a second component to the recognition that allows discrimination between identical binding handles with different hosts. This macromolecule binding selectivity can also be applied to supported lipid bilayer array systems, and can be visualized in real time using SPRi. Further investigation into the construction of complex cell membrane mimicking environments with multiple components using membrane-embedded synthetic receptors is underway in our laboratory.
Supplementary Material
Acknowledgments
The authors would like to thank the National Science Foundation (CHE-1151773 to RJH, CHE-1413449 to QC) for support of this work. L. P. acknowledges support through a US Department of Education GAANN Award #P200A120170. S. S. H. acknowledges support through an NIEHS T32 training grant (T32 ES018827).
Footnotes
Electronic supplementary information (ESI) available: New molecule synthesis and characterization; SPR sensorgrams not shown in the text. See DOI: 10.1039/c7sm00192d
Notes and references
- 1.Conner SD, Schmid SL. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 2.Hymel D, Peterson BR. Adv Drug Delivery Rev. 2012;64:797–810. doi: 10.1016/j.addr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cremer PS, Boxer SG. J Phys Chem B. 1999;103:2554–2559. [Google Scholar]
- 4.Castellana ET, Cremer PS. Surf Sci Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiller SM, Naumann R, Lovejoy K, Kunz H, Knoll W. Angew Chem, Int Ed. 2003;42:208–211. doi: 10.1002/anie.200390080. [DOI] [PubMed] [Google Scholar]
- 6.Sackmann E, Tanaka M. Trends Biotechnol. 2000;18:58–64. doi: 10.1016/s0167-7799(99)01412-2. [DOI] [PubMed] [Google Scholar]
- 7.Kellam B, De Bank PA, Shakesheff KM. Chem Soc Rev. 2003;32:327–337. doi: 10.1039/b211643j. [DOI] [PubMed] [Google Scholar]
- 8.Perez JB, Martinez KL, Segura JM, Vogel H. Adv Funct Mater. 2006;16:306–312. [Google Scholar]
- 9.Chung JK, Lee YK, Lam HYM, Groves JT. J Am Chem Soc. 2016;138:1800–1803. doi: 10.1021/jacs.5b12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamat NP, Tobé S, Hill IT, Szostak JW. Angew Chem, Int Ed. 2015;54:11735–11739. doi: 10.1002/anie.201505742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jakobsen U, Simonsen AC, Vogel S. J Am Chem Soc. 2008;130:10462–10463. doi: 10.1021/ja8030054. [DOI] [PubMed] [Google Scholar]
- 12.Zope HR, Versluis F, Ordas A, Voskuhl J, Spaink HP, Kros A. Angew Chem, Int Ed. 2013;52:14247–14251. doi: 10.1002/anie.201306033. [DOI] [PubMed] [Google Scholar]
- 13.Krishnamurthy VR, Wilson JT, Cui W, Song X, Lasanajak Y, Cummings RD, Chaikof EL. Langmuir. 2010;26:7675–7678. doi: 10.1021/la101192v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godula K, Umbel ML, Rabuka D, Botyanszki Z, Bertozzi CR, Parthasarathy R. J Am Chem Soc. 2009;131:10263–10268. doi: 10.1021/ja903114g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabuka D, Forstner MB, Groves JT, Bertozzi CR. J Am Chem Soc. 2008;130:5947–5953. doi: 10.1021/ja710644g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Godula K, Rabuka D, Nam KT, Bertozzi CR. Angew Chem, Int Ed. 2009;48:4973–4976. doi: 10.1002/anie.200805756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boonyarattanakalin S, Hu J, Dykstra-Rummel SA, August A, Peterson BR. J Am Chem Soc. 2007;129:268–269. doi: 10.1021/ja067674f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boonyarattanakalin S, Martin SE, Dykstra SA, Peterson BR. J Am Chem Soc. 2004;126:16379–16386. doi: 10.1021/ja046663o. [DOI] [PubMed] [Google Scholar]
- 19.Janout V, Lanier M, Regen SL. J Am Chem Soc. 1997;119:640–647. [Google Scholar]
- 20.Janout V, DiGiorgio C, Regen SL. J Am Chem Soc. 2000;122:2671–2672. [Google Scholar]
- 21.Triola G, Waldmann H, Hedberg C. ACS Chem Biol. 2012;7:87–99. doi: 10.1021/cb200460u. [DOI] [PubMed] [Google Scholar]
- 22.Brunsveld L, Waldmann H, Huster D. Biochim Biophys Acta, Rev Biomembr. 2009;1788:273–288. doi: 10.1016/j.bbamem.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Brunsveld L, Kuhlmann K, Alexandrov K, Wittinghofer A, Goody RG, Waldmann H. Angew Chem, Int Ed. 2006;45:6622–6646. doi: 10.1002/anie.200600855. [DOI] [PubMed] [Google Scholar]
- 24.Biros SM, Ullrich EC, Hof F, Trembleau L, Rebek J., Jr J Am Chem Soc. 2004;126:2870–2876. doi: 10.1021/ja038823m. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Liao P, Cheng Q, Hooley RJ. J Am Chem Soc. 2010;132:10383–10390. doi: 10.1021/ja102252d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghang YJ, Lloyd JJ, Moehlig MP, Arguelles JK, Mettry M, Zhang X, Julian RR, Cheng Q, Hooley RJ. Langmuir. 2014;30:10161–10166. doi: 10.1021/la502629d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghang YJ, Perez L, Morgan MA, Si F, Hamdy OM, Beecher CN, Larive CK, Julian RR, Zhong W, Cheng Q, Hooley RJ. Soft Matter. 2014;10:9651–9656. doi: 10.1039/c4sm02347a. [DOI] [PubMed] [Google Scholar]
- 28.Perez L, Ghang YJ, Williams PB, Wang Y, Cheng Q, Hooley RJ. Langmuir. 2015;31:11152–11157. doi: 10.1021/acs.langmuir.5b03124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Taira T, Young MC, Ajami D, Rebek J, Jr, Cheng Q, Hooley RJ. Langmuir. 2012;28:1391–1398. doi: 10.1021/la2039398. [DOI] [PubMed] [Google Scholar]
- 30.Hof F, Trembleau L, Ullrich EC, Rebek J., Jr Angew Chem, Int Ed. 2003;42:3150–3153. doi: 10.1002/anie.200351174. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Perez L, Mettry M, Gill AD, Byers SR, Easley CJ, Bardeen CJ, Zhong W, Hooley RJ. Chem Sci. 2017;8:3960–3970. doi: 10.1039/c7sc00865a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uttamchandani M, Wang J, Yao SQ. Mol BioSyst. 2006;2:58–68. doi: 10.1039/b513935j. [DOI] [PubMed] [Google Scholar]
- 33.Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- 34.Rusten TE, Stenmark H. Nat Methods. 2006;3:251–258. doi: 10.1038/nmeth867. [DOI] [PubMed] [Google Scholar]
- 35.Harland CW, Botyanszki Z, Rabuka D, Bertozzi CR, Parthasarathy R. Langmuir. 2009;25:5193–5198. doi: 10.1021/la804007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinman SS, Ruiz CJ, Drakakaki G, Wilkop TE, Cheng Q. ACS Appl Mater Interfaces. 2015;7:17122–17130. doi: 10.1021/acsami.5b03809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rafai Far A, Shivanyuk A, Rebek J., Jr J Am Chem Soc. 2002;124:2854–2855. doi: 10.1021/ja012453p. [DOI] [PubMed] [Google Scholar]
- 38.Soberats B, Sanna E, Martorell G, Rotger C, Costa A. Org Lett. 2014;16:2480. doi: 10.1021/ol403612e. [DOI] [PubMed] [Google Scholar]
- 39.Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang YM, Chang CE, Fan L, Dunn MF, Marsella MJ, Mueller LJ. J Am Chem Soc. 2014;136:12824–12827. doi: 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ballester P, Shivanyuk A, Rafai Far A, Rebek J., Jr J Am Chem Soc. 2002;124:14014–14016. doi: 10.1021/ja0282689. [DOI] [PubMed] [Google Scholar]
- 41.Trembleau L, Rebek J., Jr Science. 2003;301:1219–1220. doi: 10.1126/science.1086644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.