

Abstract
White matter (WM) damage following a stroke underlies a majority of the neurological disability that is subsequently observed. Although ischemic injury mechanisms are age-dependent, conserving axonal mitochondria provides consistent post-ischemic protection to young and aging WM. Nitric oxide synthase (NOS) activation is a major cause of oxidative and mitochondrial injury in gray matter during ischemia; therefore, we used a pure WM tract, isolated male mouse optic nerve, to investigate whether NOS inhibition provides post-ischemic functional recovery by preserving mitochondria. We show that pan-NOS inhibition applied before oxygen-glucose deprivation (OGD) promotes functional recovery of young and aging axons and preserves WM cellular architecture. This protection correlates with reduced nitric oxide (NO) generation, restored glutathione production, preserved axonal mitochondria and oligodendrocytes, and preserved ATP levels. Pan-NOS inhibition provided post-ischemic protection to only young axons, whereas selective inhibition of NOS3 conferred post-ischemic protection to both young and aging axons. Concurrently, genetic deletion of NOS3 conferred long-lasting protection to young axons against ischemia. OGD upregulated NOS3 levels in astrocytes, and we show for the first time that inhibition of NOS3 generation in glial cells prevents axonal mitochondrial fission and restores mitochondrial motility to confer protection to axons by preserving Miro-2 levels. Interestingly, NOS1 inhibition exerted post-ischemic protection selectively to aging axons, which feature age-dependent mechanisms of oxidative injury in WM. Our study provides the first evidence that inhibition of glial NOS activity confers long-lasting benefits to WM function and structure and suggests caution in defining the role of NO in cerebral ischemia at vascular and cellular levels.
SIGNIFICANCE STATEMENT White matter (WM) injury during stroke is manifested as the subsequent neurological disability in surviving patients. Aging primarily impacts CNS WM and mechanisms of ischemic WM injury change with age. Nitric oxide is involved in various mitochondrial functions and we propose that inhibition of glia-specific nitric oxide synthase (NOS) isoforms promotes axon function recovery by preserving mitochondrial structure, function, integrity, and motility. Using electrophysiology and three-dimensional electron microscopy, we show that NOS3 inhibition provides a common target to improve young and aging axon function, whereas NOS1 inhibition selectively protects aging axons when applied after injury. This study provides the first evidence that inhibition of glial cell NOS activity confers long-lasting benefits to WM structure and function.
Keywords: ischemia, Miro-2, mitochondria, nitric oxide synthase, oxidative stress, white matter
Introduction
Oxidative stress is one of the injury pathways that converge with excitotoxicity, leading to mitochondrial impairment during white matter (WM) ischemic injury (Baltan, 2014). We have shown that aging WM produces excessive oxidative stress markers, primarily due to alterations in axonal mitochondrial ultrastructure and function (Stahon et al., 2016). Aging axons are characterized by longer and thicker mitochondria that correlate with lower ATP levels and increased generation of nitric oxide (NO), protein nitration, and lipid peroxidation (Stahon et al., 2016). Predictably, this increase in oxidative stress contributes to the increased vulnerability of aging WM to ischemia (Baltan, 2009, 2014). Remarkably, conserving axonal mitochondria before or after ischemia promotes axon function recovery in both young and aging WM, providing a universal target to protect WM structural and functional integrity against ischemia (Baltan et al., 2011; Brunet et al., 2016). Because NO is involved in different mitochondrial signaling pathways that control energy metabolism and mitochondrial function (Peinado et al., 2014; Litvinova et al., 2015) such that excessive formation of reactive oxygen species (ROS; Kowaltowski and Vercesi, 1999) contributes to mitochondrial dysfunction, we hypothesized that activation of nitric oxide synthase (NOS) isoforms expressed in glial cells contributes to mitochondrial dysfunction and oxidative injury in WM.
NO, ROS, and reactive nitrogen species (RNS) are key mediators of oxidative injury that are produced as a result of NOS activation (Moncada et al., 1991; Lander, 1997). NO serves several physiological functions (Christopherson and Bredt, 1997); however, unrestricted accumulation of ROS and/or RNS above a critical concentration results in damage to nearby cellular components. For instance, NO can react with superoxide free radicals to form peroxynitrite (ONOO−) free radicals, which nitrate proteins and damage lipids and DNA (Radi et al., 1991; Ischiropoulos et al., 1992; Douki and Cadet, 1996). In addition, NO and peroxynitrite can react with mitochondrial enzymes to inhibit oxidative phosphorylation (Drapier and Hibbs, 1986; Cassina and Radi, 1996; Welter et al., 1996; Brown, 2001), thus limiting ATP production. The three isoforms of NOS, all derived from separate genes, are NOS1, NOS2, and NOS3 (Nathan and Xie, 1994b). NOS1 and NOS3 are constitutively expressed and are calcium-dependent, whereas NOS2 is inducible and calcium-independent (Nathan and Xie, 1994a). Despite extensive investigation into the role of NOS isoform activation in neurons in mediating preconditioning or post-ischemic protection, reports regarding the contribution of glial cells have been limited to NOS2 overactivation in the rat middle cerebral occlusion model (Iadecola et al., 1995a,b,c; Peng et al., 1996; Zhang et al., 1996). Indeed, glial cells have been viewed mainly as potential reservoirs of l-arginine, which is the substrate for NOS, rather than a source of NO production (Simmons and Murphy, 1993). It is now established that WM is injured during stroke and injury to glial cells and axons primarily contribute to neurological dysfunction (Fern et al., 1995; Garthwaite et al., 1999; Matute et al., 2007; Tekkök et al., 2007; Cho et al., 2015). Therefore, we hypothesize that glial cells play an extensive role in WM ischemic injury by activating NOS in an isoform- and age-specific manner, which damages mitochondria and impairs functional recovery of axons. We found that pan-NOS inhibition promoted axon function recovery in young and aging WM when applied before a period of oxygen-glucose deprivation (OGD), whereas selective inhibition of NOS3 in young axons and NOS3 or NOS1 in aging axons conferred post-ischemic protection. Interestingly, inhibition of NOS3 upregulation in astrocytes promoted post-ischemic recovery of axon function by preserving axonal mitochondrial integrity and motility, suggesting a role for NO in mediating glia-axon interactions. At the vascular level, NOS3 confers protection during ischemia (Iadecola, 1997; Iadecola et al., 2011; Li et al., 2014) primarily by causing vasodilatation and improving cerebral circulation (Iadecola, 1997; Yamada et al., 2000; Iadecola et al., 2011; Li et al., 2014). Therefore, activation of NOS3 can be protective or destructive, depending upon the cellular source and the stage of evolution of the ischemic process in gray matter and WM portions of the brain.
Materials and Methods
Animals and chemicals.
All experimental procedures were approved by The Institutional Animal Care and Use Committee of the Cleveland Clinic. C57BL/6J male mice were purchased from The Jackson Laboratory and were used at 3 or 12 months of age. Mice expressing mitochondrial-targeted CFP on a C57BL/6J background (Thy-1 mito-CFP mice; Misgeld et al., 2007) were originally obtained at the University of Washington and were later relocated to and bred at the Cleveland Clinic Foundation. All mice were maintained on a 12 h light/dark cycle with food and water provided ad libitum. All chemicals used for electron microscopy were purchased from Electron Microscopy Sciences, except for aspartic acid (Sigma-Aldrich). N(ω)-Nitro-l-arginine methyl ester hydrochloride (l-NAME), N5-(1-iminoethyl)-l-ornithine dihydrochloride (l-NIO), (S)-methylisothiourea sulfate (SMT), and 3-bromo-7-nitroindazole (3Br7NI) were all purchased from Tocris Bioscience. The sources for other chemicals used were described previously (Baltan et al., 2008; Stahon et al., 2016).
Optic nerve preparation, electrophysiological recordings, and OGD.
Following CO2 asphyxiation, mouse optic nerves (MONs) were obtained from C57BL/6J mice or Thy-1 mito-CFP mice at 3 or 12 months of age. MONs were gently cleared from their dural sheaths and transferred to a Haas-top perfusion chamber (Harvard Apparatus) and superfused with artificial cerebrospinal fluid (ACSF) containing the following (in mmol/L): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose. MONs were allowed to equilibrate for at least 15 min in the recording chamber in normal ACSF bubbled with a 95% O2/5% CO2 mixture. All electrophysiological recordings were performed at 37°C. Suction electrodes back-filled with ACSF were used for stimulation and for recording compound action potentials (CAPs). The stimulation electrode was connected to a stimulus isolation unit (A365R stimulus isolator; WPI) and elicited CAPs every 30 s. Stimulus pulse (30 μs duration) strength was adjusted to evoke the maximum CAP possible and then increased another 25% for supramaximal stimulation. The recording electrode was connected to an Axoclamp 900A amplifier (Molecular Devices) and the signal was amplified 20 or 50 times, filtered at 3 kHz (SR560; Stanford Research Systems), and acquired at 20–30 kHz. During experiments, the supramaximal CAP was elicited every 30 s. OGD was induced as previously described (Baltan et al., 2008, 2010, 2011; Murphy et al., 2014; Stahon et al., 2016) by switching to glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. OGD was applied for 60 min, glucose-containing ACSF and O2 were restored, and CAPs were recorded for up to 5 h after the end of OGD. All MONs were collected and fixed for subsequent immunohistochemical analysis.
Immunohistochemistry.
Perfusion-fixed (4% paraformaldehyde and 0.025% glutaraldehyde) MONs were used to determine the cellular location and expression pattern of NOS isoforms to compare young mice with aging mice. The effects of OGD were assessed in MONs collected at the end of electrophysiological recordings (5 h after the end of OGD). MONs were cryoprotected in a 30% sucrose solution for 16–18 h at 4°C. Using a cryostat (Leica, CM3050S), all MONs were sectioned at a thickness of 16 μm, blocked in 5% normal goat/donkey serum (50% by volume; Sigma-Aldrich), and permeabilized in 0.3% Triton X-100 (Sigma-Aldrich) for 1 h at room temperature. To achieve optimal adenomatous polyposis coli (APC) labeling of mature oligodendrocytes, MONs were blocked in 20% normal goat/donkey serum (by volume) for 1 h at room temperature. MON sections were incubated in various primary antibodies overnight at 4°C (Table 1). The specificities of the NOS isoform antibodies were verified by Western blot analysis of MON lysates (see Fig. 5B) and, in addition, expression patterns were verified in brain slices containing hippocampus and corpus callosum (data not shown). Donkey anti-rabbit Cy5, anti-mouse Cy5, anti-mouse Cy3, and anti-rabbit Cy3 (Jackson ImmunoResearch) at 1:100 dilutions were prepared in 2% normal goat serum (2 h at room temperature). Sytox green (ThermoFisher Scientific) was used to label cell nuclei. Sections were double- or triple-labeled.
Table 1.
List of primary antibodies used
| Antigen | Host | Catalog # | Dilution | Source |
|---|---|---|---|---|
| NOS1 | Rabbit | BML-SA227 | 1:100 | Enzo |
| NOS1 | Mouse | 37-2800 | 1:100 | Invitrogen |
| NOS2 | Rabbit | ADI-905-431 | 1:100 | Enzo |
| NOS2 | Mouse | NBP2-22119 | 1:100 | Novus Biologicals |
| NOS3 | Rabbit | ADI-KAP-NO020 | 1:100 | Enzo |
| NOS3 | Mouse | 610296 | 1:100 | BD Transduction Laboratories |
| GFAP-Cy3 | Mouse | C9205 | 1:1000 | Sigma-Aldrich |
| GFAP | Rabbit | 22522 | 1:15 | Immunostar |
| APC | Mouse | OP80 | 1:100 | Calbiochem |
| SMI-31 | Mouse | SMI31R | 1:100 | BioLegend |
| Sytox Green | 57020 | 1:25000 | EMD Millipore | |
| Iba-1 | Rabbit | 019-19741 | 1:100 | Wako |
| Miro-2 | Rabbit | AB154946 | 1:500 | Abcam |
Figure 5.
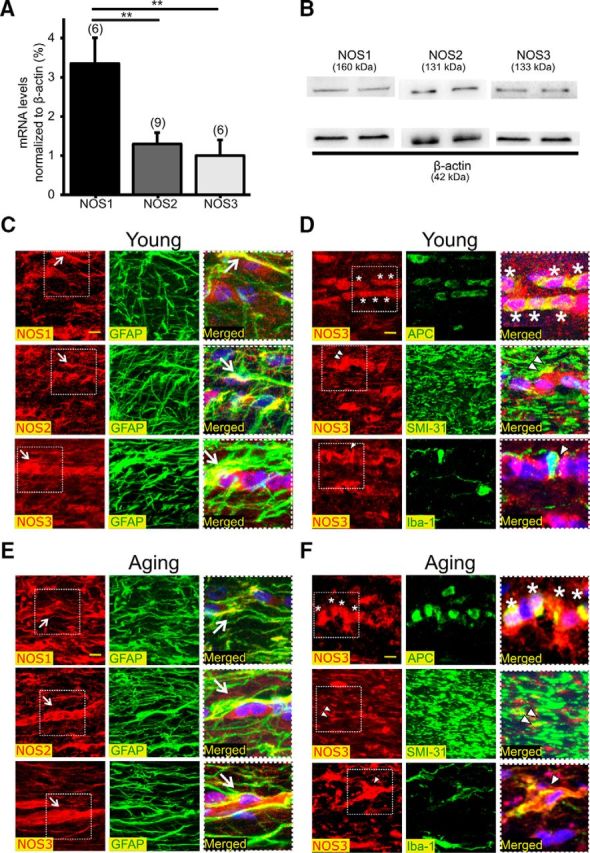
NOS isoforms are abundantly expressed in MONs in an age- and cell-specific manner. A, NOS1, NOS2, and NOS3 mRNAs were readily detected in MONs. NOS1 mRNA levels were the highest compared with either NOS2 or NOS3. **p < 0.01; one-way ANOVA with Bonferonni's post hoc test. B, All three NOS isoforms were detected in MONs by Western blotting. C, Immunolabeling for NOS1, NOS2, or NOS3 in young WM showed colocalization with GFAP(+) astrocytes (white arrows) for all three isoforms. D, NOS3 also colocalized with APC(+) oligodendrocyte soma (top, asterisks), SMI-31(+) axons (middle, white double arrowheads), and Iba-1(+) microglia (bottom, white arrowhead). Scale bars, 10 μm. E, Immunolabeling for NOS1, NOS2, or NOS3 in aging WM showed colocalization with GFAP(+) astrocytes. NOS1 and NOS3 showed extensive expression in astrocytic cell bodies (white arrows), processes, and end-feet of astrocytes. F, NOS3 colocalized with APC(+) oligodendrocyte soma (top, asterisks), SMI-31(+) axons (middle, white double arrowheads), and Iba-1(+) microglia (bottom, white arrowhead). Note that the merged images on the right are enlarged areas indicated by the squares with dashed lines on the left. Scale bars, 10 μm.
Confocal imaging.
A Leica TCS SP5 upright confocal microscope was used for digital image acquisition except for CFP imaging, for which a Leica TCS SP511 upright multiphoton laser scanning microscope was used (see next section). Argon and He–Ne lasers were used to excite Sytox Green/488 nm, Cy3/543 nm, and Cy5/647 nm. Two to three adjacent sections from each MON were imaged for a total of five areas of interest. A total of 10–16 optical sections of 1 μm thickness at 1024 × 1024 pixel size were collected in the z-axis from a single microscopic field using a 63× objective lens (HCX PLAN APO, oil-immersion; numerical aperture 1.40; Leica) under fixed gain, laser power, pinhole, and photomultiplier tube settings.
Multiphoton imaging and pixel intensity analysis of Thy-1 mito-CFP(+) mice.
As we previously described (Stahon et al., 2016), expression of CFP was imaged using a Leica TCS SP511 upright multiphoton laser-scanning microscope. Two to three adjacent sections for each MON were imaged with the wavelength set at 900 nm. A total of 10 optical sections of 1 μm thickness at 512 × 512 pixel size were collected in the z-axis from a single microscopic field using the 25× objective lens (HCX IRAPO, water-immersion; numerical aperture, 0.95; free working distance, 2.5 mm) under fixed gain, laser power, pinhole, and photomultiplier tube settings. To compare pixel intensity, all sections were processed concurrently with Leica imaging software (LAS-AF v2.7) using a single channel. The Z-stacks were projected into a single plane image before analysis and assessment of pixel intensity.
NOS m-RNA levels.
RNA from MON samples was isolated using RNeasy Mini Kits (74104, Qiagen). Reverse transcription was performed to obtain cDNA using moloney murine leukemia virus reverse transcriptase (28025013, Invitrogen), 10 mmol/L deoxynucleoside triphosphate mix, and Promega Random and Oligo (dT) Primers (PRC1101 and PRC1181, Fisher Scientific) for 1 h at 37°C. TaqMan Gene Expression Assay probes specific for NOS1 (Mm00435175_m1), NOS2 (Mm00440502_m1), NOS3 (Mm00435217), and β-actin (Mm00607939_s1) were purchased from Invitrogen. TaqMan Fast Universal PCR Master Mix (2X) (4352042, Invitrogen) was used in a 7500 Fast Real-time PCR system (Applied Biosystems). Cycle threshold values were normalized to β-actin levels, from which normalized gene expression levels were calculated.
NOS isoforms and miro-2 protein expression.
MON samples (two pairs) were homogenized (W-220F sonicator, Qsonica) on ice in 75 μl of RIPA buffer (EMD Millipore) or Cell Lysis buffer (Cell Signaling Technology) with protease inhibitor and phosphatase inhibitor cocktails (Sigma-Aldrich). Samples were then centrifuged at 14,000 rpm for 10 min, the supernatant was collected, and the protein concentration was estimated using a bicinchoninic acid assay (BCA; ThermoFisher Scientific). Protein lysates were prepared in 4× laemmli sample buffer (Bio-Rad) and 2-mercaptoethanol (Sigma-Aldrich) at a 10:1 ratio and incubated at 95°C for 10 min. Equal amounts of protein were loaded into each well (10–22 μg) of stain-free 4–20% Mini-Protean TGX gels (Bio-Rad). After gel electrophoresis and transfer, blots were blocked in 5% milk powder in TBS-Tween (0.1%) and incubated with rabbit NOS3 (1:250) and mouse β-actin (1:20,000) antibodies overnight at 4°C. Blots were incubated with secondary antibodies (goat anti-mouse HRP and goat anti-rabbit HRP) for 2 h at room temperature. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad), imaged using a Bio-Rad Chemidoc MP, and analyzed using Image Lab software v4.1.
Reduced glutathione assay.
MONs were sonicated (W-220F sonicator, Qsonica) in 100 μl of PBS with 2 mmol/L EDTA and then centrifuged at 14,000 rpm for 10 min at 4°C. The supernatant was collected and 30 μl were immediately used for glutathione measurements with a glutathione assay kit (V6911, Promega) and for BCA protein assays. Luminescence was measured using a 200 gain filter and 5 s integration on a Biotek Synergy4 system and Gen5 software. Reduced glutathione concentrations were calculated from a standard curve, normalized to protein concentration, and expressed as fold-change relative to control conditions.
NO generation assays.
MONs were sonicated (W-220F sonicator, Qsonica) within 75 μl RIPA buffer, vortexed every 2 min for 10 total min, and centrifuged at 14,000 rpm for 10 min at 4°C. After estimating the protein concentration of the supernatant (23225; ThermoFisher), 20–30 μg of protein were used for NO generation assays (NB78, Oxford Biomedical). Ultrasensitive colorimetric NOS Assay Kits use recombinant nitrate reductase for conversion of nitrate to nitrite before quantification of nitrite using Griess Reagent, thus providing an accurate determination of total NOS activity. The completed reaction is read at 540 nm. The amount of NO generated was calculated from a log–log plot of the standard curve, normalized to protein concentration, and expressed as fold-change relative to control conditions.
3-D electron microscopy.
The method used for 3-D electron microscopy (3-D EM) of MONs was as previously published with slight modifications (Ohno et al., 2011; Stahon et al., 2016). MONs were immersion-fixed in 0.1 m sodium cacodylate buffer containing 4% paraformaldehyde and 2.5% glutaraldehyde, pH 7.4. Samples were processed sequentially in 2% osmium tetroxide/potassium ferrocyanide, 1% thiocarbohydrazide, and 2% osmium tetroxide solution, and then left overnight in saturated uranyl acetate solution. Samples were then incubated in lead aspartate solution for 30 min at 60°C and dehydrated over an ethanol gradation series of freshly-prepared, ice-cold solutions of 20, 60, 80, 90, 95, and 100% ethanol and anhydrous ice-cold 100% acetone. Samples were embedded in epon resin (formulated as 10 ml EMBed-812, 8 ml dodecenyl succinic anhydrid, 4 ml methyl-5-norbornene-2, 3-dicarboxylic anhydride, and 0.4 ml 2,4,6-tri(dimethylaminomethyl)-phenol). Sample blocks were trimmed and mounted on a pin. The block face was sectioned serially by a diamond knife alternating with image capture at 100 nm thickness and 4000× magnification using a Zeiss Sigma VP scanning electron microscope equipped with a Gatan 3View in-chamber ultramicrotome and low KV backscattered electron detector. Image sets were registered using the Fiji (ImageJ) plugin for linear alignment with SIFT (Schindelin et al., 2012). Macros written for Fiji (ImageJ) were used to randomly select axons for 3-D image analysis. Investigators analyzing the data were blinded to the experimental conditions. Reconstruct softwarev1.1.00 (Fiala and Harris, 2001) was used to trace axons and to label mitochondria (Fiala, 2005). Traces were made either manually or using the “wildfire” tool. From each sample set, 20 randomly selected axons were traced and the mitochondria were labeled using predefined grid traces as normal or abnormal using the Grid Tool specified in the Grids tab. Data generated by Reconstruct software were imported to Excel 2016 (Microsoft) to obtain the corrected volume of axons and mitochondrial counts for quantification.
ATP assays.
ATP assays were performed as described previously (Baltan et al., 2011; Stahon et al., 2016). MONs were sonicated in 75 μl of 10% HClO4 and centrifuged at 4500 rpm for 10 min at 4°C. The supernatant was neutralized with 30 μl of 2.5 mol/L KOH and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatant was used to measure total cellular ATP using a kit (11699709001, Roche Diagnostics). The absorbance was measured at 560 nm on a Biotek Synergy4 system using Gen5 software. ATP concentrations were calculated from a log–log plot of the standard curve and normalized to protein concentration. The values were calculated as μmole ATP/mg protein and are expressed as relative to control conditions. Protein content was quantified in one nerve (23225; ThermoFisher) and ATP levels were measured in the other.
NOS3−/− mouse genotyping.
DNA extraction was performed on tail samples collected from NOS3 complete knock-out animals (B6.129P2-Nostm1Unc/J) and C57BL/6J animals using a REDExtract-N-Amp tissue PCR kit (XNAT-1KT, Sigma-Aldrich) according to the manufacturer's protocol. Wild-type (WT) forward 5′-AGGGGAACAAGCCCAGTAGT-3′, Mutant forward 5′-AATTCGCCAATGACAAGACG-3′, and common reverse 5′-CTTGTCCCCTAGGCACCTCT-3′ primers (Integrated DNA Technologies) were used. PCR was performed in a T100 Thermal Cycler (Bio-Rad). Gel analysis of amplicons was performed using a 1.5% agarose gel prepared with 1× tris borate-EDTA (TBE) and SYBR Safe DNA gel stain (S33102, Invitrogen) and electrophoresed (150 V, 30 min) in 1× TBE running buffer and imaged using a ChemiDoc MP (Bio-Rad) using Image Lab software v4.1. Molecular weights of the PCR products were determined by comparison to a 1kb Plus DNA ladder (10787-018, Invitrogen).
Time-lapse live imaging of mitochondrial fission and motility.
MONs obtained from Thy-1 mito-CFP(+) mice were transferred to a C-Stim CMC Microscope Chamber System (IonOptix) attached to an in-line heater (Cell micro controls MTCII temperature controller and heater, IonOptix) to keep the chamber at 37°C. MONs were superfused with ACSF bubbled with a 95% O2/5% CO2 mixture at a flow rate of 3 ml/min. OGD was induced by switching to glucose-free ACSF saturated with 95% N2/5% CO2. To determine mitochondrial motility in both the anterograde and retrograde directions, Thy-1 mito-CFP(+) MONs were imaged every 1 s at 1024 × 256 resolution from a single microscopic field using an inverted confocal microscope (Leica, DMI6000; 40× water-immersion objective; numerical aperture, 0.80). The imaging area was set to contain at least three axons (confirmed during analysis) in which there would be at least 10 mitochondria moving in either direction. Time-lapse images were captured during the last 5 min of baseline (20 min), during OGD (60 min), and during the recovery period (20 min). Imaging parameters were chosen and set to minimize photobleaching. Control experiments consisting of imaging MONs using the same parameters without OGD, which showed <10% CFP fluorescent loss over the same duration (data not shown). Mitochondrial motility, direction, and changes in mitochondrial size were analyzed using ImageJ software combined with the KymoTool Box plugin (Zala et al., 2013). Stationary and motile mitochondria, as well as the direction of motility, were identified and kymographs were generated to measure the anterograde and retrograde movements during baseline, OGD (60 min), and recovery.
Data acquisition and statistical analysis.
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP using Clampfit v10.2 (Molecular Devices). CAP area is a complex spatiotemporal summation of action potentials from individual axons (Stys et al., 1991). To compare CAP area among experimental groups, data from each group of experiments were pooled and CAP areas were normalized to baseline conditions. Data were normalized by setting the mean of initial baseline values (measured over 15 min) to a value of 1 as previously described (Baltan et al., 2008, 2011; Stahon et al., 2016). All data are presented as mean ± SEM (see Figs. 1C, 2C, 3D–F, 4D, 5A, 6, 7, 8A,D, 9B,D) or median values (see Figs. 3C, 4C). GraphPad Prism v4.0c was used for statistical analysis. For normally-distributed data, statistical significance was determined by unpaired Student's two-tailed t tests for comparing two groups (see Fig. 8D) or one-way ANOVAs followed by Bonferroni's post hoc tests for three or more groups (see Figs. 1C, 2C, 3D–F, 4D, 5A, 6, 7, 8A,D, 9B,D). Nonparametric Kruskall–Wallis tests followed by Dunn's multiple-comparison tests were used for analysis for comparing the medians of non-normally distributed data (Figs. 3C, 4C). The n values indicate the number of axons in Figures 3C and 4C, whereas in all other figures they indicate the number of optic nerves except in Figures 5A, 8A, and 9D, where they represent the number of pairs of optic nerves. The p values and significance values are indicated individually for each figure in the text.
Figure 1.
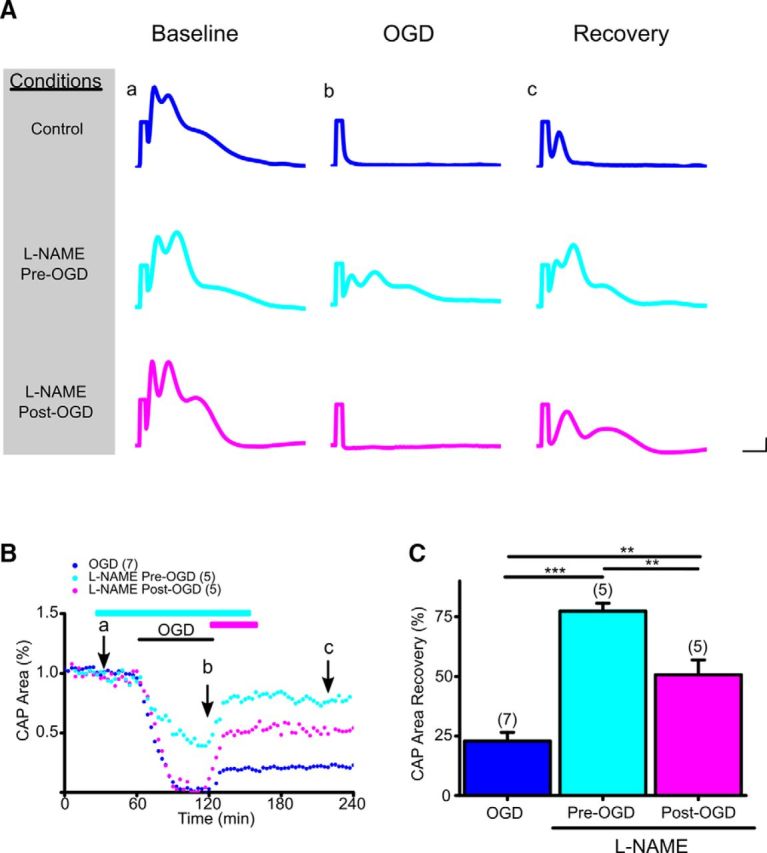
Pan-NOS inhibition promotes functional recovery of young WM when applied before or after OGD. A, Representative CAP traces at baseline for control ACSF (Aa), 60 min OGD (Ab), and recovery (Ac) conditions are shown for control (blue), pan-NOS inhibitor l-NAME applied pre-OGD (cyan), and l-NAME applied post-OGD (magenta). Scale bars: 0.5 ms, 0.5 mV. B, At baseline, CAP area remained stable over time, but was completely lost after OGD (Bb, blue), followed by a small but sustained recovery (Bc, blue; 23 ± 4%). l-NAME (200 μm) application before OGD (cyan) preserved CAP area during OGD and improved CAP area recovery during reperfusion. l-NAME post-OGD application (magenta) similarly conferred protection to axon function during reperfusion, albeit to a lesser extent. Cyan and magenta horizontal lines represent l-NAME pretreatment and post-treatment applications, respectively. C, Pan-NOS inhibition in young WM preserved axon function independent of onset of application, although pretreatment with l-NAME was more beneficial to axon function. CAP area recovery after 60 min of OGD was robust when MONs were pretreated with l-NAME (78 ± 3%), whereas CAP area recovered to 51 ± 6% when MONs were post-treated with l-NAME. n = number of MONs. **p < 0.01, ***p < 0.001; one-way ANOVA followed by Bonferonni's post hoc test. Error bars indicate SEM.
Figure 2.
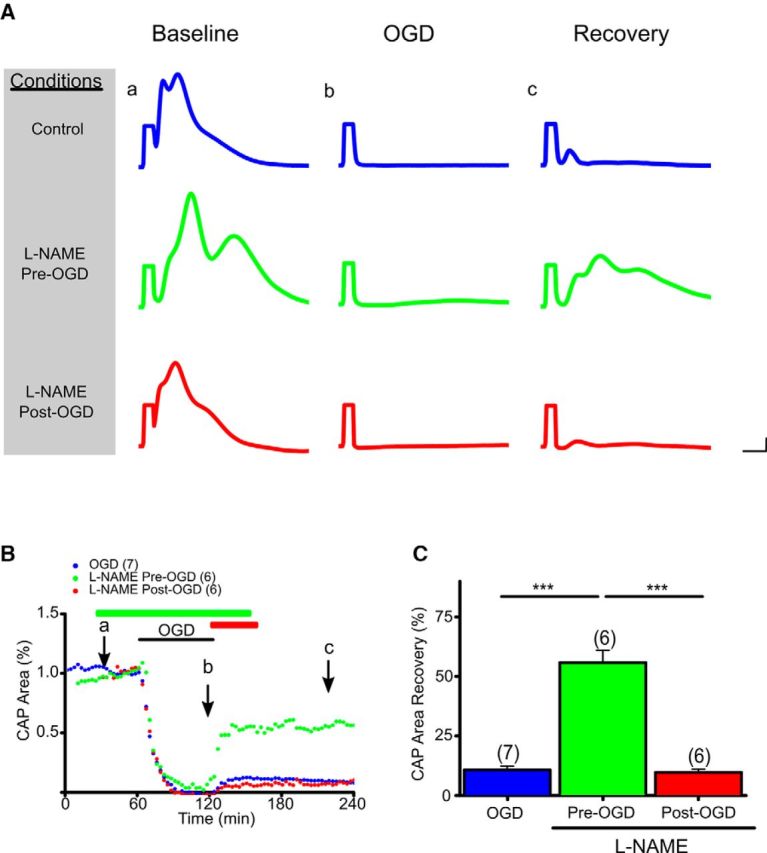
Pan-NOS inhibition promotes functional recovery of aging WM only when applied before OGD. A, Representative CAP traces at baseline for control ACSF (Aa), 60 min OGD (Ab), and recovery (Ac) conditions are shown for control (blue), pan-NOS inhibitor l-NAME applied pre-OGD (green), and l-NAME applied post-OGD (red). Scale bars: 1 ms, 1 mV. B, CAP areas showed minimal recovery after OGD (Ba–Bc, blue; 10 ± 2%). l-NAME (200 μm) application before OGD (green) preserved CAP area during OGD and improved CAP area recovery during reperfusion. l-NAME post-OGD application (red) failed to promote aging axon function recovery during reperfusion. Green and red horizontal lines represent l-NAME pretreatment and post-treatment applications, respectively. C, Pan-NOS inhibition in aging WM preserved axon function only with pretreatment with l-NAME; post-treatment was ineffective. CAP area recovery after 60 min of OGD was robust when MONs were pretreated with l-NAME (56 ± 5%), whereas CAP area recovered to 9 ± 1% when MONs were post-treated with l-NAME. n = number of MONs. ***p < 0.001; one-way ANOVA followed by Bonferonni's post hoc test. Error bars indicate SEM.
Figure 3.
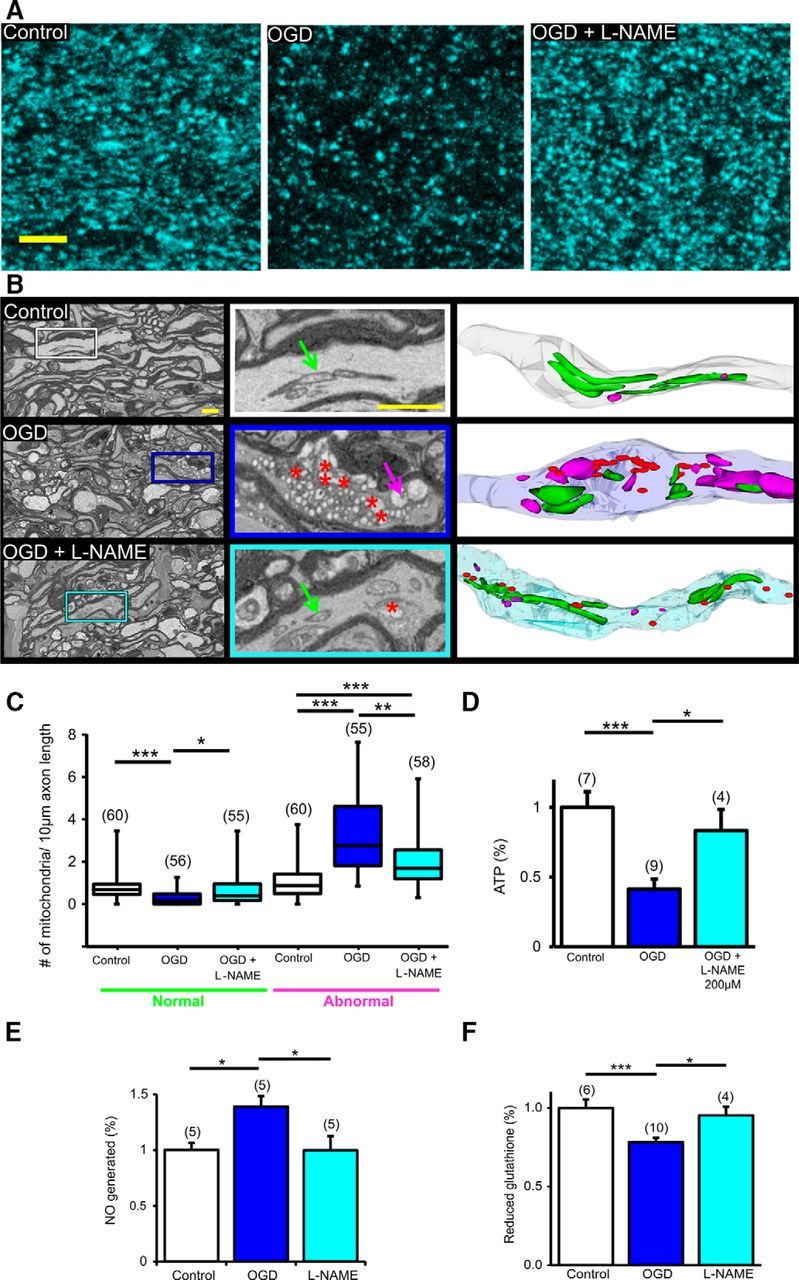
Pan-NOS inhibition protects axonal mitochondrial structure and function and restores NOS and glutathione levels in young WM. A, Two-photon microscopy images show that OGD drastically reduced CFP fluorescence in MONs from Thy-1 mito-CFP(+) mice. l-NAME application conserved CFP(+) mitochondria. Note that mitochondrial numbers declined and mitochondria became smaller and more round with OGD. Scale bar, 5 μm. B, l-NAME preserved mitochondrial morphology. Representative electron micrographs obtained from 3-D EM stacks of MONs (left and middle) under baseline conditions and OGD with or without l-NAME application. OGD was associated with myelin thinning, swelling of axons, abnormal mitochondria (magenta arrow), and formation of degradation products (red asterisks). l-NAME preserved mitochondrial structure (normal mitochondria, green arrow) and axons. Scale bar, 1 μm. Middle, Magnified views of representative axons from the left and right panels show 3-D EM reconstruction of these axons displaying normal (green) and abnormal (magenta) mitochondria and degradation products (red) in axons following OGD. C, OGD decreased the number of normal mitochondria and increased the number of abnormal mitochondria, whereas l-NAME ameliorated mitochondrial damage. n = number of MONs. *p < 0.05, **p < 0.01, ***p < 0.001; Kruskal–Wallis test followed by Dunn's multiple-comparison test. D, l-NAME preserved MON ATP levels following OGD. *p < 0.05, ***p < 0.001; one-way ANOVA with a Bonferonni's post hoc test. E, l-NAME treatment attenuated NOS activity during OGD and (F) l-NAME treatment preserved reduced glutathione levels in MONs exposed to OGD. *p < 0.05, ***p < 0.001; one-way ANOVA with Bonferonni's post hoc test.
Figure 4.
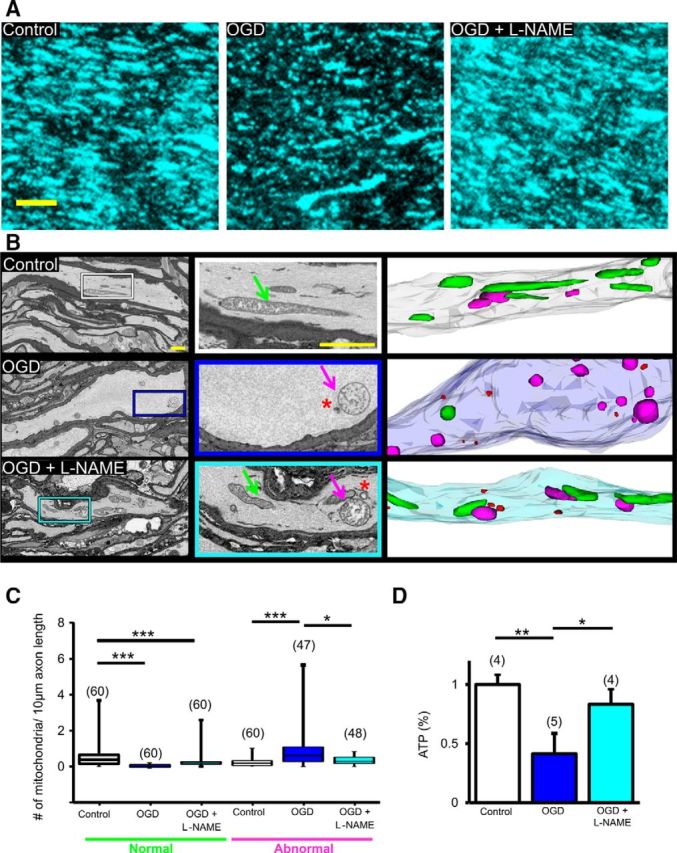
Pan-NOS inhibition protects axonal mitochondrial structure and function in aging WM. A, Two-photon microscopy images show that OGD drastically reduced CFP(+) fluorescence in MONs from Thy-1 mito-CFP(+) mice. l-NAME application conserved CFP(+) mitochondria. Note that aging axons display longer and thicker mitochondria, which became smaller in size and fewer in number when exposed to OGD. Scale bar, 5 μm. B, l-NAME preserved mitochondrial morphology. Representative electron micrographs obtained from 3-D EM stacks of MONs (left and middle) under baseline, OGD, and OGD with l-NAME pretreatment. OGD was associated with myelin thinning, marked swelling of axons, abnormal-appearing swollen mitochondria (magenta arrow), and few degradation products (red asterisks). l-NAME preserved mitochondrial structure (normal mitochondria, green arrow) and axons profiles. Scale bar, 1 μm. Middle, Magnified views of representative axons from left and right panels show 3-D EM reconstructions of these axons displaying normal (green) and abnormal (magenta) mitochondria and degradation products (red) in axons following OGD. C, OGD increased the number of abnormal mitochondria, whereas l-NAME attenuated mitochondrial numbers. n = number of MONs. *p < 0.05, ***p < 0.001; Kruskal–Wallis test followed by Dunn's multiple-comparison test. D, l-NAME preserved MON ATP levels following OGD. *p < 0.05, **p < 0.01, one-way ANOVA with Bonferonni's post hoc test.
Figure 6.
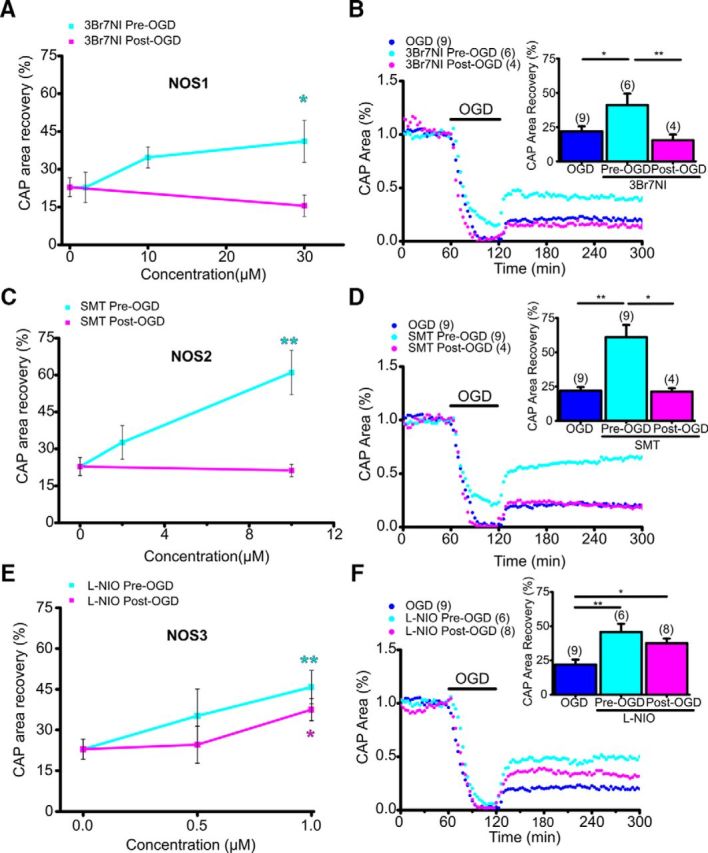
NOS3 inhibition promotes young axon function recovery following OGD. A, The NOS1 inhibitor 3Br7NI showed maximum protection with pretreatment (cyan) at 30 μm. Post-treatment (magenta) was not protective at any dose tested. B, In young WM, 3Br7NI (30 μm) pretreatment preserved axon function during OGD and improved axon function recovery following OGD; post-treatment was not protective. n = number of MONs. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferonni's post hoc test. C, The NOS2 inhibitor SMT showed maximum protection with pretreatment (cyan) at 10 μm. Post-treatment (magenta) was not protective at any dose tested. D, In young WM, SMT (10 μm) pretreatment preserved axon function during OGD and improved axon function recovery following OGD; post-treatment was not protective. n = number of MONs. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferonni's post hoc test. E, The NOS3 inhibitor l-NIO showed maximum protection with pretreatment (cyan) at 1 μm. Remarkably, l-NIO 1 μm treatment post-OGD (magenta) also provided protection. F, In young WM, l-NIO (1 μm) pretreatment and post-treatment improved axon function recovery following OGD. n = number of MONs. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferonni's post hoc test.
Figure 7.
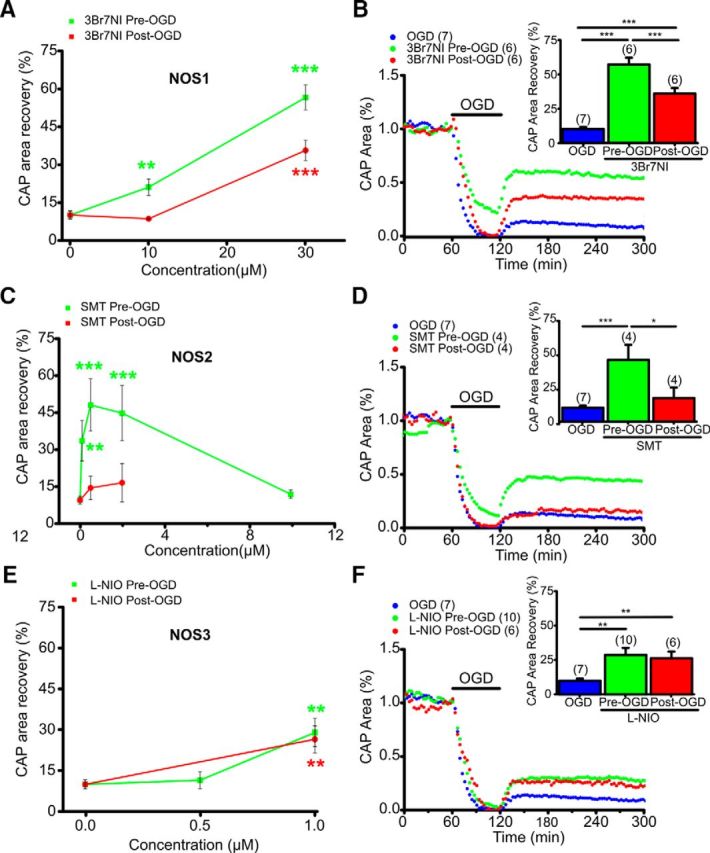
NOS1 or NOS3 inhibition promotes aging axon function recovery following OGD. A, The NOS1 inhibitor 3Br7NI showed maximum protection to axon function with pretreatment (green) at 30 μm. Post-treatment (red) was similarly protective. B, In aging WM, 3Br7NI (30 μm) pretreatment preserved axon function during OGD and improved axon function recovery following OGD; post-treatment also promoted axon function recovery. n = number of MONs. ***p < 0.001; one-way ANOVA followed by Bonferonni's post hoc test. C, The NOS2 inhibitor SMT showed maximum protection with pretreatment (green) at 0.5 μm. Post-treatment (red) was not protective at any dose tested. D, In aging WM, SMT (0.5 μm) pretreatment preserved axon function during OGD and improved axon function recovery following OGD, whereas post-treatment was not protective. n = number of MONs. *p < 0.05, ***p < 0.001; one-way ANOVA followed by Bonferonni's post hoc test. E, The NOS3 inhibitor l-NIO showed maximum protection with pretreatment (green) at 1 μm. Remarkably, l-NIO (1 μm) post-OGD (red) also conferred protection. F, In aging WM, l-NIO (1 μm) pretreatment as well as post-treatment improved axon function recovery following OGD. n = number of MONs. **p < 0.01; one-way ANOVA followed by Bonferonni's post hoc test.
Figure 8.
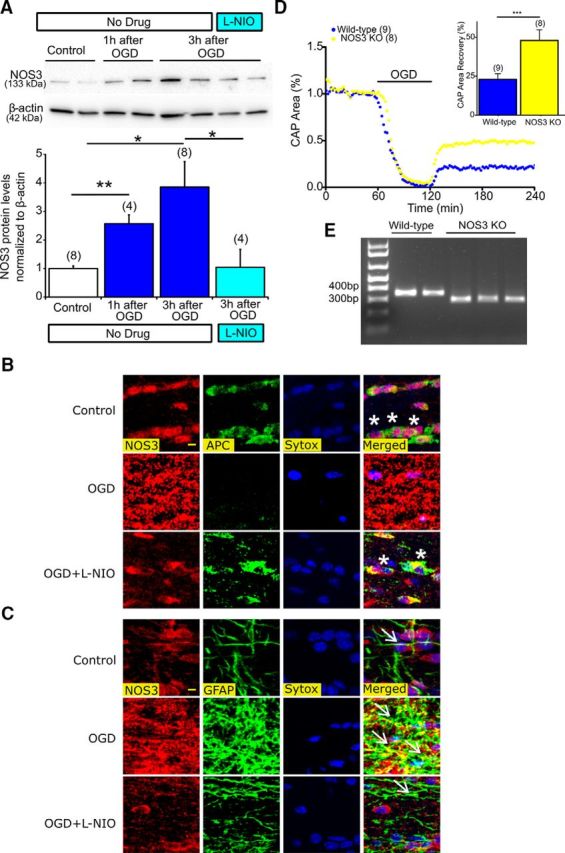
Inhibition of NOS3 upregulation in astrocytes protects oligodendrocytes and deletion of NOS3 promotes axon function against ischemia. A, MONs exposed to 60 min of OGD showed a progressive increase in NOS3 expression levels, which was attenuated with l-NIO (3 h following OGD: 3.8 ± 0.9% vs Control: 1.0 ± 0.1%), n = number of MONs. *p < 0.05, **p < 0.01; one-way ANOVA followed by Bonferonni's post hoc test. B, Colocalization of APC(+) oligodendrocytes, NOS3 (red), and Sytox (blue, nuclear staining) showed upregulation of NOS3 with OGD and loss of oligodendrocytes (white asterisks, right, merged). l-NIO (1 μm) application attenuated NOS3 upregulation and preserved oligodendrocytes. Scale bar, 5 μm. C, Colocalization of GFAP(+) astrocytes, NOS3 (red), and Sytox (blue, nuclear staining) showed upregulation of NOS3 mostly colocalized to activated astrocytic soma and processes (white arrows, right, merged). l-NIO (1 μm) application attenuated NOS3 upregulation in astrocytes and preserved oligodendrocytes. D, Axon function in MONs obtained from NOS3 knock-out mice showed improved axon function recovery following 60 min of OGD compared with control (WT) mice. n = number of MONs. ***p < 0.001; unpaired Student's two-tailed t test. E, Genotyping confirmation results for amplified DNA from tail samples. Note that the mutant band (NOS3−/−) was observed at 330 bp and the WT band (NOS3+/+) was observed at 337 bp on a 1.5% agarose gel.
Figure 9.
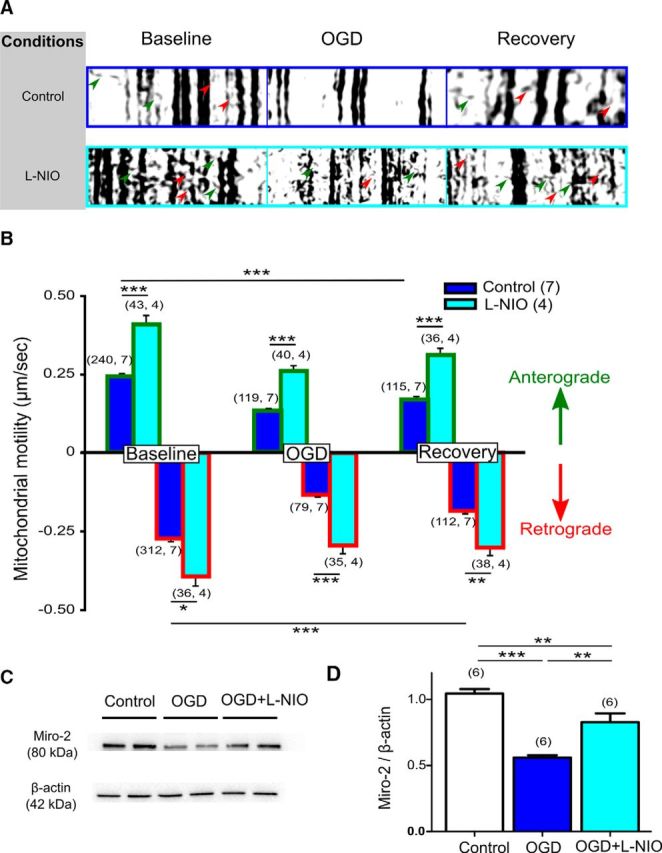
NOS3 inhibition preserves mitochondrial motility and Miro-2 levels against ischemia. A, Representative kymographs created from mito-CFP(+) live mitochondrial imaging show immotile mitochondria as vertical lines. Note the diagonal lines representing mitochondrial movement in axons in the anterograde direction (green arrows) as well as the retrograde direction (red arrows). B, Quantification of mitochondrial motility from kymographs revealed a 50% reduction in mitochondrial motility, both in the anterograde (blue with green border histograms) and retrograde directions with OGD (blue with red border histograms). l-NIO Pre-OGD application caused a prominent increase in mitochondrial motility in both directions within 10 min of application (cyan histograms), suggesting that NOS3 inhibition enhanced mitochondrial transport within the myelinated axons under control conditions. This increase in mitochondrial motility persisted during OGD (60 min) and improved further during recovery (20 min) in both directions. n = number of mitochondria, number of MONs. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Bonferonni's post hoc test. C, D, MONs exposed to 60 min of OGD showed a decrease in Miro-2 protein levels. Miro-2 plays a crucial role in regulating mitochondrial motility. l-NIO application (1 μm) preserved Miro-2 levels. **p < 0.01, ***p < 0.001; one-way ANOVA with Bonferonni's post hoc test.
Results
Pan-NOS inhibition selectively promotes axon function recovery in young WM
The isolated optic nerve, which is a purely-myelinated CNS tract, has been successfully used to study mechanisms of ischemic injury in young and aging WM (Tekkök et al., 2007; Baltan et al., 2008, 2011; Baltan, 2012, 2016; Murphy et al., 2014; Stahon et al., 2016). The effects of OGD on axon function recovery were determined in young and aging C57BL/6J (Figs. 1 and 2, respectively) or Thy-1 mito-CFP mice (Misgeld et al., 2007), which allowed fluorescent identification of mitochondrial distribution, shape, and size (Figs. 3A, 4A). The functional integrity of young (Fig. 1A,B) and aging (Fig. 2A,B) optic-nerve axons was monitored by quantifying the area under CAPs evoked by a supramaximal stimulus. To determine whether blocking pan-NOS activity protects axon function, l-NAME, which is a pan-NOS inhibitor, was introduced for 30 min after a 30 min baseline recording period. Superfusion conditions were maintained through 60 min of OGD and for the initial 30 min of the reperfusion period (Fig. 1B, cyan line). Evoked CAPs in MONs were then recorded for a total of 5 h in the absence of l-NAME. After OGD, the CAP area recovered to 23 ± 4% of the maximum recorded CAP area (Fig. 1A,B, dark blue), whereas l-NAME (200 μm) improved CAP area recovery to 77 ± 3% (p < 0.001; Fig. 1A, cyan traces, B, cyan line, C, cyan histogram). The l-NAME concentration was selected based on maximal protection conferred as previously reported (Zaleski et al., 2013). Furthermore, the presence of l-NAME preserved CAP area during OGD compared with OGD alone (Fig. 1A–C, dark blue vs cyan traces, time courses, and histograms). Because poststroke protection is more clinically relevant for stroke patients, l-NAME was also applied starting right after the end of OGD for 30 min before switching to regular ACSF (Fig. 1A,B, magenta traces and time course). Whereas OGD predictably suppressed CAP area completely, l-NAME (200 μm) application post-OGD rapidly improved CAP area recovery to 51 ± 6% (p < 0.01; Fig. 1A–C, magenta), albeit not to the same extent as application before OGD. This sustained improvement conferred by l-NAME application after OGD suggests a therapeutic window of opportunity to attenuate axon injury in young WM.
The risk of a stroke increases with age and CNS WM becomes intrinsically more vulnerable to injury with age (Baltan et al., 2008; Baltan, 2012), despite the fact that the distribution of glial cell types remains constant in different WM tracts and across different age groups (Tekkök and Goldberg, 2001; Baltan, 2014). Therefore, the effects of pan-NOS inhibition on axon function after OGD were determined in MONs obtained from 12-month-old C57BL/6J mice (Fig. 2A–C). In these aging MONs, CAP area showed minimal recovery (10 ± 2%) following OGD (60 min; Fig. 2A, dark blue traces, B, dark blue time course, C, dark blue histogram). Administration of l-NAME (200 μm) as a pretreatment failed to preserve CAP area during OGD (Fig. 2B, green time course), but following OGD it improved CAP area recovery to 56 ± 5% (p < 0.001; Fig. 2B, green time course, C, green histogram). In contrast, l-NAME application after the end of OGD did not promote CAP area recovery in aging MONs (9 ± 1%; Fig. 2B, red time course, C, red histogram).
NOS inhibition preserves axonal mitochondrial structure and function
The protective outcome of pan-NOS inhibition revealed by electrophysiological analysis in young and aging WM was further confirmed in a series of imaging studies (Figs. 3, 4). Mitochondria are essential for normal axonal function; however, they can also serve as a source of ROS/RNS. We evaluated the morphology and ultrastructure of mitochondria by monitoring CFP fluorescence in MONs from Thy-1 mito-CFP(+) mice, where axonal mitochondria can be selectively imaged (Figs. 3A, 4A) or using 3-D EM (Figs. 3B, 4B) in MONs from C57BL/6J mice under control conditions and OGD with or without NOS inhibition. OGD consistently caused loss of mitochondrial CFP(+) fluorescence compared with control conditions in young MONs (Fig. 3A, left and middle) as previously reported (Baltan et al., 2011, 2013; Baltan, 2012, 2014; Murphy et al., 2014). Application of l-NAME (200 μm) considerably attenuated the loss of CFP(+) fluorescence (Fig. 3A, right).
To validate that the changes in CFP fluorescence observed were related to mitochondrial structural changes, 3-D EM studies were performed on MONs that were immersion-fixed at the conclusion of electrophysiology experiments. Axonal profiles together with their enwrapping myelin sheaths and their respective mitochondria (Fig. 3B, top left, white rectangle) were clearly visible in 2-D images (Fig. 3B, top middle, green arrow) and reconstructed 3-D EM axons and mitochondria (Fig. 3B, top right). OGD caused structural changes to MONs such as swollen axons, thinning of myelin sheaths, shrunken axoplasm, and mitochondrial degradation (Fig. 3B, left middle, blue rectangle). In addition, abnormal mitochondria were frequently observed when nerves were exposed to OGD and were characterized by short, round swollen mitochondria with diminished or abnormal cristae (Fig. 3B, middle center, magenta arrow) surrounded by numerous degradation products (Fig. 3B, middle center, red asterisks). This was in clear contrast to the long mitochondria (Fig. 3B, top right, green) with distinct cristae that were seen in control conditions. There were few normal-looking mitochondria (Fig. 3B, middle right, green) in swollen axons, which consisted mainly of injured mitochondria (Fig. 3B, middle right, magenta) and degradation products (Fig. 3B, middle right, red). Application of l-NAME attenuated these mitochondrial changes after OGD (Fig. 3B, bottom), conserved the elongated shape of mitochondria (Fig. 3B, bottom middle, green arrows), reduced degradation products (Fig. 3B, bottom middle, red asterisks), and conserved axonal profiles (Fig. 3B, bottom right). Quantification of mitochondrial numbers based on mitochondrial morphology revealed a significant loss of normal mitochondria [Fig. 3C; control and OGD normal median mitochondrial numbers were 0.68 per 10 μm (white) and 0.18 per 10 μm (blue), p < 0.001, Kruskal–Wallis test followed by Dunn's multiple-comparison test] and an increase in the number of abnormal mitochondria in axons exposed to OGD [Fig. 3C; control and OGD abnormal median mitochondrial number were 0.87 per 10 μm (white) and 2.8 per 10 μm (blue), p < 0.001, Kruskal–Wallis test followed by Dunn's multiple-comparison test], whereas l-NAME application effectively reduced mitochondrial damage [Fig. 3C; l-NAME damaged median mitochondrial number 1.7 per 10 μm (cyan), p < 0.01 vs OGD, Kruskal–Wallis test followed by Dunn's multiple-comparison test].
To determine whether the morphological changes observed at the fluorescent and ultrastructural levels were associated with functional changes, ATP levels were quantified and compared among control, OGD, and l-NAME-treated MONs. MONs exposed to OGD showed a ∼60% decrease in levels of ATP, indicating major mitochondrial dysfunction (Fig. 3D; 0.47 ± 0.07%, p < 0.001). ATP levels were preserved following l-NAME application (0.9 ± 0.2%, p < 0.05) compared with OGD alone (0.5 ± 0.1%). Damaged mitochondria following OGD may be a major source of ROS/RNS generation, affecting axon recovery. Expectedly, NOS activity and NO generation increased with OGD (1.4 ± 0.1%; p < 0.05) compared with control MONs (Fig. 3E; 1.0 ± 0.1%). l-NAME treatment attenuated NOS activity during OGD such that it was comparable to control levels (1.0 ± 0.1%; p < 0.05). In addition, we examined levels of glutathione (GSH), which is a key free radical scavenger. GSH levels were significantly decreased in MONs exposed to OGD (0.8 ± 0.1%; p < 0.01) compared with control levels (Fig. 3F; 1.0 ± 0.1%). Application of l-NAME preserved levels of GSH (Fig. 3F; 0.9 ± 0.1%; p < 0.05). These results suggest that the maintenance of structural and functional integrity of mitochondria resulting from NOS inhibition can promote axon function recovery.
Consistent with improved axonal recovery in aging MONs, CFP(+) mitochondrial fluorescence in 12-month-old Thy-1 mito-CFP(+) mice was conserved by l-NAME application (Fig. 4A). Aging axons characteristically exhibited longer and thicker mitochondria [Fig. 4A, left, B, top left, (white rectangle), top center (green arrow); Baltan, 2012; Stahon et al., 2016]. OGD nevertheless caused extensive mitochondrial fission (Fig. 4A, middle) associated with mitochondrial swelling (Fig. 4B, middle left, blue rectangle), loss of cristae, formation of degradation products (Fig. 4B, middle center, magenta arrow and red asterisk), and marked axonal swelling (Fig. 4B, middle right). Treating aging MONs with l-NAME preserved mitochondrial structure (Fig. 4B, bottom left, cyan rectangle), reduced mitochondrial damage (Fig. 4B, bottom center, green arrow), and prevented axonal swelling (Fig. 4B, bottom right). Quantification of mitochondrial ultrastructure confirmed that OGD-induced increases in abnormal mitochondria in axons was consistently reduced by l-NAME application [Fig. 4C; OGD and l-NAME abnormal median mitochondrial numbers were 0.58 per 10 μm (blue) and 0.31 per 10 μm (cyan), respectively; p < 0.05, Kruskal–Wallis test followed by Dunn's multiple-comparison test]. Note that with aging, the number of axonal mitochondria is lower compared with mitochondrial numbers in young axons, as we previously reported (Stahon et al., 2016). Furthermore, ATP levels were reduced with OGD (Fig. 4D; 0.5 ± 0.9%, p < 0.01) compared with control conditions (1.0 ± 0.1%), but was restored by l-NAME application in aging WM (0.8 ± 0.1%, p < 0.05). The effect of l-NAME application on restoring ATP levels was consistent between young and aging optic nerves, despite the fact that ATP levels in aging axons are considerably lower compared with younger MONs (Stahon et al., 2016).
All three NOS isoforms are readily expressed in MONs
Because NOS inhibition selectively exerted protection to young axons when applied after injury, we reasoned that the cellular origins of specific NOS isoforms that mediate ischemic injury in WM change with age. To characterize NOS isoforms present in WM, we evaluated levels of isoform-specific NOS mRNAs. Quantification showed that mRNAs for NOS1, NOS2, and NOS3 were readily detected in WM (Fig. 5A). NOS1 mRNA levels were the highest (3.4 ± 0.7-fold, p < 0.01) compared with either NOS2 (1.3 ± 0.3-fold) or NOS3 (1.0 ± 0.4-fold). Because mRNA levels do not necessarily predict protein levels (Greenbaum et al., 2003), we further assessed NOS protein levels by Western blotting using isoform-specific antibodies (Fig. 5B). Protein levels of all three NOS isoforms were present in MONs. Next, we evaluated the expression pattern and localization of NOS isoforms using immunohistochemistry and isoform-specific antibodies in conjunction with confocal imaging, to support a biological basis for NOS inhibition in young (Fig. 5C,D) and aging WM (Fig. 5E,F). Glial cells were identified by GFAP for astrocytes, APC for mature oligodendrocytes, Iba-1 for microglia, and SMI-31 for axonal neurofilaments (Table 1). In young WM, the expression of NOS3 (red) colocalized with GFAP(+) astrocytes (green), in both processes and cell bodies (Fig. 5C, white arrow, bottom right, merged), APC(+) oligodendrocytes (green; Fig. 5D, white asterisks, top right, merged) and in Iba-1(+) microglia cell bodies and processes (green; Fig. 5D, white arrowhead, bottom right, merged). The expression of NOS2 (red) was restricted to astrocytes (green; Fig. 5C, middle right, merged) and oligodendrocytes (Table 2). NOS1 (red) was observed predominantly on astrocyte soma and processes (Fig. 5C, white arrow, top right, merged), but minimally on APC(+) oligodendrocytes (Table 2). SMI-31(+) axons (green) selectively expressed NOS3 (red) in young WM (Fig. 5D, white double arrowheads, middle right, merged). In aging WM, the expression of NOS3 colocalized with GFAP(+) astrocyte cell bodies along the entire length of their processes and end-feet outlining the blood vessels (Fig. 5E, white arrow, bottom right, merged). Similar to young WM, oligodendrocytes (Fig. 5F, white asterisks, top right, merged), axons (Fig. 5F, white double arrowheads, middle right, merged), and microglia (Fig. 5F, white arrowhead, bottom right, merged) extensively expressed NOS3 in aging WM. In contrast to young WM, NOS2 expression was abundant in microglia, while astrocytes and oligodendrocytes also expressed NOS2 (Table 2). The expression of NOS1 was limited to astrocytes in aging WM such that neither oligodendrocytes nor microglia expressed this isoform (Table 2). However, the expression of NOS1 was evident in aging axons (Fig. 5E, white arrow, top right, merged). Labeling characteristics and the antibodies that were used are all summarized in Figure 10 and Tables 1 and 2. The labeling characteristics and the specificity of these antibodies were also validated on brain slices that contained hippocampus and corpus callosum (data not shown). The extensive expression of these NOS isoforms in glial cells and axons implicated them as cellular targets for l-NAME, but also raised the question as to whether specific NOS isoforms expressed by specific glial cells could mediate ischemic injury via localized generation of ROS/RNS to cause age- and isoform-specific WM injury.
Table 2.
Cell-specific expression of NOS isoforms in WM differ in an age-dependent manner
| Young |
Aging |
|||||
|---|---|---|---|---|---|---|
| NOS1 | NOS2 | NOS3 | NOS1 | NOS2 | NOS3 | |
| Astrocyte | + | + | + | +++ | + | ++ |
| Oligodendrocyte | + | ++ | ++ | − | ++ | ++ |
| Axon | − | − | + | + | − | + |
| Microglia | − | − | + | − | +++ | + |
NOS1 was expressed in astrocyte somata and processes, and in oligodendrocytes in young MONs. Expression of NOS3 was observed in both processes and cell bodies of astrocytes, oligodendrocytes, and in microglia. The expression of NOS2 was restricted to astrocytes and oligodendrocytes. Axons selectively expressed NOS3 in young WM. In contrast to young WM, expression of NOS1 was limited to astrocytes in aging WM such that neither oligodendrocytes nor microglia expressed this isoform. In addition, the expression of NOS1 was evident in aging axons. NOS2 expression was strikingly abundant in microglia in aging WM compared to young WM, whereas astrocytes and oligodendrocytes also expressed NOS2. NOS3 was expressed in astrocyte cell bodies along the entire length of their processes and end-feet, outlining blood vessels in aging WM. Similar to young WM, oligodendrocytes, axons, and microglia expressed NOS3 in aging WM.
Figure 10.

Age- and isoform-dependent distribution of NOS in young and aging WM and the mechanisms of NOS3-mediated WM injury. Schematic represents localization of the three NOS isoforms in glial and axonal WM components as a function of age and injury. A, In young WM, NOS3 is expressed in astrocytes, oligodendrocytes, axons, and microglia. NOS1 and NOS2 expression were observed in oligodendrocytes and astrocytes. Inset, CAP with three distinct peaks recorded from a young MON. Miro-2 regulates mitochondrial movement along microtubules. B, With aging, astrocyte processes are more defined and are arranged in a transverse orientation relative to the axons. NOS3 showed a similar pattern of expression as observed in the young WM; however, it was also extensively present in astrocytes. In contrast to young WM, NOS2 was localized to microglia in addition to astrocytes and oligodendrocytes in aging WM. Expression of NOS1 was limited to astrocytes in aging WM such that neither oligodendrocytes nor microglia expressed this isoform. NOS2 was not expressed in axons in young and aging WM. C, When exposed to OGD, NOS3 levels were upregulated in astrocytes, resulting in increased NO generation, which led to oligodendrocytic death and interruption of mitochondrial motility through downregulation of Miro-2 levels, leading to mitochondrial fission and degradation. Note the myelin thinning and axonal swelling with OGD. D, l-NIO application preserves NOS3 levels during OGD, thus preserving mitochondrial structure, function, and motility to improve axon function recovery (inset, CAP).
Isoform-specific NOS inhibition promotes post-ischemic recovery in an age-dependent manner following OGD
To assess specific contributions from each NOS isoform in mediating ischemic damage, isoform-specific NOS inhibitors were applied to young (Fig. 6) and aging (Fig. 7) MONs exposed to OGD, as either a pre- or post-treatment. For each drug, dose–response curves were plotted to study CAP area recovery following OGD, which identified the optimal drug concentration necessary to protect MONs for post-treatment applications (Fig. 6A,C,E). Caution was taken to choose drug concentrations based on their IC50 values. Pre-OGD application of a NOS3-selective blocker, l-NIO, improved CAP area recovery at a concentration of 1 μm (Fig. 6F, cyan; 46 ± 6%, p < 0.01) compared with OGD alone (blue; 22 ± 3%). A NOS2 blocker, SMT, and a NOS1 blocker, 3Br7NI, were most effective at concentrations of 10 μm (Fig. 6D, cyan; 61 ± 9%, p < 0.01) and 30 μm (Fig. 6B, cyan; 41 ± 8%, p < 0.05), respectively. Interestingly, l-NIO preserved axon function when used either as pretreatment or post-treatment at 1 μm (Fig. 6F, magenta; 38 ± 4%, p < 0.05; Fig. 6F). On the other hand, SMT (Fig. 6D) and 3Br7NI (Fig. 6B) did not improve CAP area recovery when applied post-OGD [SMT: 21 ± 3%, p = 0.95; (D, magenta); 3Br7NI: 16 ± 4%, p = 0.2; (B, magenta)]. The specificity of l-NIO has been challenged by other studies, where l-NIO was also proposed to block NOS1 (Moore and Handy, 1997). Based on Ki values, we used l-NIO at a 1 μmol/L concentration to specifically target NOS3 (inhibitory constant Ki for NOS3 = 0.2 μmol/L, NOS1 = 3.9 μmol/L and NOS2 = 2.2 μmol/L; Mulligan et al., 1992; Boucher et al., 1999; Jiang et al., 2002; Pritchett and Shippy, 2014). 3Br7NI, the NOS1-specific blocker, did not demonstrate preservation of axon function post-OGD. To validate that l-NIO protection was mediated specifically through NOS3 inhibition, we combined l-NIO and 3Br7NI to determine whether any additive/synergistic effects of blocking both NOS3 and NOS1 could be observed. The combination of l-NIO and 3Br7NI promoted axon function to 53 ± 7% (p = 0.001) when applied as a pre-application, which was comparable to the maximum protection conferred by either of the drugs alone (data not shown). On the other hand, application of l-NIO and 3Br7NI after OGD showed improvement in CAP area recovery that was identical to that of l-NIO alone (40 ± 5% vs 38 ± 4%, p = 0.6). These results suggest that all of the NOS isoforms contribute to injury during ischemia in young MONs, but that only NOS3 contributes to the post-ischemic injury period in MONs, suggesting that NOS3 is a potential therapeutic target to preserve young WM following ischemic injury (Fig. 8D).
Parallel experiments in aging MONs revealed that l-NIO at a 1 μm concentration (Fig. 7E) improved CAP area recovery to a similar extent when applied before or after OGD (Fig. 7F, green; 29 ± 5%, p < 0.01 vs red, 27 ± 5%, p < 0.01) compared with OGD (blue; 10 ± 2%) in aging MONs (Fig. 7F). Interestingly, SMT at a 0.5 μm concentration (Fig. 7C), which is a much lower dose compared with that used in young axons (Fig. 6C), promoted CAP area recovery only when applied before OGD (Fig. 7D, green; 48 ± 11%, p < 0.001), but failed to confer any protection when applied after OGD (red; 15 ± 5%, p = 0.76) compared with OGD alone in aging MONs (11 ± 2%). The most extensive protection to aging axons was conferred by 3Br7NI (Fig. 7A; 30 μm) when applied before (B, green; 57 ± 5%, p < 0.001) or after OGD (B, red; 36 ± 4%, p < 0.001). To our surprise, both l-NIO and 3Br7NI provided post-ischemic protection to aging axons, suggesting that NOS3 and NOS1 both contribute to oxidative injury in aging WM, whereas NOS3 emerged as the universal NOS isoform to mediate post-ischemic oxidative injury in young and aging MONs.
Ischemia upregulates NOS3 levels in WM
Previous studies have shown that transient ischemia upregulates NOS3 levels, which confers neuroprotection in gray matter (Zhang et al., 1993; Nagafuji et al., 1994). To provide proof-of-principle that ischemia also upregulates NOS3 levels in WM, we exposed young MONs to OGD for 1 h and subsequently MONs were collected at 1 or 3 h after the conclusion of OGD. Western blots showed a time-dependent upregulation of NOS3 protein levels, which reached a peak at 3 h after the end of OGD (Fig. 8A; 3 h following OGD: 3.8 ± 0.9-fold vs Control: 1.0 ± 0.1, p < 0.05). Expectedly, l-NIO application effectively prevented upregulation of NOS3 protein levels following OGD. To verify that the increase in protein levels was correlated with increased immunoreactivity, MONs were labeled for NOS3 (Fig. 8B,C, red), glial cell-specific markers, and nuclei with Sytox (blue). Under control conditions, NOS3 is expressed in APC(+) oligodendrocytes (Fig. 8B, first row, control) and GFAP(+) astrocytes (Figs. 8C, first row, control; Table 2). OGD caused widespread loss of APC(+) oligodendrocytes (Fig. 8B, second row, OGD), but the expression levels of NOS3 labeling increased and displayed a diffuse pattern mostly colocalizing to GFAP(+) astrocytes following OGD (Fig. 8C, second row, OGD). l-NIO application attenuated NOS3 reactivity and restored the expression pattern akin to control conditions (Fig. 8B,C last row; OGD + l-NIO). To further verify that attenuation of cellular upregulation of NOS3 levels in WM confers functional protection, we monitored axon function recovery in MONs obtained from NOS3 knock-out (NOS3−/−) mice (Fig. 8D). NOS3 knock-out mice were confirmed by performing genotyping PCR on DNA from tail samples of NOS3−/− animals (B6.129P2-Nostm1Unc/J, The Jackson Laboratory) against WT animals (Fig. 8E; mutant band 300 bp, WT band 337 bp). CAP area of NOS3−/− consistently recovered better (48 ± 9%, p < 0.001) compared with WT (23 ± 4%). These results suggest that ischemia upregulates NOS3 protein levels in WM astrocytes, leading to oligodendrocyte death and compromising axon function recovery following ischemia (see Fig. 10).
NOS3 inhibition preserves axonal mitochondrial motility following OGD
Our recent studies showed that mitochondria undergo extensive fission during ischemia, and that mitochondrial fragmentation correlated with loss of mitochondrial motility (Bastian et al., 2018). More importantly, preventing mitochondrial fission maintained mitochondrial motility during OGD and improved it further after OGD, which promoted axon function recovery (Bastian et al., 2018). Because pan-NOS inhibition preserved mitochondrial structural integrity, we anticipated that functional protection conferred by NOS3 inhibition in WM is mediated via conservation and promotion of mitochondrial motility during and after OGD. Time-lapse live imaging of mitochondria, using optic nerves obtained from Thy-1 mito-CFP(+) mice, showed mitochondrial movement in the anterograde (Fig. 9A, Control, green arrows) and retrograde (Control, red arrows) directions as identified using kymographs (Fig. 9A), which represent 2-D images of stationary and motile mitochondria during the entire time-lapse sequence. Vertical lines represent the stationary mitochondria, while diagonal lines represent motile mitochondria (Fig. 9A). Note that the majority of mitochondria were stationary, and that mitochondria maintained a stable speed under control conditions. Onset of OGD caused a 50% reduction in mitochondrial motility both in the anterograde (Fig. 9A,B, blue histograms green border; Control OGD 0.13 ± 0.01 μm/s vs Control Baseline 0.24 ± 0.01 μm/s, p < 0.001) and retrograde directions (Fig. 9A,B, blue histograms red border; Control OGD 0.13 ± 0.01 μm/s, vs Control Baseline 0.27 ± 0.01 μm/s, p < 0.001). Recovery in the anterograde direction (Fig. 9 A,B, Control Recovery, blue histograms green border; 0.17 ± 0.01 μm/s, p < 0.05) and retrograde directions were significant (Fig. 9A,B, Control Recovery, blue histograms red border; 0.18 ± 0.01 μm/s, p < 0.05). Application of 1 μm l-NIO for 10 min caused a prominent increase in mitochondrial motility under control conditions in the anterograde direction (Fig. 9B, Anterograde, l-NIO Baseline, cyan histograms with green border, 0.35 ± 0.03 μm/s, p < 0.001) and retrograde direction (Fig. 9B; Retrograde: l-NIO Baseline, cyan histograms with red border; 0.36 ± 0.04 μm/s, p < 0.05). This increase in mitochondrial motility persisted during OGD (60 min; Fig. 9B; Anterograde: l-NIO OGD, cyan histograms with green border: 0.26 ± 0.02 μm/s vs Control OGD, blue histograms with green border: 0.13 ± 0.01 μm/s, p < 0.001; Retrograde: l-NIO OGD, cyan histograms with red border: 0.27 ± 0.02 μm/s vs Control OGD, blue histograms with red border: 0.13 ± 0.01 μm/s, p < 0.001) and recovery (20 min; Fig. 9 B; Anterograde: l-NIO Recovery, cyan histograms with green border: 0.32 ± 0.02 μm/s vs Control Recovery, blue histograms with green border: 0.17 ± 0.01 μm/s, p < 0.001; Retrograde: l-NIO Recovery, cyan histograms with red border: 0.28 ± 0.03 μm/s vs Control Recovery, blue histograms with red border: 0.17 ± 0.01 μm/s, p < 0.01) in both directions. Mitochondrial motility along microtubules is regulated by protein complexes, of which ATP and the calcium-dependent protein Miro-2 play a major role (Guo et al., 2005; Saotome et al., 2008; Russo et al., 2009; Melkov et al., 2016). Miro-2 has been shown to be increasingly associated with neurodegenerative diseases that are characterized with mitochondrial dysfunction (Tang, 2015). Dysregulation of Miro-2 leads to mitochondrial arrest in movement and clearance (Wang et al., 2011). Miro also affects both anterograde (Macaskill et al., 2009; Wang and Schwarz, 2009) and retrograde motility (Morlino et al., 2014) and the fusion-fission dynamics of mitochondria (Misko et al., 2010; Tang, 2015). We observed that OGD resulted in a decrease in Miro-2 protein levels in optic nerves (Fig. 9C,D; Control 1.0 ± 0.03-fold vs OGD 0.5 ± 0.02, p < 0.001); 1 μm l-NIO application preserved Miro-2 levels in MONs exposed to OGD (0.8 ± 0.07, p < 0.01 compared with OGD). Together with the electrophysiology and CFP imaging results, these findings suggest that NOS3 inhibition promotes axon function recovery by preventing mitochondrial fission and by preserving mitochondrial structure and motility during ischemia because of a decrease in Miro-2 levels (Figs. 9, 10).
Discussion
We showed that inhibition of NOS activity, specifically inhibition of NOS3 activity, before or after an ischemic episode promotes functional recovery of axons in young and aging optic nerves and preserves WM architecture. The main correlates of these effects included preservation of oligodendrocytes, attenuated NOS3 upregulation, a reduction in NO generation, improved glutathione levels, maintenance of mitochondrial structure and motility, and preservation of ATP levels. Our findings suggest that NOS3 inhibition in glial cells targets mitochondrial energy regulation and oxidative injury in young and aging WM, thus promoting recovery following ischemia (Fig. 10).
The central finding in our study was that NOS inhibition with l-NAME preserved axon structure and function of WM. This effect was apparent in young and aging WM when l-NAME was applied before ischemia, whereas post-ischemic l-NAME application was only effective in young WM. Axon function directly correlates with tissue energy reserves, since Na+-K+ ATPase activity is closely dependent upon ATP levels. As a result, OGD caused a significant reduction in ATP levels and a drastic loss of CFP fluorescence, which was profoundly restored after NOS inhibition in young and aging optic nerves. Expectedly, increased NO generation and reduced glutathione levels caused by OGD were also restored to control levels by NOS inhibition. Consequently, the optic nerves were found to abundantly express NOS1, NOS2, and NOS3, which showed a glial cell-specific pattern that differed with aging. Subsequently, isoform-specific inhibition of NOS identified NOS3 in young and aging WM as a universal target to attenuate post-ischemic injury. Consistent with these results, NOS3 cellular expression patterns remained similar between young and aging WM, in contrast to NOS1 and NOS2. NOS3 was consistently expressed by young and aging oligodendrocytes, astrocytes, axons, and microglia and NOS3 inhibition consistently provided significant protection of axon function that correlated with preservation of mitochondria at the ultrastructural level, suggesting that intervention in the oxidative pathway provides post-ischemic protection to WM.
Interestingly, NOS1 inhibition with 3Br7NI provided similar post-ischemic protection selectively in aging WM. NOS1 is selectively expressed by aging axons, but not by young axons, suggesting that NOS isoforms expressed in axons may dictate the amount of post-ischemic functional recovery. Additional confirmation was provided when NOS2 inhibition, which was expressed neither in young nor in old axons, was ineffective in conferring post-ischemic functional protection to young or aging WM. NOS2 was abundantly expressed in aging microglia, in contrast to young microglia, and it is possible that failure of l-NAME application to exert post-ischemic protection to aging axons may be due to its broad inhibition of all NOS isoforms, including NOS2, in aging WM. This suggestion presumes that microglial NOS2 activation following OGD is protective of aging WM (Fig. 10; Neumann et al., 2006; Kawano et al., 2007; Li et al., 2008; Kato et al., 2016). Protective effects of specific NOS isoforms in microglia during ischemia remain to be discovered.
A novel aspect of our study was the demonstrated post-ischemic efficacy of NOS3 inhibition to protect axon function and structure against ischemia. Improved axon function recovery in optic nerves from NOS3 knock-out mice provided further confirmation of our pharmacological results. NOS3 activation in in vivo ischemia limits the degree of flow reduction by promoting vasodilatation and by inhibiting microvascular plugging by platelets and leukocytes, thereby conferring protection in the initial few hours following ischemia (Toda et al., 2009). Subsequently, ischemia in NOS3 knock-out mice produces a larger infarct size, because of the loss of beneficial hemodynamic effects of endothelial NO synthesis (Huang et al., 1996). In addition, pharmacological interventions that upregulate NOS3 (Laufs et al., 2002; Asahi et al., 2005; Tsai et al., 2007) and/or increase cerebral blood flow acting via endothelial mechanisms (Jiang et al., 2002; Rikitake et al., 2005; Shin et al., 2007) have been shown to be protective following ischemic stroke. Together with our results showing that NOS3 inhibition protects axon function after ischemia, these results suggest using caution when defining the role of NOS3 in cerebral ischemia at the vascular and cellular levels once the blood–brain barrier is compromised after a stroke. We propose that NOS3 inhibition is a universal target to protect WM independent of age, whether applied before or after ischemia, and that this is a result of the drug targeting the oxidative pathway during spatiotemporal progression of WM injury. Expectedly, NOS inhibition attenuated oxidative stress markers, preserved CFP(+) fluorescent mitochondria, and ultimately conserved mitochondrial ultrastructure and ATP production. Spatiotemporal progression of injury mechanisms in WM varies with age and approaches that protect young WM are often ineffective in, or even harmful to, aging WM (Baltan et al., 2008; Baltan, 2009, 2014). Among a number of molecular and structural changes that are responsible for dictating age-specific injury mechanisms in WM, alterations in mitochondrial structure and function underlie the increased vulnerability of aging WM to ischemia. It is plausible that the number of mitochondria and/or the capacity to scavenge products of oxidative injury may underlie the differential vulnerability of glial cells and axons to ischemia. Specifically, aging mitochondria produce elevated oxidative stress markers, predisposing aging axons to subsequent injury (Stahon et al., 2016). Therefore, conserving mitochondrial structure, and subsequently function, provides a common target to attenuate injury in young and aging WM. Excitotoxicity initiates the oxidative injury pathway during ischemia, but oxidative stress becomes the primary injury mechanism during reperfusion. Therefore, interventions that conserve mitochondria and attenuate oxidative stress provide post-ischemic protection to WM function. These results suggest that NO generation contributes to the destruction of mitochondria either by disrupting the electron transport chain and/or by peroxynitrites, which are generated by the combination of NO and ROS. It is also possible that NOS3 inhibition may affect downstream components of oxidative injury. Alternatively, NOS3 could also be regulated post-transcriptionally by influencing mRNA stability (Laufs and Liao, 1998) and post-translationally by modulation of protein stability or protein phosphorylation (Dimmeler et al., 1999; Fulton et al., 1999). These post-translational modifications might provide a powerful mechanism for downregulation of NOS3 activity, similar to NOS1 phosphorylation, which decreases NOS1 catalytic activity and might be a useful strategy for neuroprotection (Hayashi et al., 1999). These plausible mechanisms in WM injury are currently under investigation.
A novel finding in our study was that Miro-2, which is a calcium sensing protein that couples mitochondria to microtubules and regulates anterograde and retrograde motility (Guo et al., 2005; Russo et al., 2009; Wang and Schwarz, 2009; Melkov et al., 2016), was attenuated following WM ischemic injury. NOS3 inhibition promoted mitochondrial motility by preserving Miro-2 levels when MONs were exposed to OGD. Interestingly, even under baseline conditions, NOS3 inhibition increased mitochondrial motility in both the anterograde and retrograde directions. This is the first report on the effects of NOS3 inhibition on mitochondrial motility. NO acts via soluble guanylyl cyclase and second messenger cGMP to regulate intracellular calcium in a cell-specific manner (Clementi and Meldolesi, 1997; Yao and Huang, 2003). It is possible that attenuation of NO and ROS following NOS3 inhibition causes a decrease in cytosolic calcium, promoting Miro-2 binding to the cytoskeletal motor proteins kinesin and dynein, either directly or via adaptor proteins to promote mitochondrial motility (Saotome et al., 2008; Cai and Sheng, 2009; Macaskill et al., 2009; Wang and Schwarz, 2009).
Another significant finding in our study was the demonstration that the axon-protective action associated with NOS3 inhibition correlated with preservation of oligodendrocytes in WM. Oligodendrocytes myelinate axons and support axon function (Lubetzki et al., 1993; Tekkök and Goldberg, 2001; Arai and Lo, 2009; Baltan et al., 2011). We observed NOS3 labeling in mature oligodendrocytes. WM ischemia resulted in loss of oligodendrocytes and axon function, which was attenuated by NOS3 inhibition. The presence of NOS3 in mature oligodendrocytes warrants further studies to understand the cell-specific role of NOS3 in the regulation of oligodendrocyte differentiation and maturation. Whether application of a NOS3 blocker preserving oligodendrocyte precursors will prove to be therapeutically relevant in conditions such as human periventricular WM injury that leads to cerebral palsy in preterm infants, which is a condition that predominantly affects late oligodendrocyte precursors (Back et al., 2012; Back and Miller, 2014; Back, 2015), will be an interesting area to investigate in future studies. OGD caused widespread loss of oligodendrocytes, which correlated with upregulation of NOS3 levels in astrocytes. NOS3 inhibition attenuated NOS3 upregulation, reduced astrocyte activation, and ameliorated oligodendrocyte loss. Because NOS3 levels were elevated in astrocytes, it is possible that diffusion of astrocytic NO (Gibson et al., 2005) contributed to oligodendrocyte death (Fig. 10). Astrocytes and neuronal elements can also cross talk through glutamate (Shimizu-Sasamata et al., 1998), the NO/d-serine system (Darra et al., 2009), and other diffusible molecules (Quirié et al., 2013); however, these remain unexplored in WM. It is also plausible that microglial NOS3 activation may damage oligodendrocytes in a NO-dependent manner or via activation of inflammatory pathways (Merrill et al., 1993; Murphy et al., 1993). Importantly, preservation of oligodendrocytes provided significant protection of axon function that correlated with preservation of CFP(+) mitochondria and ultrastructural integrity and function of mitochondria. These results further support the novel concept that oligodendrocytes support axons structurally and metabolically and regulate axon function by providing energy metabolites (Saab et al., 2016).
It is intriguing that despite having a deleterious effect on gray matter, NOS3 inhibition preserves WM against ischemia. Devising ways for targeted delivery of l-NIO directly to WM and bypassing the blood–brain barrier is technically challenging; however, if successful, it can be beneficial in disorders such as vascular dementia, where cognitive deficits are caused by extensive WM lesions, and similar to stroke, the risk increases with aging (Englund, 1998; Barber et al., 1999; Rosenberg et al., 2001). Because NOS3 inhibition promoted axon function in both young and aging WM, it may have a potential role in treating this disorder. Furthermore, the protective effect of preconditioning WM with NOS3 inhibition has never been studied and may have possible therapeutic significance to decrease the cognitive decline associated with WM lesions in aging (Wong et al., 2002).
Our studies conclude that ischemic WM injury leads to axonal dysfunction through NOS-mediated mitochondrial disruption and oxidative injury (Fig. 10). WM components diffusely express all three isoforms of NOS in a cell-specific manner that changes with age, establishing a prominent “nitrinergic role” for glial cells (Murphy et al., 1993) in contrast to previous reports (Garthwaite et al., 2002, 2005). Furthermore, NOS3 provides a universal target for post-ischemic benefit to young and aging WM (Fig. 10). Activation of NOS3 can be protective or destructive, depending on its effects on the vascular versus cellular sites and on the stage of evolution of ischemic processes in gray matter (Iadecola et al., 1994; Wiencken and Casagrande, 1999; Li et al., 2014). The WM is injured in most strokes, contributing to the disability associated with clinical deficits. Therefore, defining the role of NOS3 in cerebral ischemia remains an important step for the development of new targeted neuroprotective strategies for the post-ischemic brain.
Footnotes
This work was supported by Grants from NIA (AG033720) to S.B and NINDS (NS094881) to S. Brunet and S. Baltan. We thank Dr. Chris Nelson for help editing this paper.
The authors declare no competing financial interests.
References
- Arai K, Lo EH (2009) Oligovascular signaling in white matter stroke. Biol Pharm Bull 32:1639–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Huang Z, Thomas S, Yoshimura S, Sumii T, Mori T, Qiu J, Amin-Hanjani S, Huang PL, Liao JK, Lo EH, Moskowitz MA (2005) Protective effects of statins involving both eNOS and tPA in focal cerebral ischemia. J Cereb Blood Flow Metab 25:722–729. 10.1038/sj.jcbfm.9600070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA. (2015) Brain injury in the preterm infant: new horizons for pathogenesis and prevention. Pediatr Neurol 53:185–192. 10.1016/j.pediatrneurol.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Miller SP (2014) Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol 75:469–486. 10.1002/ana.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Riddle A, Dean J, Hohimer AR (2012) The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics 9:359–370. 10.1007/s13311-012-0108-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S. (2009) Ischemic injury to white matter: an age-dependent process. Neuroscientist 15:126–133. 10.1177/1073858408324788 [DOI] [PubMed] [Google Scholar]
- Baltan S. (2012) Histone deacetylase inhibitors preserve function in aging axons. J Neurochem 123:108–115. 10.1111/j.1471-4159.2012.07949.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S. (2014) Age-dependent mechanisms of white matter injury after stroke. In: White matter injury in stroke and CNS disease (Baltan S, Carmichael ST, Matute C, Xi G, Zhang JH, eds), pp 373–403. New York, NY: Springer. [Google Scholar]
- Baltan S. (2016) Age-specific localization of NMDA receptors on oligodendrocytes dictates axon function recovery after ischemia. Neuropharmacology 110:626–632. 10.1016/j.neuropharm.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR (2008) White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci 28:1479–1489. 10.1523/JNEUROSCI.5137-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Inman DM, Danilov CA, Morrison RS, Calkins DJ, Horner PJ (2010) Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci 30:5644–5652. 10.1523/JNEUROSCI.5956-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS (2011) Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci 31:3990–3999. 10.1523/JNEUROSCI.5379-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Morrison RS, Murphy SP (2013) Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics 10:798–807. 10.1007/s13311-013-0201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P, Perry R, O'Brien J (1999) White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer's disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatry 67:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian C, Politano S, Day J, McCray A, Brunet S, Baltan S (2018) Mitochondrial dynamics and preconditioning in white matter. Cond Med 1:64–72. [PMC free article] [PubMed] [Google Scholar]
- Boucher JL, Moali C, Tenu JP (1999) Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for l-arginine utilization. Cell Mol Life Sci 55:1015–1028. 10.1007/s000180050352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC. (2001) Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta 1504:46–57. 10.1016/S0005-2728(00)00238-3 [DOI] [PubMed] [Google Scholar]
- Brunet S, Bastian C, Baltan S (2016) Ischemic injury to white matter: an age-dependent process. In: Non-neuronal mechanisms of brain damage and repair after stroke (Chen J, Zhang J, Hu X, eds), pp 327–343. Switzerland: Springer. [Google Scholar]
- Cai Q, Sheng ZH (2009) Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron 61:493–496. 10.1016/j.neuron.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina A, Radi R (1996) Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328:309–316. 10.1006/abbi.1996.0178 [DOI] [PubMed] [Google Scholar]
- Cho AH, Kim HR, Kim W, Yang DW (2015) White matter hyperintensity in ischemic stroke patients: it may regress over time. J Stroke 17:60–66. 10.5853/jos.2015.17.1.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Bredt DS (1997) Nitric oxide in excitable tissues: physiological roles and disease. J Clin Invest 100:2424–2429. 10.1172/JCI119783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E, Meldolesi J (1997) The cross-talk between nitric oxide and Ca2+: a story with a complex past and a promising future. Trends Pharmacol Sci 18:266–269. 10.1016/s0165-6147(97)01087-0 [DOI] [PubMed] [Google Scholar]
- Darra E, Ebner FH, Shoji K, Suzuki H, Mariotto S (2009) Dual cross-talk between nitric oxide and D-serine in astrocytes and neurons in the brain. Cent Nerv Syst Agents Med Chem 9:289–294. 10.2174/187152409789630415 [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999) Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature 399:601–605. 10.1038/21224 [DOI] [PubMed] [Google Scholar]
- Douki T, Cadet J (1996) Peroxynitrite mediated oxidation of purine bases of nucleosides and isolated DNA. Free Radic Res 24:369–380. 10.3109/10715769609088035 [DOI] [PubMed] [Google Scholar]
- Drapier JC, Hibbs JB Jr (1986) Murine cytotoxic activated macrophages inhibit aconitase in tumor cells: inhibition involves the iron-sulfur prosthetic group and is reversible. J Clin Invest 78:790–797. 10.1172/JCI112642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund E. (1998) Neuropathology of white matter changes in Alzheimer's disease and vascular dementia. Dement Geriatr Cogn Disord 9 Suppl 1:6–12. 10.1159/000051183 [DOI] [PubMed] [Google Scholar]
- Fern R, Ransom BR, Waxman SG (1995) Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol 74:369–377. 10.1152/jn.1995.74.1.369 [DOI] [PubMed] [Google Scholar]
- Fiala JC. (2005) Reconstruct: a free editor for serial section microscopy. J Microsc 218:52–61. 10.1111/j.1365-2818.2005.01466.x [DOI] [PubMed] [Google Scholar]
- Fiala JC, Harris KM (2001) Extending unbiased stereology of brain ultrastructure to three-dimensional volumes. J Am Med Inform Assoc 8:1–16. 10.1136/jamia.2001.0080001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase akt. Nature 399:597–601. 10.1038/21218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite G, Brown G, Batchelor AM, Goodwin DA, Garthwaite J (1999) Mechanisms of ischaemic damage to central white matter axons: a quantitative histological analysis using rat optic nerve. Neuroscience 94:1219–1230. 10.1016/S0306-4522(99)00389-9 [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Goodwin DA, Batchelor AM, Leeming K, Garthwaite J (2002) Nitric oxide toxicity in CNS white matter: an in vitro study using rat optic nerve. Neuroscience 109:145–155. 10.1016/S0306-4522(01)00447-X [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Batchelor AM, Goodwin DA, Hewson AK, Leeming K, Ahmed Z, Cuzner ML, Garthwaite J (2005) Pathological implications of iNOS expression in central white matter: an ex vivo study of optic nerves from rats with experimental allergic encephalomyelitis. Eur J Neurosci 21:2127–2135. 10.1111/j.1460-9568.2005.04062.x [DOI] [PubMed] [Google Scholar]
- Gibson CL, Coughlan TC, Murphy SP (2005) Glial nitric oxide and ischemia. Glia 50:417–426. 10.1002/glia.20143 [DOI] [PubMed] [Google Scholar]
- Greenbaum D, Colangelo C, Williams K, Gerstein M (2003) Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol 4:117. 10.1186/gb-2003-4-9-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE (2005) The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47:379–393. 10.1016/j.neuron.2005.06.027 [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Nishio M, Naito Y, Yokokura H, Nimura Y, Hidaka H, Watanabe Y (1999) Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J Biol Chem 274:20597–20602. 10.1074/jbc.274.29.20597 [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA (1996) Enlarged infarcts in endothelial nitric oxide synthase knock-out mice are attenuated by nitro-l-arginine. J Cereb Blood Flow Metab 16:981–987. 10.1097/00004647-199609000-00023 [DOI] [PubMed] [Google Scholar]
- Iadecola C. (1997) Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci 20:132–139. 10.1016/S0166-2236(96)10074-6 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Pelligrino DA, Moskowitz MA, Lassen NA (1994) Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab 14:175–192. 10.1038/jcbfm.1994.25 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Xu X, Zhang F, el-Fakahany EE, Ross ME (1995a) Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab 15:52–59. 10.1038/jcbfm.1995.6 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu S, Casey R, Ross ME (1995b) Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 15:378–384. 10.1038/jcbfm.1995.47 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Xu X (1995c) Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol 268:R286–R292. 10.1152/ajpregu.1995.268.1.R286 [DOI] [PubMed] [Google Scholar]
- Iadecola C, Kahles T, Gallo EF, Anrather J (2011) Neurovascular protection by ischaemic tolerance: role of nitric oxide. J Physiol 589:4137–4145. 10.1113/jphysiol.2011.210831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS (1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 298:431–437. 10.1016/0003-9861(92)90431-U [DOI] [PubMed] [Google Scholar]
- Jiang MH, Kaku T, Hada J, Hayashi Y (2002) Different effects of eNOS and nNOS inhibition on transient forebrain ischemia. Brain Res 946:139–147. 10.1016/S0006-8993(02)02870-6 [DOI] [PubMed] [Google Scholar]
- Kato G, Inada H, Wake H, Akiyoshi R, Miyamoto A, Eto K, Ishikawa T, Moorhouse AJ, Strassman AM, Nabekura J (2016) Microglial contact prevents excess depolarization and rescues neurons from excitotoxicity. eNeuro 3:ENEURO.0004–16.2016. 10.1523/ENEURO.0004-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Kunz A, Abe T, Girouard H, Anrather J, Zhou P, Iadecola C (2007) iNOS-derived NO and Nox2-derived superoxide confer tolerance to excitotoxic brain injury through peroxynitrite. J Cereb Blood Flow Metab 27:1453–1462. 10.1038/sj.jcbfm.9600449 [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE (1999) Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 26:463–471. 10.1016/S0891-5849(98)00216-0 [DOI] [PubMed] [Google Scholar]
- Lander HM. (1997) An essential role for free radicals and derived species in signal transduction. FASEB J 11:118–124. 10.1096/fasebj.11.2.9039953 [DOI] [PubMed] [Google Scholar]
- Laufs U, Liao JK (1998) Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by rho GTPase. J Biol Chem 273:24266–24271. 10.1074/jbc.273.37.24266 [DOI] [PubMed] [Google Scholar]
- Laufs U, Gertz K, Dirnagl U, Böhm M, Nickenig G, Endres M (2002) Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res 942:23–30. 10.1016/S0006-8993(02)02649-5 [DOI] [PubMed] [Google Scholar]
- Li QF, Zhu YS, Jiang H (2008) Isoflurane preconditioning activates HIF-1alpha, iNOS and Erk1/2 and protects against oxygen-glucose deprivation neuronal injury. Brain Res 1245:26–35. 10.1016/j.brainres.2008.09.069 [DOI] [PubMed] [Google Scholar]
- Li ST, Pan J, Hua XM, Liu H, Shen S, Liu JF, Li B, Tao BB, Ge XL, Wang XH, Shi JH, Wang XQ (2014) Endothelial nitric oxide synthase protects neurons against ischemic injury through regulation of brain-derived neurotrophic factor expression. CNS Neurosci Ther 20:154–164. 10.1111/cns.12182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinova L, Atochin DN, Fattakhov N, Vasilenko M, Zatolokin P, Kirienkova E (2015) Nitric oxide and mitochondria in metabolic syndrome. Front Physiol 6:20. 10.3389/fphys.2015.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubetzki C, Demerens C, Anglade P, Villarroya H, Frankfurter A, Lee VM, Zalc B (1993) Even in culture, oligodendrocytes myelinate solely axons. Proc Natl Acad Sci USA 90:6820–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:541–555. 10.1016/j.neuron.2009.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Sánchez-Gómez MV, Pérez-Samartín A, Rodríguez-Antigüedad A, Pérez-Cerdá F (2007) Excitotoxic damage to white matter. J Anat 210:693–702. 10.1111/j.1469-7580.2007.00733.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkov A, Baskar R, Alcalay Y, Abdu U (2016) A new mode of mitochondrial transport and polarized sorting regulated by dynein, Milton and Miro. Development 143:4203–4213. 10.1242/dev.138289 [DOI] [PubMed] [Google Scholar]
- Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE (1993) Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol 151:2132–2141. [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo. Nat Methods 4:559–561. 10.1038/nmeth1055 [DOI] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30:4232–4240. 10.1523/JNEUROSCI.6248-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43:109–142. [PubMed] [Google Scholar]
- Moore PK, Handy RL (1997) Selective inhibitors of neuronal nitric oxide synthase: is no NOS really good NOS for the nervous system? Trends Pharmacol Sci 18:204–211. 10.1016/S0165-6147(97)90624-6 [DOI] [PubMed] [Google Scholar]
- Morlino G, Barreiro O, Baixauli F, Robles-Valero J, González-Granado JM, Villa-Bellosta R, Cuenca J, Sánchez-Sorzano CO, Veiga E, Martín-Cófreces NB, Sánchez-Madrid F (2014) Miro-1 links mitochondria and microtubule dynein motors to control lymphocyte migration and polarity. Mol Cell Biol 34:1412–1426. 10.1128/MCB.01177-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MS, Moncada S, Ward PA (1992) Protective effects of inhibitors of nitric oxide synthase in immune complex-induced vasculitis. Br J Pharmacol 107:1159–1162. 10.1111/j.1476-5381.1992.tb13423.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S, Simmons ML, Agullo L, Garcia A, Feinstein DL, Galea E, Reis DJ, Minc-Golomb D, Schwartz JP (1993) Synthesis of nitric oxide in CNS glial cells. Trends Neurosci 16:323–328. 10.1016/0166-2236(93)90109-Y [DOI] [PubMed] [Google Scholar]
- Murphy SP, Lee RJ, McClean ME, Pemberton HE, Uo T, Morrison RS, Bastian C, Baltan S (2014) MS-275, a class I histone deacetylase inhibitor, protects the p53-deficient mouse against ischemic injury. J Neurochem 129:509–515. 10.1111/jnc.12498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuji T, Sugiyama M, Matsui T (1994) Temporal profiles of Ca2+/calmodulin-dependent and -independent nitric oxide synthase activity in the rat brain microvessels following cerebral ischemia. Acta Neurochir Suppl (Wien) 60:285–288. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW (1994a) Nitric oxide synthases: roles, tolls, and controls. Cell 78:915–918. 10.1016/0092-8674(94)90266-6 [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie QW (1994b) Regulation of biosynthesis of nitric oxide. J Biol Chem 269:13725–13728. [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K (2006) Microglia provide neuroprotection after ischemia. FASEB J 20:714–716. 10.1096/fj.05-4882fje [DOI] [PubMed] [Google Scholar]
- Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, Trapp BD (2011) Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of ranvier. J Neurosci 31:7249–7258. 10.1523/JNEUROSCI.0095-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado JR, Diaz-Ruiz A, Frühbeck G, Malagon MM (2014) Mitochondria in metabolic disease: getting clues from proteomic studies. Proteomics 14:452–466. 10.1002/pmic.201300376 [DOI] [PubMed] [Google Scholar]
- Peng ZC, Pietra C, Sbarbati A, Ziviani L, Yan XB, Osculati F, Bentivoglio M (1996) Induction of NADPH-diaphorase activity in the rat forebrain after middle cerebral artery occlusion. Exp Neurol 138:105–120. 10.1006/exnr.1996.0051 [DOI] [PubMed] [Google Scholar]
- Pritchett JS, Shippy SA (2014) Monitoring of in vivo manipulation of nitric oxide synthases at the rat retina using the push-pull perfusion sampling and capillary electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci 955–956:81–85. 10.1016/j.jchromb.2014.02.022 [DOI] [PubMed] [Google Scholar]
- Quirié A, Demougeot C, Bertrand N, Mossiat C, Garnier P, Marie C, Prigent-Tessier A (2013) Effect of stroke on arginase expression and localization in the rat brain. Eur J Neurosci 37:1193–1202. 10.1111/ejn.12111 [DOI] [PubMed] [Google Scholar]
- Radi R, Beckman JS, Bush KM, Freeman BA (1991) Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 288:481–487. 10.1016/0003-9861(91)90224-7 [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Kim HH, Huang Z, Seto M, Yano K, Asano T, Moskowitz MA, Liao JK (2005) Inhibition of rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 36:2251–2257. 10.1161/01.STR.0000181077.84981.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Sullivan N, Esiri MM (2001) White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke 32:1162–1168. [DOI] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, Zinsmaier KE (2009) Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci 29:5443–5455. 10.1523/JNEUROSCI.5417-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab ASTzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Möbius W, Goetze B, Jahn HM, Huang W, Steffens H, Schomburg ED, Pérez-Samartín A, Pérez-Cerdá F, Bakhtiari D, Matute C, Löwel S, Griesinger C, Hirrlinger J, Kirchhoff F, Nave KA, et al. (2016) Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 91:119–132. 10.1016/j.neuron.2016.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the miro GTPase. Proc Natl Acad Sci U S A 105:20728–20733. 10.1073/pnas.0808953105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Sasamata M, Bosque-Hamilton P, Huang PL, Moskowitz MA, Lo EH (1998) Attenuated neurotransmitter release and spreading depression-like depolarizations after focal ischemia in mutant mice with disrupted type I nitric oxide synthase gene. J Neurosci 18:9564–9571. 10.1523/JNEUROSCI.18-22-09564.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, Huang PL, Boas DA, Liao JK, Moskowitz MA, Ayata C (2007) Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab 27:998–1009. 10.1038/sj.jcbfm.9600406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons ML, Murphy S (1993) Cytokines regulate l-arginine-dependent cyclic GMP production in rat glial cells. Eur J Neurosci 5:825–831. 10.1111/j.1460-9568.1993.tb00934.x [DOI] [PubMed] [Google Scholar]
- Stahon KE, Bastian C, Griffith S, Kidd GJ, Brunet S, Baltan S (2016) Age-related changes in axonal and mitochondrial ultrastructure and function in white matter. J Neurosci 36:9990–10001. 10.1523/JNEUROSCI.1316-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR (1991) Na+-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol 30:375–380. 10.1002/ana.410300309 [DOI] [PubMed] [Google Scholar]
- Tang BL. (2015) MIRO GTPases in mitochondrial transport, homeostasis and pathology. Cells 5:e1. 10.3390/cells5010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Goldberg MP (2001) AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci 21:4237–4248. 10.1523/JNEUROSCI.21-12-04237.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR (2007) Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab 27:1540–1552. 10.1038/sj.jcbfm.9600455 [DOI] [PubMed] [Google Scholar]
- Toda N, Ayajiki K, Okamura T (2009) Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev 61:62–97. 10.1124/pr.108.000547 [DOI] [PubMed] [Google Scholar]
- Tsai SK, Hung LM, Fu YT, Cheng H, Nien MW, Liu HY, Zhang FB, Huang SS (2007) Resveratrol neuroprotective effects during focal cerebral ischemia injury via nitric oxide mechanism in rats. J Vasc Surg 46:346–353. 10.1016/j.jvs.2007.04.044 [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL (2009) The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136:163–174. 10.1016/j.cell.2008.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906. 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter R, Yu L, Yu CA (1996) The effects of nitric oxide on electron transport complexes. Arch Biochem Biophys 331:9–14. 10.1006/abbi.1996.0276 [DOI] [PubMed] [Google Scholar]
- Wiencken AE, Casagrande VA (1999) Endothelial nitric oxide synthetase (eNOS) in astrocytes: another source of nitric oxide in neocortex. Glia 26:280–290. 10.1002/(SICI)1098-1136(199906)26:4%3C280::AID-GLIA2%3E3.0.CO%3B2-W [DOI] [PubMed] [Google Scholar]
- Wong TY, Klein R, Sharrett AR, Couper DJ, Klein BEK, Liao D-P, Hubbard LD, Mosley TH (2002) Cerebral white matter lesions, retinopathy, and incident clinical stroke. J Am Med Assoc 288:67–74. [DOI] [PubMed] [Google Scholar]
- Yamada M, Huang Z, Dalkara T, Endres M, Laufs U, Waeber C, Huang PL, Liao JK, Moskowitz MA (2000) Endothelial nitric oxide synthase-dependent cerebral blood flow augmentation by l-arginine after chronic statin treatment. J Cereb Blood Flow Metab 20:709–717. 10.1097/00004647-200004000-00008 [DOI] [PubMed] [Google Scholar]
- Yao X, Huang Y (2003) From nitric oxide to endothelial cytosolic Ca2+: a negative feedback control. Trends Pharmacol Sci 24:263–266. 10.1016/S0165-6147(03)00122-6 [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Saudou F (2013) Huntingtin's function in axonal transport is conserved in Drosophila melanogaster. PLoS One 8:e60162. 10.1371/journal.pone.0060162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaleski J, Bachleda A, Runkle A, Baltan S (2013) Age-dependent activity of nitric oxide synthase during ischemic white matter injury. July 3–6, 2013 EuroGlia, Berlin Germany. [Google Scholar]
- Zhang F, Casey RM, Ross ME, Iadecola C (1996) Aminoguanidine ameliorates and l-arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke 27:317–323. 10.1161/01.STR.27.2.317 [DOI] [PubMed] [Google Scholar]
- Zhang ZG, Chopp M, Zaloga C, Pollock JS, Förstermann U (1993) Cerebral endothelial nitric oxide synthase expression after focal cerebral ischemia in rats. Stroke 24:2016–2021; discussion 2021–2022. 10.1161/01.STR.24.12.2016 [DOI] [PubMed] [Google Scholar]