

Abstract
Background
Dietary habits have been linked with variability of gut microbiota composition and disease risk.
Objective
The aim of this study was to evaluate the effect of feeding a cocoa powder with or without a probiotic on the composition and function of the fecal microbiome of pigs.
Methods
Four groups of 8 pigs each were fed a standard growth diet supplemented with cocoa powder, Lactobacillus rhamnosus (LGG), cocoa powder + LGG, or an equal amount of fiber similar to that found in cocoa powder (control group). Fecal samples were collected prior to and 4 wk after initiation of the dietary intervention. Microbiota composition was determined after amplification of the first 2 variable regions of the 16S ribosomal DNA (rDNA). Predictions of metagenomic function were calculated using 16S rDNA sequence data through Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt).
Results
After 4 wk of treatment, bacterial abundance analysis demonstrated a prebiotic effect of cocoa powder on endogenous Bifidobacteriaceae and Lactobacillaceae and increased abundance of saccharolytic butyrate-producing bacteria like Roseburia. An increased bacterial evenness, Shannon diversity index, and diverse metabolic profile were detected in microbiomes of pigs fed the cocoa powder + LGG (P < 0.05) but not in pigs in the other 3 groups.
Conclusion
The data generated from this work demonstrated that 4-wk dietary treatment with cocoa powder alone or in combination with LGG probiotic had an impact on the composition and function of the fecal microbiota of healthy pigs.
Keywords: microbiota, metagenomic function, 16S rDNA, sequencing, PICRUSt, cocoa powder, Lactobacillus rhamnosus LGG, Acticoa
Introduction
Analysis of metagenomic data has shown that diet can modulate microbial communities and related metabolites to promote health or affect disease (1, 2). Epidemiological studies have indicated that consumption of diets rich in polyphenols derived from fruits and vegetables is associated with reduced risk of chronic diseases as plant-derived dietary fiber and/or flavanoids may mediate the observed protective effects through their interaction with the microbiome (3, 4). Flavanoid-rich cocoa products have been described as prebiotics that can positively affect the growth of beneficial bacterial species from the genera Lactobacillus and Bifidobacterium in humans and pigs (5–7) or reduce the prevalence of pathogenic species in rodents (8). It has been suggested that cocoa flavanols and associated fiber in cocoa products may modulate levels and activities of certain
bacterial species in the gut microbiome (9); however, identification of the bacterial taxa associated with dietary cocoa intervention needs further investigation. We designed a study to test the effect of cocoa powder on the overall composition and function of the host microbiome using a swine model. In addition, we wanted to validate the previously observed prebiotic effect of cocoa powder on the endogenous host microbiome in combination with feeding Lactobacillus rhamnosus (LGG) as an exogenous probiotic strain which is widely consumed by humans. Swine, as an animal model, share many more anatomic and physiologic similarities with humans than do rodents or large domestic animals (10). Therefore, we designed a study to examine changes in composition and function of the fecal microbiome after feeding young pigs a corn-, alfalfa-, and soy-based diet supplemented with cocoa powder, LGG, cocoa powder + LGG, or an equal amount of fiber similar to that found in cocoa powder and of maltodextrin which was used as the vehicle for the LGG preparation (control group). We integrated the information gathered from phylogenetic analysis and predicted metagenomic changes in the microbiome to validate the prebiotic effect of cocoa powder in growing pigs.
Methods
Ethics statement
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (NRC 2011). All animal procedures for this specific study have been approved by Institutional Animal Care and Use Committee, Beltsville Animal Care and Use Committee (BACUC), under Principal Investigator protocol No. 13-028. Collection of all samples complied with regulations for animal welfare. The pigs utilized for this work did not become ill based on daily observation of food intake and biweekly measurements of body weight prior to collection of samples.
Animals and experimental design
Thirty-two white Yorkshire-Landrace crossbred barrows procured from Oak Hill Genetics (Ewing, IL) were chosen from litters born within the same week. After weaning at 3 wk of age, pigs were transported to Beltsville, MD in a climate-controlled truck with bedding and access to water at all times. Pigs were housed individually in a confinement facility with pen separations that prevent oral contact among pigs. The facility is equipped with heat lamps and airflow to maintain a comfortable temperature (24°C) and follows a 12-h light and dark cycle with access to water at all times. After 3 wk of quarantine, the average weight of the pigs was 14.61 ± 1.6 kg (mean ± SD). Pigs were stratified by weight and randomly assigned to the 4 treatment groups (n = 8 pigs/group), so that there was no difference in weight between the 4 groups at the start of the study. Body weights were collected biweekly during the 4-wk dietary intervention. The experimental design was a 2 × 2 design, including 2 levels for each independent variable (cocoa and probiotic). Pigs were given twice daily a pre-weighed amount of standard 17% protein-corn, alfalfa, and soy-based diet (Supplementary Table 1) according to the requirements of growing pigs and supplemented with either (1) a vehicle-placebo (maltodextrin) and fiber, (2) 1 × 1010 cfu LGG and fiber, (3) 26 g of cocoa powder, or (4) LGG + 26 g of cocoa powder. Analysis of cocoa powder (26 g) found that it contained 1.24 g of soluble fiber and 6.42 g of insoluble fiber (Supplementary Table 1). Either cocoa powder or soluble (dextrin 1.24 g) and insoluble (6.42 g cellulose) fiber was dissolved in 30 mL of water and the suspension was applied to the dry feed given to pigs every morning. Lyophilized LGG in maltodextrin or the maltodextrin placebo alone were mixed with 5 mL of phosphate buffered saline (PBS) until dissolved and delivered orally to each pig using individual disposable syringes. Treatment groups were allocated to each side of the building according to their probiotic treatment and separated by a raised floor to minimize possible cross-contamination after daily washing of the pens. Pelleted diets contained no antimicrobials or growth promoters. Cocoa powder was derived from non-roasted cocoa beans (commercially available as Acticoa, Barry Callebaut, Switzerland). Equivalent amounts of dietary cellulose (Nutricology, Innovative Nutrition, Alameda, CA) and Benefiber contains dextrin (Benefiber, Novartis, NJ) were commercially acquired and incorporated in the control diet. LGG or maltodextrin vehicle control were provided by Chr. Hansen Denmark and given to the LGG or control treatment groups, respectively.
Analysis of total flavanols in cocoa powder was determined by 4-dimethylaminocinnamaldehyde (DMAC), a validated spectrophotometric assay (11) (Brunswick Laboratories, Southborough, MA) that uses a commercially available B-type proanthocyanidin dimer standard to determine milligram proanthocyanidins (PACs) equivalents. Individual flavanol analysis up to 5 polymers (5P) was done with the ultra-high performance liquid chromatography with high resolution mass spectrometry (UHPLC-HRMS)-Orbitrap MS system (ThermoFisher Scientific, San Jose, CA) (6) using catechin and epicatechin, procyanidin B1, and procyanidin C1 as standards for flavanol monomers, dimers, and trimers, respectively. Flavanol tetramers and pentamers were measured using catechin (Sigma Aldrich, St Louis, MO) as a reference standard with relative response factors as previously described (12). Insoluble and soluble fiber AOAC (Association of Official Agricultural Chemistry) analysis was done by Covance Laboratories (Madison, WI). LGG viability was assessed by culture in MRS-Agar plates (Anaerobe Systems, Morgan Hill, CA). All pigs were sacrificed by intravenous injection with Euthasol (50 mg sodium pentobarbital/kg of body weight) (Virbac Animal Health, Inc., Fort Worth, TX) at the end of the dietary intervention.
Fecal specimen collection and processing for 16S ribosomal DNA amplicon multi-tag sequencing
Fresh fecal samples were directly collected from each pig using a cotton swab to stimulate defecation. Five-gram aliquots were collected in sterile 50 mL plastic tubes before (week 0) and after (week 4) the dietary intervention. One-gram aliquots were stored at –80°C until further processing. DNA was extracted using the QIAamp DNA stool kit (Qiagen, Germantown, MD) according to the manufacturer's instructions but involving an initial Precellys disruption with ceramic beads (Krackeler Scientific, NY). DNA concentration was determined by Nanodrop (ThermoFisher Scientific, Wilmington, DE). A 10 ng aliquot of extracted DNA was used to determine the relative abundance of L. rhamnosus using a species-specific real-time PCR assay (222-Forward primer: 5′-CGGTTCTGCCTTGAAAGCA-3′, 327-Reverse primer 5′-GGTTTCACGAACTGGGGTTG-3′, and tuf-261-revT-TET-labeled probe 5′-CTGTTCAGGATCGCCTTC-3′), against a 106 base pair (bp) fragment of the single copy tuf gene encoding the elongation factor Tu that facilitates polypeptide elongation during translation and that has been used for identification of L. rhamnosus species as previously described (13). The DNA was also amplified for length heterogeneity PCR (LH-PCR) fingerprinting and then sequenced using Ion Torrent technology (14, 15). The fingerprinting step was done as a quality control. A fusion forward primer 27F (5′-AGAGTTTGATCCTGGCTCA G-3′), which contained different 8 bp tags and an adapter for sequencing with personal genome machine (PGM), and a 6-carboxyfluorescein (FAM) labeled reverse primer 355R′ (5′-GCTGCCTCCCGTAGGAGT-3′), that included the Ion Torrent reverse adapter, were used in duplicate PCRs. Both of these primers are universal primers for bacteria and amplify the first 2 variable regions of the 16S ribosomal DNA (rDNA). For fingerprinting, the PCR products were diluted based on their band intensity on agarose gel electrophoresis using a Gel Logic 112 Camera with molecular imaging software (Carestream Health Inc., Woodbridge, CT). Duplicate LH-PCR fingerprinting products were mixed (in 1:10) with a 1:20 ratio of the internal lane standard 600 (ILS600, Promega Corporation, Madison, WI) and HiDi Formamide (Applied Biosystems, Foster City, CA). The ILS600 standard containing fragments from 60 to 600 bp was used as a reference to measure the size of the PCR product. The diluted samples were separated on an ABI 3130 xl fluorescent capillary sequencer (Applied Biosystems, Foster City, CA) and analyzed using the Genemapper™ software package (Applied Biosystems, Foster City, CA). The LH-PCR fingerprinting step was used to check the reproducibility of the PCR and allow us to choose the DNA dilution that has the optimal intensity of all the peaks in samples before pooling and sequencing with PGM Ion Torrent (Applied Biosystems, Foster City, CA). This step is necessary to assure quantitative amplification of the components in a sample and prevent kinetic bias (15). We used the Ion PGM Hi-Q kits and followed manufacturer protocol (https://tools.thermofisher.com/content/sfs/manuals/MAN0010902_PGM_HiQ_OT2_Kit_UG.pdf).
The Naïve Bayesian Classifier from the Ribosomal Database Project version 11 (RDP11) was used to identify the taxa present in each sample. Additionally, Operational Taxonomic Units (OTUs) were also defined using the Quantitative Insights into Microbial Ecology (QIIME) pipeline and taxonomic information for each OTU was annotated with the UCLUST tool based on the Green Genes database. The fourth level of taxa assignment (family) was used in the calculation of diversity and abundance. Beta diversity was analyzed using unweighted Unifrac distances, which is a method for comparing microbial communities by measuring phylogenetic distances between communities and represented in a 2-dimensional principal coordinate analysis (PCoA) plot using the QIIME pipeline (16). Alpha diversity, which is a measure of within-sample diversity, was measured using the Shannon diversity index (17) which encompasses bacterial species abundance (i.e., the total number of bacterial species) and evenness (i.e., the distribution of those species) using alpha diversity script in QIIME (http://qiime.org/scripts/alpha_diversity.html).
Predicted metabolic profile
OTUs were normalized for the number of 16S rDNA gene copies and used to predict metagenome functional content using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (18). PICRUSt uses existing annotations of genes based on bacterial evolutionary information as well as 16S gene copy numbers from the Integrated Microbial Genomes database to predict metagenomes from 16S rDNA data (19). This methodology predicts metabolic function of the microbiome with an average correlation of approximately R = 0.8 between inferred and measured gene content (18).
Statistical analyses
Bacterial abundance distributions were modeled using a generalized linear mixed-effects model with multinomial distribution and generalized logit link for each of the 6 observed bacterial classifications. Correlation among time measures from the same subject was modeled using the smallest corrected Akaike's Information Criterion (AICC) fit statistic value to choose among the covariance structures: heterogeneous compound symmetry (csh), compound symmetry, heterogeneous variance component (un (1)), first-order Toeplitz (toep (1)), or first-order heterogeneous auto-regressive (arh (1))—by specifying each structure in the TYPE = option in the RANDOM statement of SAS PROC GLIMMIX (SAS Institute, Inc., Cary, NC). At each bacterial classification level and scale, an individual bacterium was used as its own “bacterial category” in the model if, for any of the 8 treatments, the bacteria total reads exceeded 5% of the total reads for all bacteria observed in that sample, when combining all subjects. Bacteria below this arbitrarily chosen cutoff were combined into a single “all others” bacterial category. Each bacterium exhibited a specific abundance, which was expressed as a percentage of the total bacterial abundance observed. An OR provided a natural way to simultaneously characterize the relationship between 2 bacteria in the bacterial profile and to determine whether this relationship changed between 2 experimental conditions. Comparisons among the bacterial abundance distributions included all pairs of the 4 treatments at a given week (among-treatment effect) and all pairs for 4 wk of a given treatment (within-treatment effect). The bacteria that exhibit the greatest abundance across all treatments were used as a reference category for the calculation of ORs. Significance of an OR was determined by whether one is included in or excluded from the OR’s 95% CI. An OR of 1 indicates that the 2 entities being compared “occur with equal likelihood”. An asterisk (*) was placed next to each OR that was statistically significant. All comparisons with different letters denote significant differences present after pairwise comparisons. Diversity metrics were produced using QIIME scripts on the normalized read counts. A cocoa treatment × probiotic × weeks ANOVA was fit to L. rhamnosus copies per gram (cpg) data with SAS PROC GLIMMIX, using negative binomial distribution, log link function, and Laplace optimization.
The linear discriminant analysis effect size (LEfSe), an algorithm for biomarker discovery that identifies enrichment of abundant taxa or function between ≥2 groups, was used to compare all taxa at different taxonomic levels simultaneously (i.e., phylum, class, order, family, genus) between treatment groups. The nonparametric Kruskal-Wallis statistical test was used to compute differences among treatment groups and then paired Wilcoxon’s Rank Sum tests among subgroups. This method uses a linear discriminant analysis (LDA) model which utilizes continuous independent variables to predict 1 dependent variable and provides an effect size for the significantly different taxa or metabolic functions based on relative differences between 2 conditions—taking into account both variability and discriminatory power (20). Unless stated otherwise, alpha values of 0.05 were used for the nonparametric Kruskal-Wallis and paired Wilcoxon's Rank sum test, and a threshold of >2.0 was chosen for logarithmic LDA score display. A series of bar graphs were constructed to show the relationship between significantly different metabolic functions or taxa at different phylogenetic levels differentiating clades with a common ancestor (20).
Results
Cocoa flavanols
Analysis of the cocoa powder fed to pigs showed that the major components were flavanols, theobromine, caffeine, and caffeoyl aspartic acid as previously described (21). The total flavanol concentration was 137.19 mg/g expressed as B-type proanthocyanidin dimer equivalent (11), containing 15.70 mg/g of flavanols up to 5 polymers as determined by UHPLC-HRMS (12). A 26 g cocoa powder treatment provided an average of 160.5 mg/kg body weight (bw) of total flavanols (range 140.2–187.5 mg/kg bw) to the pigs. This dose was chosen because it had previously demonstrated a prebiotic effect on the pig microbiome with detectable metabolites in tissues but with no significant effect on pig growth (6). Analysis of cocoa powder indicated that 26 g of cocoa powder contained 1.24 g of soluble fiber and 6.42 g of insoluble fiber. A comparable amount of these types of fibers was added to the diet of the pigs in the control and LGG-only groups so that all pigs had an equivalent amount of soluble and insoluble fiber.
Pig body weights
Pig body weight was measured before and after the dietary intervention. Pigs in the cocoa + LGG-fed group gained 1.98 more kg (7.5 ± 0.33, mean ± SE) compared to pigs in the control group (5.48 ± 0.56) (P = 0.01), but were not different than pigs fed cocoa powder alone (6.25 ± 0.65) or LGG alone (6.51 ± 0.45) after the 4-wk intervention. No clinical signs of disease such as diarrhea or change in appetite and behavior were observed throughout the 4-wk dietary intervention.
Cocoa powder affects bacterial diversity when combined with the probiotic LGG
Bacterial evenness (relative abundance of species) was increased from 0.66 ± 0.02 (mean ± SE) to 0.73 ± 0.01 in the cocoa powder + LGG-treated pigs (Figure 1A) (P = 0.01) and the mean ± SE Shannon diversity index was also increased to 4.17 ± 0.10 compared to the baseline level of 3.70 ± 0.12 (Figure 1B) (P = 0.01) after 4 wk of dietary intervention. Neither evenness nor Shannon diversity index were changed in the other 3 groups. Using a 2D PCoA plot, clusters that differentiate the microbial population among collection times showed a partial clustering of samples by diet 4 wk after consumption of the control (4/7 pigs), cocoa powder + LGG (5/8 pigs), or LGG (5/7 pigs) diets (P < 0.05, Bonferroni adjusted) with the first 2 components representing 34% and 13% of the total variation. Non-significant separation was seen in pigs fed cocoa powder (Supplementary Figure 1).
FIGURE 1.
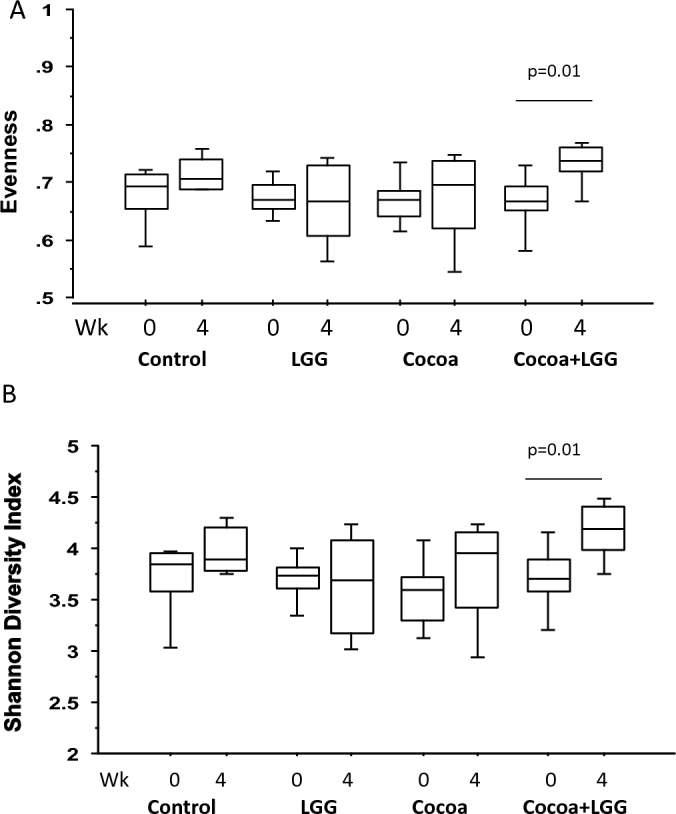
Fecal microbiota abundance. Changes in relative abundance of each species, evenness (A); microbial species diversity, Shannon diversity index (B), in fecal microbiome derived from week 0 and week 4 fecal samples collected from pigs fed fiber control, cocoa powder, LGG, or LGG + cocoa powder. Values are means ± SEs. Significant difference between weeks is denoted with P values. LGG, Lactobacillus rhamnosus.
Cocoa powder does not affect abundance of L. rhamnosus LGG
L. rhamnosus relative abundance expressed as cpg was calculated in fecal samples collected at baseline and week 4 after the dietary intervention. L. rhamnosus cpg were increased in LGG (P < 0.0001) and cocoa-powder + LGG (P < 0.001)-fed pigs with no change in control or cocoa-fed pigs after 4 wk of dietary intervention. No differences in cpg were detected among the 2 groups given LGG (P ≥ 0.64) (Supplementary Figure 2). However, inherent Lactobacillus species abundance was increased in response to the cocoa powder intervention.
Diet-induced changes in bacterial abundance after 4 weeks
After sequencing, an average of 14,901 ± 5812 (mean ± SD) high-quality reads per each of the 64 fecal samples were generated for computational analysis with a 0.1% abundance cutoff to normalize for the variation in their depth of coverage (22). Estimated changes in bacterial abundance calculated by OR analysis are summarized in Supplementary Tables 2–6. Dietary effects were evaluated by comparing week 0 (baseline) to week 4 for each dietary group. The microbiome phylogenetic phylum ratio (r) of Bacteroides to Firmicutes (B:F) in fecal samples at week 0 (baseline) was increased at week 4 after continuous feeding with control (r = 0.27 compared with r = 0.87), cocoa powder (r = 0.24 compared with r = 0.58), or the combination of cocoa powder + LGG (r = 0.24 compared with r = 0.71) (P < 0.05, Wilcoxon’s Signed Rank test). No change was detected in the B:F ratio of pigs fed LGG after 4 wk (r = 0.30 compared with r = 0.26) (Figure 2A). Differences in bacterial abundance were also observed at class (Figure 2B) and order (Figure 2C) levels. At the family (Figure 2D) and genus (Figure 2E) levels, the Porphyromonadaceae_other, Prevotellaceae_other, Lachnospiraceae_other, and Ruminococcaceae_other were 2.57–6.55 times more abundant at week 4 compared to week 0 for pigs fed the control, cocoa powder, or cocoa powder + LGG diets. The family_genus Lactobacillaceae_Lactobacillus was 18.88 (95% CI values 3.23, 110.44) times more abundant compared to week 0 only in pigs fed the cocoa powder diet (Figure 2D, E, Supplementary Tables 5 and 6) with no significant differences in bacterial abundances in pigs fed the LGG diet between weeks 0 and 4.
FIGURE 2.


Estimated distribution of bacterial abundance among treatments from multinomial distribution model. Abundances for phylum (A), class (B), order (C), family (D), and genus (E) in the fecal microbiome of pigs fed fiber control, cocoa powder, LGG, or cocoa powder + LGG were compared between weeks for each diet to determine diet treatment effects after 4 wk. Means sharing the same letter are not significantly different from each other. B/F, Bacteroides-to-Firmicutes ratio; LGG, Lactobacillus rhamnosus.
No changes at the phylum level were detected between all assigned treatment groups at week 0 (Supplementary Table 2). However, at a lower phylogenetic level, the class Bacilli and order Lactobacillales were more abundant in pigs assigned to the LGG group than in pigs assigned the control diet or cocoa powder, respectively (Supplementary Table 3), with more Streptococcus abundance in pigs assigned to the LGG group compared to the control or cocoa powder diets (Supplementary Table 6). The increased abundance in Streptococcus consistently reflected at genus, family, order, and class level in the group of pigs initially selected to be fed the LGG diet at week 0, indicated a bias in the abundance of Streptococcaceae in this group that was not corrected by the process of randomization.
Bacterial taxa enriched by diet
Linear discriminant analysis effect size (LEfSe), a method designed for biomarker discovery (20, 23–25), was used to determine relative enrichment or depletion in bacterial taxa in response to treatment with control, cocoa powder, or cocoa powder + LGG diets for 4 wk. The pigs fed the LGG diet were not included in this analysis as the basal sample was enriched in Streptococcaceae family members when compared to the pigs assigned to the control and cocoa powder diets at week 0 (P < 0.05) (Supplementary Figure 3). Cladograms indicating the taxa showing significant differences in the fecal microbiome were created for integration of results at different phylogenetic levels (Figure 3). Based on our LEfSe analysis, we can predict that consumption of cocoa powder induces an enrichment of Bifidobacteriaceae, Bacteroidiaceae, and Anaeroplasmataceae, a reduction in Streptococcaceae, with some change in distribution of Clostridiales including a reduction in Clostridiaceae and an enrichment in Lachnospiraceae genera Dorea and Roseburia, Eubacteriaceae, Anaerovorax from Clostridiales Incertae sedis XIII, and Succinivibrio from Gammaproteobacteria (Figure 3B). The addition of LGG to cocoa powder increased the diversity of the fecal microbiome indicated by an enrichment of Flavobacteriaceae, Sphingobacteriaceae, Spirochaetaceae, and certain Proteobacteria like Desulfovibrionaceae with a reduction of several Clostridiales including Lactobacillus, Streptococcus, Blautia, and Faecalibacterium but maintenance of Lachnospiraceae (Figure 3C). The control diet also induced enrichment of Bacteroidales, but with a major depletion of members from class Bacilli including Lactobacillales and class Clostridia including Clostridiales (Figure 3A).
FIGURE 3.

Linear discriminant analysis effect size (LEfSe) cladogram representing differentially abundant taxa in fecal microbiome. Fecal samples derived from pigs fed control (A), cocoa powder (B), or cocoa powder + LGG (C). Only taxa with linear discriminant analysis scores >2 are presented. Each color in the pie chart represents the corresponding bacterial taxa in the legend. The LEfSe method was performed to determine individual taxa that were enriched (green) or depleted (red) within each dietary treatment. LGG, Lactobacillus rhamnosus.
Bacterial metagenome function prediction based on 16S rDNA
PICRUSt was used to predict metagenomic functions from the phylogenetic profiles observed. The resulting biological pathways were analyzed and displayed through LEfSe with an LDA score ≥2.0 identifying significantly abundant bacteria within each comparison (Supplementary Figure 4). Biological pathways were organized in functional categories, including Metabolism, Genetic Information, Environmental Information, Cellular, Organ Systems, and Human Diseases, to compare the functional enrichment in each treatment group. A total of 110 biological pathways were predicted for samples taken from pigs fed the control (68), cocoa powder (13), and cocoa powder + LGG diets (29) after 4 wk of dietary intervention (Supplementary Table 7). Metabolism was the predominant functional category in pigs fed the control (46%), cocoa powder (38%), and cocoa powder + LGG diets (76%) at week 4; however, the distribution of metabolic pathways was different among treatment groups (Figure 4). In samples from pigs fed the control diet, there was a predominance of metabolism pathways associated with cofactors and vitamins (19%), glycan biosynthesis (19%), amino acids (16%), energy (13%), biosynthesis of other metabolites (10%), carbohydrates (7%), and lipids (6%). In samples derived from pigs fed cocoa powder, energy metabolism (50%), cofactors and vitamins (33%), and terpenoids and polyketide (17%) pathways were predominant. Samples from pigs fed cocoa powder + LGG also expressed a diversified metabolism with amino acids (29%), cofactors and vitamins (19%), energy metabolism (19%), biosynthesis of other metabolites (9%), nonproteinogenic amino acids (9%), carbohydrates (5%), lipids (5%), and glycan biosynthesis (5%) pathways after 4 wk of treatment (Figure 4).
FIGURE 4.

Distribution of PICRUSt predicted metabolic pathways. Pie charts summarize predicted metabolic pathway enrichment in response to diets supplemented with fiber control (A), cocoa powder (B), and cocoa powder + LGG (C) for 4 wk. LGG, Lactobacillus rhamnosus; PICRUSt, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States.
Discussion
The data generated from this work demonstrate that a 4-wk dietary treatment with cocoa powder alone or in combination with LGG probiotic had an impact on the composition and function of the fecal microbiota of healthy pigs. Results from 16S rDNA sequence analyses have shown that bacterial gut communities are similar among omnivorous mammals (26). Similarities at the phylum level between bacteria in the pig and human gut include the presence of Firmicutes and members of the Bacteroidetes as the most abundant bacteria in the gastrointestinal tract with some differences in the relative abundance of certain phylotypes. However, recent metagenomic data showed the intestinal microbiomes of pigs and humans are similar from a functional perspective in spite of the differences in microbial community structure (27). Our previous data showed that consumption of cocoa-derived flavanols increased colonic microbial metabolites derived from cocoa flavanols in most swine bio-fluids and tissues, increased the abundance of fecal Bifidobacterium and Lactobacillus species, and reduced inflammation (6). In the current study, we fed pigs an amount of cocoa powder with LGG (used at a dose of 1 × 1010 CFU/d) equivalent to a controlled human clinical study (13) compared to pigs fed cocoa powder and LGG alone to evaluate the prebiotic effect of cocoa powder on the gut microbiome composition and function. These dietary treatments were compared to pigs fed a control diet containing a similar amount of fiber compared to the cocoa powder and maltodextrin equivalent to the vehicle used in the LGG supplement.
Analysis of the gut microbiota structure done by PCR amplification of the first 2 variable regions of the 16S rDNA using a multi-tag sequencing protocol (22) identified diet-induced changes in abundance of bacteria in the fecal microbiome from the phylum to the species level. Phylogenetic structure studies of microbial communities in populations exposed to different environments have used the Bacteroides:Firmicutes (B:F) phylum ratio to describe the impact of dietary habits and their association with host inflammatory markers and metabolic risks (28, 29). Dietary treatment with control, cocoa powder alone, or in combination with LGG increased the B:F ratio, due to a significant reduction in bacterial taxa within Firmicutes and a concomitant increase in Bacteroidetes. Similar shifts in major phyla have been described in other swine studies with pigs fed cocoa husk meal (7) or diets supplemented with fiber consisting of non-starch polysaccharide fractions (30). The phylum-level profiles we observed are also similar to the profiles of healthy humans with a high B:F ratio as opposed to altered profiles observed in gut dysbiosis, specifically those seen in obesity, IBS-diarrhea subtype, IBD, and traveler's diarrhea with a lower B:F ratio (31–34). These observations suggest that diets supplemented with cocoa powder or an equivalent amount of fiber have the potential to modulate the B:F ratio and improve energy balance (31). A lower B:F ratio has been observed in Brachyspira-induced diarrhea relative to sham-treated pigs or those that do not develop clinical disease following inoculation with Brachyspira (35). Whether a cocoa powder alone or cocoa + LGG-induced increase in B:F ratio can reduce the risk of infection with other pathogens in pigs will have to be determined.
Regarding diversity analysis of fecal microbiota, the unweighted Unifrac analysis indicated that clustering and PCoA could not completely discriminate the effect among diets as there was a partial overlap of samples from baseline and week 4 of pigs fed cocoa powder with and without LGG and control diets. However, differences in the level of diversity and bacterial species composition within the community were seen in pigs fed cocoa powder combined with LGG, suggesting a possible symbiotic effect. It has been suggested that a higher level of diversity and bacterial species composition of the microbiota is observed in healthy individuals as opposed to those suffering from dysbiosis of the intestinal microbiota commonly associated with inflammation of the lower gastrointestinal tract (36, 37). Cocoa powder supplementation alone induced an overall enrichment of Bacteroidales, Bifidobacteriales, and Tenericutes in fecal samples from pigs, with a corresponding reduction in Clostridiaceae and Streptococcaceae and maintenance of Lachnospiraceae, including the Roseburia and Dorea, that together with Lactobacillus and Bifidobacterium were similarly stimulated by prebiotics fed to lean human subjects (38). The Lachnospiraceae is one of the most abundant families found in the mammalian gut and has been associated with gut health, purportedly due to a higher production of butyrate by Roseburia that provides an important and beneficial energy source for intestinal epithelial cells (39). Feeding cocoa powder increased Bacteroidales-Prevotellaceae, a family of polysaccharide-fermenting bacteria that produce SCFAs linked to improved glycemic control. Bifidobacteriales-Bifidobacteriaceae have been shown to have a consistent anti-obesity effect in humans, and members from the phylum Tenericutes have an increased abundance in healthy humans relative to individuals with metabolic syndrome (40). Specifically, Anaeroplasmataceae (within the phylum Tenericutes) was increased in pigs fed cocoa powder and cocoa powder + LGG. Their abundance has been correlated with better fiber digestibility (41) and negatively correlated with various atherosclerosis biomarkers in a mouse model (42). Therefore, our study provides new evidence supporting the enhancement of beneficial microbial populations in response to a diet with cocoa powder, as has been previously described with other phenolic compounds (43, 44). Diets with cocoa powder only, but not the other diets, promoted the enrichment of inherent Lactobacillaceae species in the gut of pigs (Figure 2D); while pigs fed diets containing LGG independently increased abundance of L. rhamnosus LGG relative to pigs fed the control or cocoa powder diets alone (P < 0.05) (Supplementary Figure 2). Feeding LGG along with cocoa powder increased the diversity and abundance of more species from the order Bacteroidales, including Porphyromonadaceae and Rikenellaceae; order Flavobacteriales, including Flavobacteriaceae; order Sphingobacteriales, including Sphingobacteriaceae and Cytophagaceae; and Spirochaetaceae, with a depletion in the abundance of Lactobacillaceae independent of feeding LGG.
Firmicutes are the most abundant phylum in pre-weaning pigs, shifting gradually to Bacteroidetes after weaning (45). The decreased abundance in Firmicutes was over-represented in fecal samples from pigs fed the control diet by a reduction in the order Lactobacillales, including the family Streptococcaceae, and order Clostridiales (Figure 3), while the reduction in Firmicutes in pigs fed cocoa powder included a similar reduction in Streptococcaceae, but maintenance of Clostridiales such as Lachnospiraceae and Eubacteriaceae.
The composition of the fecal microbiota in pigs is also likely to be shaped by environmental factors such as pen location, stress, antibiotic use, or seasonal effects (46). In the current experiment, all pigs showed a similar composition of bacteria at the phylum level when initially divided into treatment groups; however, multinomial microbiota and LEfSe analysis indicated that pigs initially allocated to the LGG-fed group had 7% and 5% more Streptococcaceae abundance relative to those allocated to the control and cocoa powder-fed groups, respectively, that was reflected in a change in Bacillales, one of the most abundant classes, at week 0. The Streptococcus genus is composed mostly of facultative anaerobes with several pathogenic species that may impact animal health (47). Even though pigs fed LGG did not present any clinical sign of disease during the study, this group was excluded from LEfSe analysis because this initial bias in microbiota composition could have affected the analysis of metagenomics differences due to the initial abundance in Streptococcaceae or explain the lack of response observed after dietary treatment with LGG.
The results of the predicted metagenome analysis in fecal samples from pigs fed the control or cocoa powder + LGG diets after 4 wk showed a diverse profile with enriched metabolism for cofactors and vitamins and amino acids, while those fed cocoa powder were more specialized in energy metabolism. The functional and microbial abundance data identified a more diverse taxonomy and enriched metabolic profile that appeared to coincide with greater weight gain in pigs fed cocoa powder + LGG. Fecal samples from pigs fed the control diet showed differences in bacterial abundance and metabolic function but no impact on diversity or weight gain. The diversity of the microbiota and weight gain was only affected when cocoa powder was combined with LGG. These observations indicated that LGG may enhance the beneficial effect of flavanols coming from cocoa powder by increasing diversity through microbial-derived SCFA production or by suppressing inflammation (48). Increased weight gain has been associated with other lactobacilli species in pigs (49). Our results suggest that pigs fed cocoa powder + LGG had significantly higher diversity in their fecal microbiome and predicted metabolic function. Based on these results, we infer that feeding cocoa powder + LGG could provide an advantage to a host with reduced microbial diversity in the event of dysbiosis. The clinical significance of these findings will have to be validated further under diet- or pathogen-induced dysbiosis.
Assessing the impact of feeding cocoa powder in the diet on fecal microbial composition and metabolic activity in pigs is relevant to humans because of the comparable physiology and metabolic processing of polyphenols. The results from this study demonstrated a prebiotic effect of cocoa powder on endogenous Bifidobacteriaceae and Lactobacillaceae and increased abundance of saccharolytic butyrate-producing bacteria like Roseburia (50). Fermentation of polysaccharides by colonic microorganisms that produce SCFAs such as acetate, propionate, and butyrate has important implications in intestinal epithelial permeability (51, 52). Production of butyrate has been shown to decrease the pH and to prevent growth of pathogenic organisms that compromise gut health (53). A reduction in Roseburia has been recently reported to contribute to the dysbiosis of ulcerative colitis patients (54). In addition, analyses of the metagenome revealed that functional genes related to energy production and conversion were increased in pigs fed cocoa powder, suggesting a more efficient capture of energy from the diet.
From a health perspective, the intake of cocoa powder either alone or in combination with LGG could be an effective approach to promote the growth of butyrogenic-type bacteria, while competitively excluding non-desirable bacteria. Future experiments to evaluate the specific effects of cocoa powder under intestinal dysbiosis or longitudinal assessments would determine the resilience of dietary-induced changes in microbiota and delineate mechanisms of action. Overall, our data indicate that cocoa powder selectively promoted the growth of butyrogenic bacteria like Roseburia and probiotic species from the Lactobacillaceae or Bifidobacteriaceae families known to positively influence host metabolism and promote health.
Supplementary Material
Acknowledgments
The authors’ responsibilities were as follows—GIS-A, SJ, JFU, and CD: designed the research; GIS-A, SL, SJ, EB, YX, AM, JFU, and CDD: conducted the research; GIS-A, MS, RG, BV, and PG: analyzed the data; GIS-A, JFU, and CDD: wrote the manuscript; GIS-A: had primary responsibility for final content; and all authors: read and approved the final manuscript.
Notes
Supported by USDA project 8040-51530-056-00 and Inter Agency Agreement 8040-51530-056-13 with the Office of Dietary Supplements, NIH. Barry Callebaut supplied cocoa powder (Acticoa). Chr. Hansen provided the probiotic and maltodextrin.
Author disclosures: GIS-A, SL, SJ, EB, YX, MS, RG, BV, AM, JFU, PG, and CDD, no conflicts of interest.
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Supplemental Tables 1–7 and Figures 1–4 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/cdn/.
Abbreviations used:
- cpg
copies per gram
- LDA
linear discriminant analysis
- LEfSe
Linear Discriminant Analysis Effect Size
- LGG
Lactobacillus rhamnosus
- LH-PCR
length heterogeneity PCR
- OTU
Operational Taxonomic Unit
- PCoA
principal coordinate analysis
- PICRUSt
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States
- QIIME
Quantitative Insights into Microbial Ecology
- rDNA
ribosomal DNA
References
- 1. Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med 2016;8(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pozuelo M, Panda S, Santiago A, Mendez S, Accarino A, Santos J, Guarner F, Azpiroz F, Manichanh C. Reduction of butyrate- and methane-producing microorganisms in patients with Irritable Bowel Syndrome. Sci Rep 2015;5:12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klinder A, Shen Q, Heppel S, Lovegrove JA, Rowland I, Tuohy KM. Impact of increasing fruit and vegetables and flavonoid intake on the human gut microbiota. Food Funct 2016;7(4):1788–96. [DOI] [PubMed] [Google Scholar]
- 4. Landete JM. Updated knowledge about polyphenols: functions, bioavailability, metabolism, and health. Crit Rev Food Sci Nutr 2012;52(10):936–48. [DOI] [PubMed] [Google Scholar]
- 5. Tzounis X, Rodriguez-Mateos A, Vulevic J, Gibson GR, Kwik-Uribe C, Spencer JP. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am J Clin Nutr 2011;93(1):62–72. [DOI] [PubMed] [Google Scholar]
- 6. Jang S, Sun J, Chen P, Lakshman S, Molokin A, Harnly JM, Vinyard BT, Urban JF Jr, Davis CD, Solano-Aguilar G. Flavanol-enriched cocoa powder alters the intestinal microbiota, tissue and fluid metabolite profiles, and intestinal gene expression in pigs. J Nutr 2016;146(4):673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Magistrelli D, Zanchi R, Malagutti L, Galassi G, Canzi E, Rosi F. Effects of cocoa husk feeding on the composition of swine intestinal microbiota. J Agric Food Chem 2016;64(10):2046–52. [DOI] [PubMed] [Google Scholar]
- 8. Massot-Cladera M, Pérez-Berezo T, Franch A, Castell M, Pérez-Cano FJ. Cocoa modulatory effect on rat faecal microbiota and colonic crosstalk. Arch Biochem Biophys 2012;527(2):105–12. [DOI] [PubMed] [Google Scholar]
- 9. Strat KM, Rowley TJ 4th, Smithson AT, Tessem JS, Hulver MW, Liu D, Davy BM, Davy KP, Neilson AP. Mechanisms by which cocoa flavanols improve metabolic syndrome and related disorders. J Nutr Biochem 2016;35:1–21. [DOI] [PubMed] [Google Scholar]
- 10. Bahr A, Wolf E. Domestic animal models for biomedical research. Reprod Domest Anim 2012;47(Suppl 4):59–71. [DOI] [PubMed] [Google Scholar]
- 11. Prior RL, Fan E, Ji H, Howell A, Nio C, Payne MJ, Reed J. Multi-laboratory validation of a standard method for quantifying proanthocyanidins in cranberry powders. J Sci Food Agric 2010;90(9):1473–8. [DOI] [PubMed] [Google Scholar]
- 12. Lin LZ, Harnly JM. Quantitation of flavanols, proanthocyanidins, isoflavones, flavanones, dihydrochalcones, stilbenes, benzoic acid derivatives using ultraviolet absorbance after identification by liquid chromatography-mass spectrometry. J Agric Food Chem 2012;60(23):5832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Solano-Aguilar G, Molokin A, Botelho C, Fiorino AM, Vinyard B, Li R, Chen C, Urban J Jr, Dawson H, Andreyeva I, et al. Transcriptomic profile of whole blood cells from elderly subjects fed probiotic bacteria Lactobacillus rhamnosus GG ATCC 53103 (LGG) in a phase I open label study. PLoS One 2016;11(2):e0147426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang DJ, Kakiyama G, Betrapally NS, Herzog J, Nittono H, Hylemon PB, Zhou H, Carroll I, Yang J, Gillevet PM, et al. Rifaximin exerts beneficial effects independent of its ability to alter microbiota composition. Clin Transl Gastroenterol 2016;7(8):e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sikaroodi M, Gillevet PM. Quality control in multi-tag pyrosequencing of microbial communities. Biotechniques 2012;53(6):381–3. [DOI] [PubMed] [Google Scholar]
- 16. Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2013;2(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shannon CE, Weaver W. The mathematical theory of communication. Urbana, IL: University of Illinois Press, 1998. [Google Scholar]
- 18. Langille MG, Zaneveld J, Gregory Caporaso J, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31(9):814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 2012;40(Database issue):D115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jang S, Lakshman S, Beshah E, Xie Y, Molokin A, Vinyard BT, Urban JF, Davis CD, Solano-Aguilar GI. Flavanol-rich cocoa powder interacts with Lactobacillus rhamnossus LGG to alter the antibody response to infection with the parasitic nematode Ascaris suum. Nutrients 2017;9(10):1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gillevet P, Sikaroodi M, Keshavarzian A, Mutlu EA. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem Biodivers 2010;7(5):1065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szafranski SP, Wos-Oxley ML, Vilchez-Vargas R, Jáuregui R, Plumeier I, Klawonn F, Tomasch J, Meisinger C, Kühnisch J, Sztajer H, et al. High-resolution taxonomic profiling of the subgingival microbiome for biomarker discovery and periodontitis diagnosis. Appl Environ Microbiol 2015;81(3):1047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bunyavanich S, Shen N, Grishin A, Wood R, Burks W, Dawson P, Jones SM, Leung DYM, Sampson H, Sicherer S, et al. Early-life gut microbiome composition and milk allergy resolution. J Allergy Clin Immunol 2016;138(4):1122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shoskes DA, Altemus J, Polackwich AS, Tucky B, Wang H, Eng C. The urinary microbiome differs significantly between patients with chronic prostatitis/chronic pelvic pain syndrome and controls as well as between patients with different clinical phenotypes. Urology 2016;92:26–32. [DOI] [PubMed] [Google Scholar]
- 26. Ley RE, Hamady M, Lozupone C, Turnbaugh P, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science 2008;320(5883):1647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamendella R, Santo Domingo JW, Ghosh S, Martinson J, Oerther DB. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol 2011;11:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franco-de-Moraes AC, de Almeida-Pititto B, da Rocha Fernandes G, Gomes EP, da Costa Pereira A, Ferreira SRG. Worse inflammatory profile in omnivores than in vegetarians associates with the gut microbiota composition. Diabetol Metab Syndr 2017;9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davis SC, Yadav JS, Barrow SD, Robertson BK. Gut microbiome diversity influenced more by the Westernized dietary regime than the body mass index as assessed using effect size statistic. Microbiologyopen 2017;6(4):e00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian G, Wu X, Chen D, Yu B, He J. Adaptation of gut microbiome to different dietary non-starch polysaccharide fractions in a porcine model. Mol Nutr Food Res 2017;61(10):1700012. [DOI] [PubMed] [Google Scholar]
- 31. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444(7122):1022–3. [DOI] [PubMed] [Google Scholar]
- 32. Jeffery IB, O'Toole PW, Öhman L, Claesson MJ, Deane J, Quigley EM, Simrén M. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut 2012;61(7):997–1006. [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y, Zhi F. Lower level of bacteroides in the gut microbiota is associated with inflammatory bowel disease: a meta-analysis. Biomed Res Int 2016;2016:5828959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Youmans BP, Ajami NJ, Jiang ZD, Campbell F, Wadsworth WD, Petrosino JF, DuPont HL, Highlander SK. Characterization of the human gut microbiome during travelers’ diarrhea. Gut Microbes 2015;6(2):110–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Costa MO, Chaban B, Harding JCS, Hill JE. Characterization of the fecal microbiota of pigs before and after inoculation with “Brachyspira hampsonii”. PLoS One 2014;9(8):e106399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loh G, Blaut M. Role of commensal gut bacteria in inflammatory bowel diseases. Gut Microbes 2012;3(6):544–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winter SE, Baumler AJ. Why related bacterial species bloom simultaneously in the gut: principles underlying the ‘Like will to like’ concept. Cell Microbiol 2014;16(2):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aguirre M, Bussolo de Souza C, Venema K. The gut microbiota from lean and obese subjects contribute differently to the fermentation of arabinogalactan and inulin. PLoS One 2016;11(7):e0159236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 2008;27(2):104–19. [DOI] [PubMed] [Google Scholar]
- 40. Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, Lee S, Song Y-M, Lee K, Sung J, Ko G. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 2016;66(6):1031–8. [DOI] [PubMed] [Google Scholar]
- 41. Niu Q, Li P, Hao S, Zhang Y, Kim SW, Li H, Ma X, Gao S, He L, Wu W, et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci Rep 2015;5:9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan YK, Singh Brar M, Kirjavainen PV, Chen Y, Peng J, Li D, Leung FC-C, El-Nezam H. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: a pilot study of high fat diet and its intervention with Lactobacillusrhamnosus GG (LGG) or telmisartan in ApoE-/- mice. BMC Microbiol 2016;16(1):264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mills CE, Tzounis X, Oruna-Concha MJ, Mottram DS, Gibson GR, Spencer JP. In vitro colonic metabolism of coffee and chlorogenic acid results in selective changes in human faecal microbiota growth. Br J Nutr 2015;113(8):1220–7. [DOI] [PubMed] [Google Scholar]
- 44. Mosele JI, Macia A, Motilva MJ. Metabolic and microbial modulation of the large intestine ecosystem by non-absorbed diet phenolic compounds: a review. Molecules 2015;20(9):17429–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pajarillo EA, Chae JP, Balolong MP, Kim HB, Seo KS, Kang DK. Pyrosequencing-based analysis of fecal microbial communities in three purebred pig lines. J Microbiol 2014;52(8):646–51. [DOI] [PubMed] [Google Scholar]
- 46. Kim HB, Isaacson RE. The pig gut microbial diversity: understanding the pig gut microbial ecology through the next generation high throughput sequencing. Vet Microbiol 2015;177(3–4):242–51. [DOI] [PubMed] [Google Scholar]
- 47. Moreno LZ, Matajira CE, Gomes VT, Silva AP, Mesquita RE, Christ AP, Sato MI, Moreno AM. Molecular and antimicrobial susceptibility profiling of atypical Streptococcus species from porcine clinical specimens. Infect Genet Evol 2016;44:376–81. [DOI] [PubMed] [Google Scholar]
- 48. Ni Y, Wong VH, Tai WC, Li J, Wong WY, Lee MM, Fong FL, El-Nezami H, Panagiotou G. A metagenomic study of the preventive effect of Lactobacillus rhamnosus GG on intestinal polyp formation in ApcMin/+ mice. J Appl Microbiol 2017;122(3):770–84. [DOI] [PubMed] [Google Scholar]
- 49. Chiang ML, Chen HC, Chen KN, Lin YC, Lin YT, Chen MJ. Optimizing production of two potential probiotic Lactobacilli strains isolated from piglet feces as feed additives for weaned piglets. Asian-Australas J Anim Sci 2015;28(8):1163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duncan SH, Aminov RI, Scott KP, Louis P, Stanton TB, Flint HJ. Proposal of Roseburia faecis sp. nov., Roseburia hominis sp. nov. and Roseburia inulinivorans sp. nov., based on isolates from human faeces. Int J Syst Evol Microbiol 2006;56(Pt 10):2437–41. [DOI] [PubMed] [Google Scholar]
- 51. Scheithauer TP, Dallinga-Thie GM, de Vos WM, Nieuwdorp M, van Raalte DH. Causality of small and large intestinal microbiota in weight regulation and insulin resistance. Mol Metab 2016;5(9):759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 2009;139(9):1619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anand S, Kaur H, Mande SS. Comparative in silico analysis of butyrate production pathways in gut commensals and pathogens. Front Microbiol 2016;7:1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Preter V, Machiels K, Joossens M, Arijs I, Matthys C, Vermeire S, Rutgeerts P, Verbeke K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2015;64(3):447–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.