

Abstract
Summary
UV cross-linking and immunoprecipitation (CLIP), followed by high-throughput sequencing, is a powerful biochemical assay that maps in vivo protein-RNA interactions on a genome-wide scale. The CLIP Tool Kit (CTK) aims at providing a set of tools for flexible, streamlined and comprehensive CLIP data analysis. This software package extends the scope of our original CIMS package.
Availability and Implementation
The software is implemented in Perl. The source code and detailed documentation are available at http://zhanglab.c2b2.columbia.edu/index.php/CTK.
1 Introduction
Specific interaction of RNA-binding proteins (RBPs) with their target transcripts is essential for many steps of gene expression regulation. RBP interaction sites can be mapped on a genome-wide scale by UV cross-linking and immunoprecipitation of protein–RNA complexes, followed by high-throughput sequencing of the isolated RNA fragments (HITS-CLIP or CLIP-Seq) (Licatalosi et al., 2008). Since its initial development, HITS-CLIP and its variations have been applied in numerous studies (Darnell, 2010) and efforts have been made to compile published datasets (Yang et al., 2015). However, most studies implemented custom analysis tools optimized for a specific application. As a result, there remains a lack of software packages that are able to provide flexible, streamlined and comprehensive analysis of CLIP regardless of the CLIP protocol used. This gap imposes challenges for researchers who are new to CLIP, and raises issues with comparing and integrating results from different studies.
We previously developed the CIMS software package for processing CLIP data and mapping protein-RNA interactions at single nucleotide resolution (Moore et al., 2014). The latter takes advantage of crosslink-induced mutation sites (CIMS), which are nucleotide deletions or substitutions introduced at the protein–RNA crosslink sites by reverse transcriptase (Zhang and Darnell, 2011). Some variations of CLIP, such as iCLIP (Konig et al., 2010) and BrdU-CLIP (Weyn-Vanhentenryck et al., 2014), allow the capture of CLIP tags that are truncated at crosslink sites, and analysis of such crosslink-induced truncation sites (CITS) was also included in the CIMS package in later releases.
The CLIP Tool Kit (CTK), named to more precisely reflect the expansion of its scope to providing comprehensive CLIP data analysis, represents a major upgrade of the CIMS software package and has many advantages over existing CLIP data analysis software. Compared to the previous version of our analysis pipeline, CTK includes several algorithmic innovations, numerous optimizations and detailed documentation that significantly improve its performance and usability.
2 Software description
2.1 CLIP data preprocessing and mapping
CTK uses Burrows Wheeler Aligner (BWA) as the standard tool for read alignment. BWA allows the user to specify mismatch parameters by rate rather than by absolute number, which both simplifies and improves handling of CLIP tags of varying sizes. In addition, CTK operates on FASTQ files, to take advantage of sequence quality scores for read mapping, and on output SAM files, the standard format for storing read mapping information. Therefore, if desired, other aligners can also be used seamlessly for alignment.
CTK applies very stringent criteria to collapse PCR duplicates, which are distinguished by a random barcode (i.e., unique molecule identifier or UMI) attached to CLIP tags in most current CLIP protocols. After read mapping, a model-based algorithm is used to identify ‘sufficiently distinct’ barcodes among reads with the same chromosome starts by modeling the sequencing errors and the copy number of each duplicate sequence. Compared to the previous CIMS package, CTK uses a sparse data representation with greatly reduced memory usage and run time.
2.2 Identifying CLIP tag clusters and peak calling
Due to the increase in CLIP library complexity and sequencing depth, multiple CLIP tag clusters or peaks might not have clear separation, especially in abundant transcripts. To address this issue, CTK performs peak calling using a novel ‘valley seeking’ algorithm. In brief, CTK calculates the number of overlapping CLIP tags at each genomic position to find local maxima. Two neighboring local maxima with peak height h1 and h2 are considered to be two different peaks only when they are separated by a valley of depth d = h − v, where h = min(h1, h2) and v is the read coverage at the valley position. The user is asked to specify the relative valley depth (e.g. v/h ≥ 0.9), so that the algorithm can accommodate transcripts of different abundance. To define a more stringent subset of CLIP tag peaks, CTK performs additional statistical assessment on whether the observed peak height is more than one would expect by chance using different background models and scan statistics.
2.3 CIMS and CITS analysis
CTK uses essentially the same statistical models for CIMS and CITS as the previous package to evaluate the reproducibility of candidate sites, but it includes several important optimizations. First, spurious mutations due to sequencing errors or low-quality mapping have been eliminated because CTK allows fewer mismatches for shorter reads. Second, because we noticed that crosslinking-induced deletions of multiple consecutive nucleotides are relatively common in CIMS analysis, and that these sites appear to show distinct properties compared to sites with single nucleotide deletions, CTK now identifies oligonucleotide deletions of different sizes and performs separate CIMS analyses.
We expect that these methods can be readily applied to data generated by different variations of CLIP. For example, CIMS analysis can be applied to PAR-CLIP data (Hafner et al., 2010), if one focuses on C→U transitions, and CITS analysis can be performed on data generated by BrdU-CLIP or iCLIP.
3 Results
We applied CTK to the Rbfox CLIP data derived from mouse brain tissues and human cells using different protocols (Van Nostrand et al., 2016; Weyn-Vanhentenryck et al., 2014) and found significant improvement compared to our previous package. Results from CTK gave a larger number of unique CLIP tags because we were able to retain shorter tags mapped with a smaller number of mismatches. In general, these shorter tags identified were reliable based on their genomic distribution and several other diagnostic measures.
We also compared CTK with several other software packages ((Clipper (Lovci et al., 2013), Piranha (Uren et al., 2012) and PIPE-CLIP (Chen et al., 2014)) for peak calling and identification of crosslink sites. For this comparison, we took advantage of the specific Rbfox binding motif, UGCAUG, which provides us with an objective measure of accuracy. CTK consistently achieved higher accuracy than the compared tools, as shown in the higher motif enrichment around peaks (Fig. 1A and B). Testing with more stringent valley depths also resulted in higher enrichment of UGCAUG, with little loss in sensitivity.
Fig. 1.
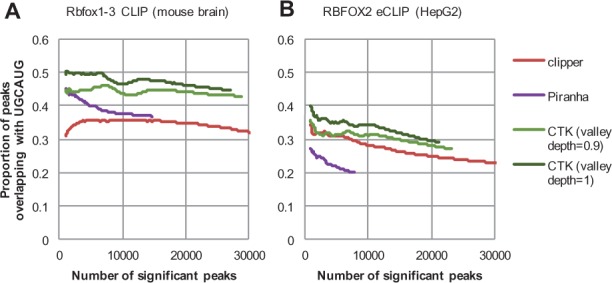
Comparison of CTK and other software packages in CLIP data analysis. (A) Rbfox1-3 CLIP (mouse brain). (B) Rbfox2 eCLIP (HepG2). In each panel, CLIP tag peaks were called by different algorithms using varying thresholds. The fraction of peaks overlapping with the Rbfox binding motif site (UGCAUG in +/−50 nt around peak center) is shown. Pipe-CLIP was not able to converge in our tests, and thus the results are not reported
Funding
This work was supported by grants from the National Institutes of Health (NIH) (R00GM95713 and R01NS89676) and the Simons Foundation Autism Research Initiative (307711).
Conflict of Interest: none declared.
References
- Chen B. et al. (2014) PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biol., 15, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell R.B. (2010) HITS-CLIP: panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip. Rev. RNA, 1, 266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M. et al. (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell, 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J. et al. (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol., 17, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi D.D. et al. (2008) HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature, 456, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovci M.T. et al. (2013) Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol., 20, 1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. et al. (2014) Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protocols, 9, 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren P.J. et al. (2012) Site identification in high-throughput RNA-protein interaction data. Bioinformatics (Oxford, England), 28, 3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L. et al. (2016) Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods, 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck S. et al. (2014) HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep., 6, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.C. et al. (2015) CLIPdb: a CLIP-seq database for protein–RNA interactions. BMC Genomics, 16, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Darnell R.B. (2011) Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol., 29, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]