

Abstract
Motivation
Protein model quality assessment (QA) plays a very important role in protein structure prediction. It can be divided into two groups of methods: single model and consensus QA method. The consensus QA methods may fail when there is a large portion of low quality models in the model pool.
Results
In this paper, we develop a novel single-model quality assessment method QAcon utilizing structural features, physicochemical properties, and residue contact predictions. We apply residue-residue contact information predicted by two protein contact prediction methods PSICOV and DNcon to generate a new score as feature for quality assessment. This novel feature and other 11 features are used as input to train a two-layer neural network on CASP9 datasets to predict the quality of a single protein model. We blindly benchmarked our method QAcon on CASP11 dataset as the MULTICOM-CLUSTER server. Based on the evaluation, our method is ranked as one of the top single model QA methods. The good performance of the features based on contact prediction illustrates the value of using contact information in protein quality assessment.
Availability and Implementation
The web server and the source code of QAcon are freely available at: http://cactus.rnet.missouri.edu/QAcon
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
With the wide application of next generation sequencing, there exists a big gap between the large number of protein sequences and the number of known protein structures (Li et al., 2015). Compared with the experimental techniques for determining protein structures, computational methods for protein structure prediction are less accurate, but much faster and cheaper, and therefore can potentially fill the gap (Hayat et al, 2015; McGuffin, 2008; Roche and McGuffin, 2016; Uziela et al, 2016; Wallner and Elofsson, 2003). During the protein structure prediction process, the quality assessment (QA) of predicted structural models is very important for model selection and ranking. There are two different kinds of QA methods in general. The multi-model QA methods (McGuffin and Roche, 2010; Wang et al, 2011) (Cao et al, 2015a, b) using the pairwise comparison between all models work well when there is enough consensus in a pool of models predicted by different protein structure prediction methods. However, it may fail when there are lot of bad models dominating the model pool (Cao et al, 2014a, b). It is also limited by requiring a sufficient number of models as input. The single-model QA methods (Cao et al, 2014a, b, 2016; Cao and Cheng, 2016; Wang et al, 2009) make protein model quality assessment based on a single model itself, without using the information of other models. So the predicted quality score is not influenced by the number and quality of the input model pool. For single-model QA methods, there is another type of QA method (quasi-single QA methods) that combines available template information usually achieves good performance on the CAMEO continuous benchmark (Haas et al, 2013), such as ModFOLD4 (McGuffin et al, 2013). The two kinds of QA methods are benchmarked biannually in the Critical Assessment of Techniques for Protein Structure Prediction (CASP) - a community-wise, worldwide experiment for blindly test protein structure prediction methods. There are two evaluation stages for QA methods in CASP: stage 1 which has 20 models with different qualities for each target, and stage 2 which has 150 top models selected by the organizers. The results of different QA predictors are evaluated after CASP releases the native structures. In this paper, we develop a single-model QA method (QAcon) based on machine learning and various protein features, which is ranked as one of the best single-model quality assessment methods according to the CASP official evaluation results (Kryshtafovych et al, 2015). Inspired by the previous research of using residue-residue contact information for selecting near-native protein models (Tress and Valencia, 2010), we develop a new contact score (Con score) that is based on contact predictions for protein model quality assessment.
2 Methods
QAcon is a new machine learning based single-model QA method. There are in total 12 input features used by QAcon. These 12 input features are: RF_CB_SRS_OD score (Rykunov and Fiser, 2007), SS score, SP score, EC score, SU score, EM score, ES score, SA score, RWplus score (Zhang and Zhang, 2010), ModelEvaluator (Wang et al, 2009), Dope score (Shen and Sali, 2006) and Con score. The detailed description of these methods is listed in Supplementary Table S1. Figure 1 illustrates the detailed flowchart for the Con score feature in QAcon. For predicting contacts, coevolution-based method PSICOV is executed and deep learning based method DNcon is used only if the number of homologous sequences for PSICOV predictions is less than 3000. The precision of the top-L predicted contacts (Con score) is used as an input feature along with other 11 features. This precision is defined as the percent of top-L predicted contacts that actually exist in a protein structural model, where L is sequence length. A two layer neural network was trained on the input features to predict the global quality (i.e. the similarity between a model and its corresponding native structure) of a model. We used CASP9 datasets to train our method, and blindly tested it in CASP11 as MULTICOM-CLUSTER server.
Fig. 1.
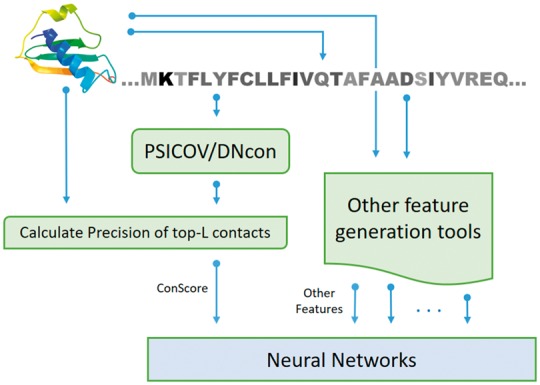
The flowchart of Con score and other input features used in QAcon
3 Results
QAcon was blindly tested in the CASP11 experiment. Table 1 shows the performance of QAcon and other top single-model QA methods on stage 1 and stage 2 of CASP11. The results show that QAcon is consistently ranked among top 3 single-model QA methods in terms of average correlation and loss on both stage 1 and stage 2. In order to show the impact of using contact information in QAcon, we re-trained the two-layer neural network on the input features without Con score and added the performance of QAcon without Con score into Table 1. Improvement on both correlation and loss metric is found by adding Con score into QAcon. We did a Wilcoxon signed ranked sum test on correlation and loss metric with and without adding Con score for QAcon. The p-value for average correlation with and without Con score is 0.005 and 0.003 for stage 1 and stage 2 respectively, 0.202 and 0.494 for loss of stage 1 and stage 2 respectively. A detail comparison with additional top performing methods (including the quasi-single model and clustering methods) is shown on Supplementary Table S2.
Table 1.
The per-target average correlation, average loss for QAcon and several state-of-art QA methods on Stage1 and Stage2 of CASP11
| Server name | Corr. | Loss. | Corr. | Loss. |
|---|---|---|---|---|
| Stage1 | Stage 1 | Stage 2 | Stage 2 | |
| ProQ2 (Uziela and Wallner, 2016) | 0.643 | 0.090 | 0.372 | 0.058 |
| QAcon | 0.639 | 0.100 | 0.395 | 0.067 |
| QAcon without Con score | 0.613 | 0.115 | 0.346 | 0.073 |
| VoroMQA (Olechnovič et al, 2011) | 0.561 | 0.108 | 0.401 | 0.069 |
| Wang_SVM (Liu et al, 2016) | 0.655 | 0.109 | 0.362 | 0.085 |
| Wang_deep_1 (Liu et al, 2016) | 0.613 | 0.128 | 0.302 | 0.089 |
Supplementary Figure S1 shows the performance of each feature on stage 1 and stage 2 of CASP11 datasets. Except that the feature SU (surface score) has negative correlation with model quality, other features (including Con – contact feature) have similar performance on both stage 1 and stage 2. On the top 25 targets with good contact prediction the average correlation of Con feature and real GDT-TS score is 0.682 and 0.221 on stage 1 and stage 2 respectively, while on the bottom 25 targets with bad contact prediction the average correlation is -0.189 and 0.056 on stage 1 and stage 2 respectively. The results demonstrate that the contact feature is useful for model quality assessment, while its impact is contingent on the accuracy of contact prediction.
Supplementary Material
Funding
The work is supported by US National Institutes of Health (NIH) grant (R01GM093123) to JC.
Conflict of Interest: none declared.
References
- Cao R. et al. (2014a) Designing and evaluating the MULTICOM protein local and global model quality prediction methods in the CASP10 experiment. BMC Struct. Biol., 14, 13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. (2014b) SMOQ: a tool for predicting the absolute residue-specific quality of a single protein model with support vector machines. BMC Bioinformatics, 15, 120.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. (2015a) Large-scale model quality assessment for improving protein tertiary structure prediction. Bioinformatics, 31, i116–i123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. (2015b) Massive integration of diverse protein quality assessment methods to improve template based modeling in CASP11. Proteins Struct. Funct. Bioinf, 84, 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. et al. (2016) DeepQA: Improving the estimation of single protein model quality with deep belief networks, arXiv preprint arXiv:1607.04379. [DOI] [PMC free article] [PubMed]
- Cao R., Cheng J. (2016) Protein single-model quality assessment by feature-based probability density functions. Sci. Rep., 6, 23990.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J. et al. (2013) The Protein Model Portal—a comprehensive resource for protein structure and model information. Database, 2013, bat031.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat S. et al. (2015) All-atom 3D structure prediction of transmembrane β-barrel proteins from sequences. Proc. Natl. Acad. Sci. U. S. A., 112, 5413–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryshtafovych A. et al. (2015) Methods of model accuracy estimation can help selecting the best models from decoy sets: assessment of model accuracy estimations in CASP11. Proteins Struct. Funct. Bioinf., 84, 349–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. (2015) A large-scale conformation sampling and evaluation server for protein tertiary structure prediction and its assessment in CASP11. BMC Bioinformatics, 16, 337.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. et al. (2016) Benchmarking Deep Networks for Predicting Residue-Specific Quality of Individual Protein Models in CASP11. Sci. Rep., 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L. (2008) The ModFOLD server for the quality assessment of protein structural models. Bioinformatics, 24, 586– 587. [DOI] [PubMed] [Google Scholar]
- McGuffin L., Roche D. (2010) Rapid model quality assessment for protein structure predictions using the comparison of multiple models without structural alignments. Bioinformatics, 26, 182–188. [DOI] [PubMed] [Google Scholar]
- McGuffin L.J. et al. (2013) The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res., gkt294.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olechnovič K. et al. (2011) Voroprot: an interactive tool for the analysis and visualization of complex geometric features of protein structure. Bioinformatics, 27, 723–724. [DOI] [PubMed] [Google Scholar]
- Roche D.B., McGuffin L.J. (2016) Toolbox for Protein Structure Prediction. Yeast Cytokinesis: Methods Protoc., 1369, 363–377. [DOI] [PubMed] [Google Scholar]
- Rykunov D., Fiser A. (2007) Effects of amino acid composition, finite size of proteins, and sparse statistics on distance‐dependent statistical pair potentials. Proteins Struct. Funct. Bioinf., 67, 559–568. [DOI] [PubMed] [Google Scholar]
- Shen M., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci., 15, 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress M.L., Valencia A. (2010) Predicted residue–residue contacts can help the scoring of 3D models. Proteins Struct. Funct. Bioinf., 78, 1980–1991. [DOI] [PubMed] [Google Scholar]
- Uziela K., Wallner B. (2016) ProQ2: estimation of model accuracy implemented in Rosetta. Bioinformatics, 32, 1411–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziela K. et al. (2016) ProQ3: Improved model quality assessments using Rosetta energy terms, arXiv preprint arXiv:1602.05832. [DOI] [PMC free article] [PubMed]
- Wallner B., Elofsson A. (2003) Can correct protein models be identified? Protein Sci., 12, 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. et al. (2011) MUFOLD-WQA: a new selective consensus method for quality assessment in protein structure prediction. Proteins, 79, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2009) Evaluating the absolute quality of a single protein model using structural features and support vector machines. Proteins, 75, 638–647. [DOI] [PubMed] [Google Scholar]
- Zhang J., Zhang Y. (2010) A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PloS One, 5, e15386.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.