

Abstract
Motivation
Alternative splicing and alternative transcription are a major mechanism for generating transcriptome diversity. Differential alternative splicing and transcription (DAST), which describe different usage of transcript isoforms across different conditions, can complement differential expression in characterizing gene regulation. However, the analysis of DAST is challenging because only a small fraction of RNA-seq reads is informative for isoforms. Several methods have been developed to detect exon-based and gene-based DAST, but they suffer from power loss for genes with many isoforms.
Results
We present PennDiff, a novel statistical method that makes use of information on gene structures and pre-estimated isoform relative abundances, to detect DAST from RNA-seq data. PennDiff has several advantages. First, grouping exons avoids multiple testing for ‘exons’ originated from the same isoform(s). Second, it utilizes all available reads in exon-inclusion level estimation, which is different from methods that only use junction reads. Third, collapsing isoforms sharing the same alternative exons reduces the impact of isoform expression estimation uncertainty. PennDiff is able to detect DAST at both exon and gene levels, thus offering more flexibility than existing methods. Simulations and analysis of a real RNA-seq dataset indicate that PennDiff has well-controlled type I error rate, and is more powerful than existing methods including DEXSeq, rMATS, Cuffdiff, IUTA and SplicingCompass. As the popularity of RNA-seq continues to grow, we expect PennDiff to be useful for diverse transcriptomics studies.
Availability and implementation
PennDiff source code and user guide is freely available for download at https://github.com/tigerhu15/PennDiff.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
RNA sequencing (RNA-seq) has revolutionized transcriptomics studies due to its ability to profile the entire transcriptome in an unbiased fashion. With RNA-seq, we can quantitatively measure gene expression, discover novel transcripts and detect single nucleotide variations. Unlike the genome, which gives a static view of the genetic and regulatory information defining a phenotype, the transcriptome is dynamic and varies in different tissues, developmental stages and disease states (Kratz and Carninci, 2014). Knowledge in transcriptomic variations is critical for understanding how genes are regulated in response to internal and external conditions.
A major mechanism for generating transcriptomic variations is alternative splicing, a biological process that occurs either co-transcriptionally or post-transcriptionally (Han et al., 2011). During this process, specific exons of a gene can be included or excluded from messenger RNA (mRNA), leading to different transcript isoforms. These isoforms are then translated into functionally unique proteins, which may respond differently across conditions. There are many forms of alternative splicing; common forms include exon skipping, intron retention, mutually exclusive exons and alternative 5′ donor site or alternative 3′ acceptor site for an included exon. Recent evidence suggests that over 90% of multi-exon human genes are alternatively spliced (Wang et al., 2008). Another mechanism for the generation of transcriptome diversity is alternative transcription, which involves the use of alternative transcriptional initiation and/or termination sites in gene transcription. Alternative transcription can give rise to different pre-mRNAs, some of which can further undergo alternative splicing. A recent study showed that alternative transcription exceeds alternative splicing in generating transcriptome diversity (Pal et al., 2011). Alternative splicing or transcription may be altered in disease cells and their mis-regulation can produce aberrant proteins that drive the development of disease (Scotti and Swanson, 2016). Differential alternative splicing or transcription (DAST), which describes different usage of transcript isoforms across different conditions, can complement differential expression in characterizing gene regulation.
The analysis of DAST is challenging because the isoform origin for only a small fraction of the sequenced reads can be determined in a typical RNA-seq dataset. Existing methods for DAST analysis often take conceptually different approaches (Hooper, 2014). Exon-based methods, such as MISO (Katz et al., 2010), MATS (Shen et al., 2012), rMATS (Shen et al., 2014), DEXSeq (Anders and Huber, 2010) and DSGSeq (Wang et al., 2013), test for differential exon usage for each individual exon or exon-trio consisting of a cassette exon and two flanking exons. In contrast, gene-based methods such as Cuffdiff (Trapnell et al., 2012), SplicingCompass (Aschoff et al., 2013), DiffSplice (Hu et al., 2013), rSeqDiff (Shi and Jiang, 2013) and IUTA (Niu et al., 2014), detect DAST at gene level rather than considering each exon individually. Gene-based methods naturally account for combined alternative splicing or transcription effects across exons and hence reduce the need for multiple testing. However, gene-based methods may suffer from power loss for genes with many isoforms because estimation uncertainty for isoform expression estimation increases dramatically as the number of isoforms increases.
Recognizing the limitations of existing methods, we propose PennDiff, a statistical approach that can be considered as a hybrid of exon- and gene-based methods. PennDiff is based on the observation that the distribution of isoform relative abundances is the most general characterization of splicing or transcription pattern because any other characteristic features such as exon-inclusion levels, can be derived from this distribution. To reduce the impact of estimation uncertainty on each individual isoform, exon-inclusion level for each alternative exon is inferred by collapsing isoforms that share the exon of interest. This allows PennDiff to make use of all aligned reads to detect DAST. Through extensive simulations and the analysis of a real RNA-seq dataset, we demonstrate that PennDiff significantly outperforms existing methods.
2 Materials and methods
2.1 Quantification of alternative splicing or transcription using exon-inclusion level
Since PennDiff is a gene-by-gene based method, throughout the rest of the text, we describe the analysis for a particular gene only. We note that multi-mapping reads are discarded from our analysis, hence recent paralogs are not considered in PennDiff. Given a gene, let I denote the set of its all known isoforms (e.g. based on refSeq, UCSC, Gencode or Ensembl gene annotation). An exon is alternatively spliced or transcribed if it is included in some isoform(s) but not in the other. Following Jiang and Wong (Jiang and Wong, 2009), when two isoforms share part of an exon, we split the exon into non-overlapping parts and treat each part as a virtual exon. Figure 1 shows an example in which the gene has three isoforms, and 14 virtual exons, among which nine are alternatively spliced or transcribed.
Fig. 1.

Partitioning biological exons into non-overlapping virtual exons in a gene with three isoforms. This gene has 14 virtual exons, of which 9 are alternative spliced or transcribed. These alternative exons can be divided into three exon groups
A vital step in PennDiff is to estimate exon-inclusion level for each alternative exon, which is defined as the proportion of transcripts that originate from isoforms with the exon included. For an alternative exon e, the exon-inclusion level for subject i can be estimated as , where Ie represents the set of isoforms that have exon e included, and θi,j is the relative abundance of isoform j in subject i. For the example in Figure 1, the exon-inclusion level for exon 4 is xi, 4 = θi, 1 + θi, 3. Estimated isoform relative abundances can be obtained by existing algorithms such as Cufflinks (Trapnell et al., 2010), PennSeq (Hu et al., 2014) or RSEM (Li and Dewey, 2011). We note that, different exons may have the same exon-inclusion level; for example, xi,2 = xi,3 = xi,5 = xi,6 = θi,2, xi,1 = xi,9 = θi,3 and xi,4 = xi,7 = xi,8 = θi,1 + θi,3. This observation prompted us to group exons according to their exon-inclusion levels. To detect DAST, we treat exon group as the analysis unit rather than considering each exon individually. In the example in Figure 1, there are nine alternative exons, but only three exon groups, indicating that grouping exons by their isoform origins can substantially reduce the need for multiple testing.
2.2 Gaussian copula regression on exon-inclusion levels
The exon-inclusion levels for exon groups within the same gene are correlated due to the sharing of certain isoforms. After exon-inclusion levels are quantified, the next step is to build a statistical model to account for such correlations in DAST analysis. Since the joint distribution of exon-inclusion levels is unknown, an alternative way is to characterize the marginal distributions for exon-inclusion levels and their correlations separately. A flexible and robust approach for such modeling is Gaussian copula regression, which has been utilized previously in gene mapping of quantitative traits and analysis of correlated data (He et al., 2012; Song et al., 2009). In Gaussian copula regression, we model the marginal distribution of each exon-inclusion level using a generalized linear model, and then apply a multivariate normal distribution to link the generalized linear models together to account for the correlations. The separation of marginal distributions and correlation structure makes Gaussian copula regression versatile in modeling non-normal dependent observations.
Since exon-inclusion level takes values between 0 and 1, it is reasonable to assume a beta distribution for it. The beta distribution, as is well known, is flexible in modeling proportions because its density can have different shapes depending on the values of the two parameters that index the distribution. Under this assumption, the distribution of exon-inclusion level for exon group in subject i is , and the density of can be written as
The expected value and variance of are
where can be interpreted as the precision parameter of the beta distribution.
In Gaussian copula regression, the marginal generalized linear model is specified by
| (1) |
where is a link function that relates the mean of and covariates that influence the mean. For the purpose of DAST detection, we include disease status indicator (1 for case; 0 for control) as a covariate, but other covariates can certainly be included in (1). We choose logit function as the link function.
The joint distribution for exon-inclusion levels across all M exon groups is given by
where is the dispersion parameter of the marginal generalized linear model for exon group m, is the cumulative beta distribution function of given , is the cumulative distribution function of a standard normal random variable, and ) is the cumulative distribution function of multivariate normal random variables with dimensions and correlation matrix . Due to the complexity of gene structure, it is difficult to explicitly derive correlations for exon-inclusion levels. Therefore, we choose to use exchangeable correlation structure for , which depends on a single parameter . A detailed description of the notation is shown in Supplementary Table S4.
2.3 Detection of DAST events
Based on the above Gaussian copula regression model, the likelihood function for a dataset with n subjects can be written as
| (2) |
where with , and is the density function of given and is an M-dimensional identity matrix.
With Gaussian copula regression, we can detect DAST both at the exon level and the gene level. In exon-based analysis, we test for exon group m to determine differential exon usage. Rejection of this null hypothesis indicates that all exons within this exon group are differentially utilized between cases and controls. In gene-based analysis, we test for any . Rejection of this null hypothesis indicates differential isoform usage of the gene. Both hypotheses can be tested using likelihood ratio test with test statistic . Under the null hypothesis, this test statistic approximately follows a distribution in which for exon-based test and for gene-based test.
2.4 RNA-seq data simulation
We conducted simulations to evaluate the performance of PennDiff and compared it with other state-of-the-art algorithms for DAST analysis based on RefSeq and Enselbml, two commonly used gene annotations in published studies. To simulate a realistic dataset with known ground truth, we used Flux Simulator to generate RNA-seq data (Griebel et al., 2012). The Flux Simulator program assigns expression value for each isoform following a mixed power/exponential law. Additionally, it simulates common sources of systematic bias in the abundance and distribution of produced reads by in silico library preparation and sequencing. The use of Flux Simulator facilitates the comparison of different methods under a more realistic setting than evaluations based on simulating directly count data and not the full RNA-seq protocol.
To simulate RNA-seq reads using Flux Simulator, the human genome sequence (hg19, NCBI build 37) was downloaded from UCSC Genome Browser (https://genome.ucsc.edu/). We simulated 76 bp paired-end reads for 20 cases and 20 controls (∼12 million reads per subject) based on RefSeq annotation and 20 cases and 20 controls (∼60 million reads per subject) based on Ensembl annotation. To make our simulated data close to those seen in real studies, the isoform relative abundances of each gene were sampled from a Dirichlet distribution in which the mean and variance parameters were determined from a real human eye RNA-seq dataset (Li et al., 2014). The simulated RNA-seq reads were mapped to the hg19 reference human genome using Tophat with default options (Trapnell et al., 2009). For each gene, isoform relative abundances were estimated using PennSeq (Hu et al., 2014), a program that we recently developed for isoform relative abundance estimation, which is robust to non-uniformity in read coverage.
In all results presented in this paper, we only considered genes with at least two isoforms. Additionally, we required a gene to have at least 20 mapped reads on average across all RNA-seq samples. We also evaluated the impact of sample size by analyzing a subset of n cases and n controls (n = 5) randomly chosen from the full simulated dataset. There were 4408 genes (19 310 alternative exons) for the RefSeq annotation and 6321 genes (180 478 alternative exons) for the Ensembl annotation, respectively.
2.5 Human induced pluripotent stem cell RNA-seq study
2.5.1 Differentiation of human induced pluripotent stem cells (iPSCs) to macrophages
Detailed protocols were described in our recent publication (Zhang et al., 2015). Briefly, to induce differentiation, embryoid bodies were generated by culturing small aggregates of feeder-depleted iPSCs in COSTAR ultra-low attachment surface multiwell plate in StemPro-34 media supplemented with different cytokine cocktails. From day-8, macrophage culture media (20% fetal bovine serum in RPMI 1640 media supplemented with 100 ng/ml M-CSF) was used to enrich for myeloid precursors. At day-15, single cells were transferred to BD PrimariaTM tissue culture plate for expansion and maturation, completed at day-22. Human protocols for this work were approved by the University of Pennsylvania and Columbia University Medical Center Human Subjects Research Institutional Review Boards. RNA samples from iPSCs and iPSC derived macrophages were extracted using All Prep DNA/RNA/miRNA Universal Kit (Qiagen, Valencia, CA). With a minimum of 300 ng input RNA, libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA), followed by 100 bp paired-end sequencing on an Illumina’s HiSeq 2000 machine. The RNA-seq data were aligned to the hg19 reference genome using STAR 2.3.0 with default options. The aligned data were filtered using the following criteria: the mapping quality score of each read was ≥30, reads from the same pair were mapped to the same chromosome with expected orientations and mapping distance between the read pair was <500 000 bp, and each read was uniquely mapped. Isoform relative abundances for RefSeq annotated isoforms were estimated using PennSeq based on the filtered alignment files.
2.5.2 RT-PCR validation
Total RNA was isolated as previously described (Zhang et al., 2015). Reverse transcription was performed from 500 ng total RNA per sample using the High Capacity RNA to cDNA Master Mix kit (Applied Biosystems, Life Technologies) following the standard protocol. PCR amplification (28 cycles) was performed using the following primers targeting the constitutive exons flanking the alternative one for SYTL2 (Forward 5′—CCACAGTGCCTACACAACCTGATA—3′; Reverse 5′—CCAGATTGCCAAAGTCTCCACTAT—3′). The amplification products were analyzed on a 3% ethidium bromide agarose gel alongside a DNA ladder that allows for resolution of small bp changes in PCR product sizes (pBR322 DNA-MspI Digest ladder, New England BioLabs). Gel images were analyzed using ImageJ.
3 Results
3.1 Exon-inclusion level estimation
Reliable estimation of exon-inclusion level is critical for DAST detection. In PennDiff, a key step is the estimation of exon-inclusion level by collapsing isoforms sharing the same alternative exon. The reliance on isoform relative abundances allows us to utilize all aligned reads to estimate exon-inclusion levels. To evaluate whether PennDiff yields more accurate estimate, we randomly selected one subject from the simulated dataset, estimated its exon-inclusion levels for all alternative exons using PennDiff. Figure 2 shows that for exons with inclusion levels estimated by PennDiff based on both annotations (RefSeq and Ensembl), the estimates have a good agreement with the true values and the Spearman correlation coefficients were 0.87 and 0.76, respectively, on the logit scale.
Fig. 2.
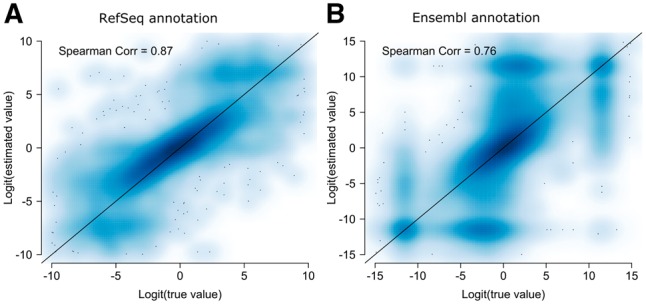
Smooth scatter plot of logit transformed estimated exon-inclusion levels versus logit transformed true values. Correlation was calculated on the logit transformed values. (A) Exon-inclusion levels estimated by PennDiff based on RefSeq annotation (8061 alternative splicing or transcription events). (B) Exon-inclusion levels estimated by PennDiff based on Ensembl annotation (49 607 alternative splicing or transcription events)
3.2 Performance of exon-based tests
Next, we evaluated the performance of PennDiff in exon-based analysis, and compared it with two other exon-based methods including DEXSeq and rMATS. All methods were run with the same input dataset. We set a threshold t1 on mean exon-inclusion level difference between cases and controls, to define the ground truth of DAST. An exon was considered to be a DAST event if the mean exon-inclusion level difference, denoted by , was greater than t1. To evaluate power with different effect sizes, the value of t1 was set at 0.1, 0.15 and 0.2, respectively. To evaluate type I error rate, a true non-DAST event was defined as an exon with . In practice, we encountered many instances of exons that showed statistically significant DAST but exhibited exon-inclusion level difference that is too small to warrant biological significance. Therefore, we required an alternative exon to show exon-inclusion level difference >0.05 in order to be declared as a DAST event.
Since different methods use different criteria to filter out exons with invalid results (failure of numerical algorithms, or small number of junction reads etc.), the numbers of tests returned by each method are quite different. To make a fair comparison, we calculated type I error rates and power in two ways. In the first approach, the calculations were based on the true number of DAST and non-DAST events in the input data, which include those failed to be analyzed by each method. Including all events in the input data allows us to better assess each method’s sensitivity and specificity. In real studies, it is desirable to have a method that yields valid results for all events. In the second approach, the type I error rates and power of each method were calculated based on its own returned test results. The denominators in these calculations can be substantially different among methods.
Figure 3 shows the comparison results based on virtual exons when PennDiff and DEXSeq were evaluated using the first approach when t1 was 0.1. For data generated with the RefSeq annotation, the number of DAST events detected by PennDiff ranged from 2876 to 3575, and only 1.59 to 2.42% of these events were false positives. In contrast, DEXSeq detected 2218 to 3107 events; however, type I error rates (5.46, 9.10%) were inflated under RefSeq annotation. For data generated with the Ensembl annotation, the number of DAST events ranged from 7726 to 9095 for PennDiff, and 2666 to 3841 for DEXSeq. Both methods had well-controlled type I error rates. To control for multiple testing, we also evaluated the false discovery rate of each method. Supplementary Table S5 shows that both PennDiff and DEXSeq had false discovery rate controlled at the 5% level.
Fig. 3.
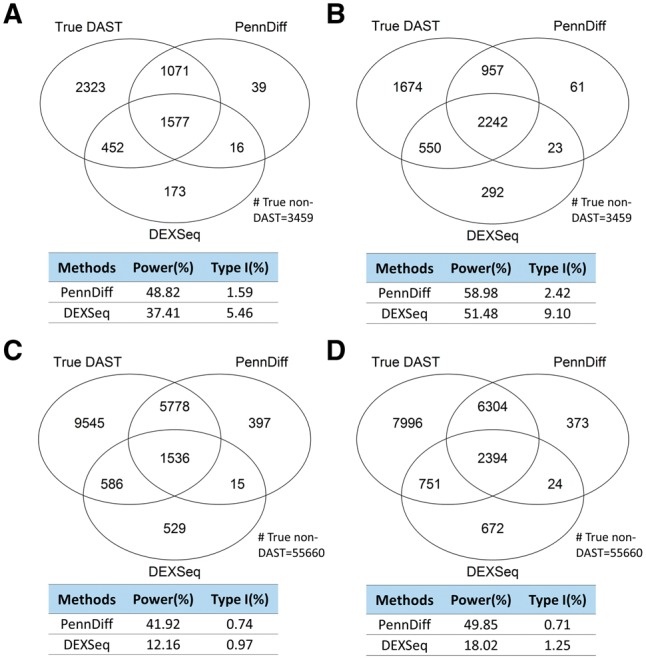
Type I error and power of exon-based methods with different sample sizes and gene annotations. Calculations were based on all DAST and non-DAST exons in the input data. Significance was evaluated at the 5% significance level. An exon with true exon-inclusion level difference > was defined as a true DAST exon. (A) 5 versus 5 based on RefSeq annotation. (B) 20 versus 20 based on RefSeq annotation. (C) 5 versus 5 based on Ensembl annotation. (D) 20 versus 20 based on Ensembl annotation
In power comparison, both methods had increased power as the threshold value t1 increased because differentially spliced or transcribed exons with larger inclusion level difference were easier to detect. The power of PennDiff was consistently higher than DEXSeq under both gene annotations (RefSeq and Ensembl) because the reliance on isoform relative abundances for exon-inclusion level estimation allows PennDiff to utilize all aligned reads, whereas count-based method such as DEXSeq only use reads mapped to the tested exon and ignore reads mapped elsewhere even if they are informative for exon-inclusion level estimation. We observed similar patterns at other threshold values of t1 (Supplementary Figs S1 and S3). Supplementary Table S1 shows the comparison results of PennDiff and DEXseq based on the second approach. The power of DEXSeq was consistently lower than PennDiff.
Next, we compared with rMATS, an exon-based method that was designed to detect differential alternative splicing events only. To make a fair comparison with rMATS, we focused on those differential alternative splicing events that were analyzed by rMATS, including cassette exon, mutually exclusive exons, retained intron, alternative 5′ splice site and alternative 3′ splice site (Supplementary Table S3). The type I error rates of rMATS exceeded the nominal level, especially when sample size was small. Additionally, PennDiff had greater power than rMATS for all scenarios we considered.
3.3 Performance of gene-based tests
Next, we evaluated the performance of PennDiff in gene-based analysis and compared it with three other gene-based methods, including IUTA, SplicingCompass and Cuffdiff. A gene was considered DAST when the mean Hellinger distance between cases and controls, denoted by , was greater than t1. Hellinger distance, which describes the similarity between two probability distributions was calculated as
where and represent the mean isoform relative abundances determined based on the human eye RNA-seq dataset (see Materials and methods section). This distance has been utilized previously (Monlong et al., 2014) to measure the splicing ratio difference between two genes with multiple isoforms. The threshold values of t1 varied at 0.1, 0.15 and 0.2, respectively. We required a gene with estimated Hellinger distance >0.05 in order to be declared as a DAST gene. We also compared the type I error rates and power among different methods using two ways, similar to those employed in exon-based comparisons.
Figure 4 (Supplementary Figs S2 and S4) shows that empirical type I error rates and power of PennDiff, IUTA, SplicingCompass and Cuffdiff based on true DAST and non-DAST genes in the input data under RefSeq and Ensembl annotations. All methods had type I error rates controlled at the 5% significance level when n was 20; however, when n was 5, the type I error rate of IUTA was slightly above 5% under the RefSeq annotation. Furthermore, PennDiff had higher power than the other methods for most situations, and method-specific results (Supplementary Table S2) indicate a similar pattern. Supplementary Table S6 shows that all methods had false discovery rate controlled at the 5% level. The power of Cuffdiff is much lower than the other gene-based methods. The poor performance of Cuffdiff is possibly due to the fact that it detects splicing change at gene level by directly comparing the relative usage of all isoforms in each gene. This requires highly accurate isoform relative abundance estimation, which is challenging especially for genes with complicated exonic structure and low read coverage. Indeed, this challenge motivated PennDiff to test differential splicing by comparing exon-inclusion levels for exons in the same group. Compared to isoform relative abundance, exon-inclusion level can be estimated more accurately by collapsing isoforms sharing the same exon while utilizing all reads mapped to the gene.
Fig. 4.
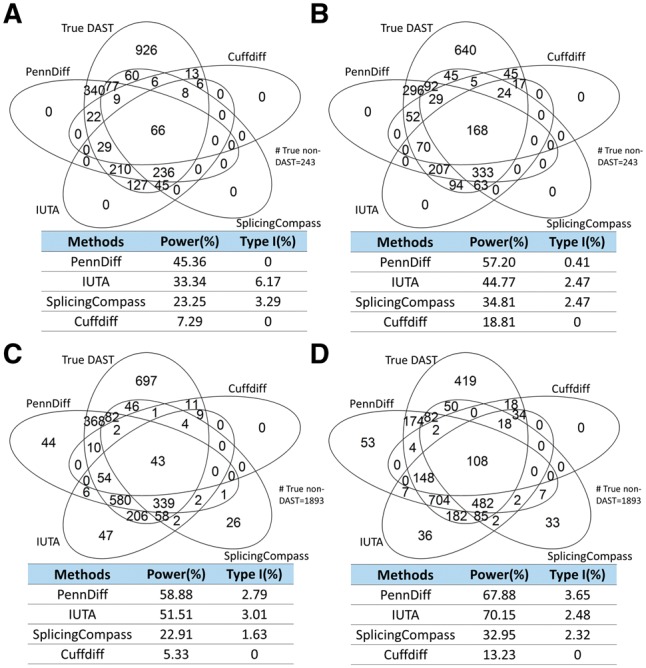
Type I error and power of gene-based methods with different sample sizes and gene annotations. Calculations were based on all DAST and non-DAST genes in the input data. Significance was evaluated at the 5% significance level. A gene with true Hellinger distance > was defined as a true DAST gene. (A) 5 versus 5 based on RefSeq annotation. (B) 20 versus 20 based on RefSeq annotation. (C) 5 versus 5 based on Ensembl annotation. (D) 20 versus 20 based on Ensembl annotation
3.4 Impact of exon grouping and gene structure
An important feature of PennDiff is grouping exons that originate from the same isoform(s). As shown in Figure 1, exon grouping can substantially reduce the need for multiple testing. Among the 4408 RefSeq genes (6321 Ensembl genes) that met our analysis criteria in the full dataset, the number of alternative exons is 19 310 (180 478 for Ensembl), but the number of exon groups is 9684 (108 997 for Ensembl), suggesting that multiple testing corrections can be reduced by about twofold. To evaluate the impact of exon grouping on power, we calculated power separately by the number of exons per group. Figure 5A shows that PennDiff performed consistently better than DEXSeq. The power of PennDiff was relatively stable as the number of exons per group increased, whereas DEXSeq had reduced power. These results demonstrate the advantage of PennDiff in exon-based analysis, especially for exon groups having many alternative exons.
Fig. 5.

The impact of gene complexity on power of different methods. (A) Power comparison between PennDiff and DEXSeq when results were stratified by the number of exons per group (≥2: 2765 exon groups, ≥3: 1103 exon groups, ≥4: 668 exon groups, ≥5: 460 exon groups, ≥6: 370 exon groups). Significance was evaluated at the 5% level. (B) Power comparison between PennDiff, IUTA and SplicingCompass when results were stratified by the number of isoforms per gene (≥2: 6321 genes, ≥5: 4232 genes, ≥10: 2102 genes, ≥15: 941 genes, ≥20: 426 genes, ≥25: 189 genes). Significance was evaluated at the 5% level
Gene-based methods tend to have reduced power for genes with a large number of isoforms. To circumvent this problem, PennDiff tests DAST at the gene level by comparing exon-inclusion levels of all exon groups of a gene. To evaluate if this grouping is effective, we compared with IUTA and SplicingCompass. We focused on the analysis of data generated based on Ensembl annotation due to its more comprehensive annotation on isoforms than RefSeq. We classified the 6321 Ensembl genes into six groups based on the number of isoforms per gene. Figure5B shows that the power of SplicingCompass dropped significantly as the number of isoforms increased. In contrast, PennDiff and IUTA were relatively stable as the number of isoforms increased. These results suggest that PennDiff and IUTA were robust to the increased complexity of gene structure.
3.5 Robustness to under- and over-annotation of isoforms
As PennDiff relies on gene annotation in isoform expression estimation and exon grouping, it is important to evaluate its robustness when isoforms are misannotated. To this end, we evaluated the performance of PennDiff with under- and over-annotated isoforms. For under-annotation, we simulated RNA-Seq reads based on 100% of the Ensembl annotation and analyzed the simulated data with various methods using 90% (10% less), 75% (25% less) and 50% (50% less) of the Ensembl annotation. Figure 6A shows that PennDiff outperformed the other methods in most settings. Compared to IUTA, PennDiff had lower power only when n = 20 and t1 = 0.2 and this gap was reduced when isoforms were more under-annotated. Also, from 100 to 75% annotation, PennDiff was clearly more robust than IUTA. For SplicingCompass, it had significantly lower power than PennDiff and IUTA, even though its power was less affected by the degree of under-annotation. Similarly, for over-annotation, we simulated data based on 66% of the Ensembl annotation and analyzed the simulated data with various methods using 73% (10% more), 83% (25% more) and 100% (50% more) of the true annotation. Figure 6B shows that the power of PennDiff was robust to over-annotation and it was generally more powerful than IUTA. SplicingCompass still had the lowest power compared to other two methods.
Fig. 6.

The impact of mis-annotation of isoforms on power of different methods. (A) Evaluation of the impact of under-annotation of isoforms. Shown are the power estimates of PennDiff, IUTA and SplicingCompass based on 100% (true), 90% (10% less), 75% (25% less) and 50% (50% less) of the Ensembl annotated isoforms. (B) Evaluation of the impact of over-annotation of isoforms. Shown are the power estimates of PennDiff, IUTA and SplicingCompass based on 66% (true), 73% (10% more), 83% (25% more) and 100% (50% more) of the Ensembl annotated isoforms
3.6 Application to a human-induced pluripotent stem cell study
To evaluate the performance of PennDiff in real settings, we analyzed a RNA-seq dataset generated from a human induced pluripotent stem cell (iPSC) study in which RNA-seq data were generated on iPSCs and iPSC-derived macrophages (iPSDMs) (Zhang et al., 2015). In this paper, we focused on DAST analysis between iPSC and iPSDM samples generated from three subjects each with two replicates. These samples were sequenced using Illumina’s HiSeq 2000 machine, yielding approximately 130 million 101 bp pair-end reads per sample, 95% mapping rate to the reference genome, and approximately 70% reads uniquely mapped and filtered. In the DAST analysis, among genes with two or more isoforms annotated by RefSeq, we considered the 4889 genes that had at least 20 mapped reads on average and performed DAST analysis using PennDiff, DEXSeq, IUTA and SplicingCompass. For PennDiff, the isoform relative abundances were estimated using PennSeq for RefSeq annotated isoforms. We did not compare with Cuffdiff and rMATS due to their lack of sensitivity or inflated type I error rates shown in the simulations (Supplementary Tables S2–S3).
Figure 7A (left panel) shows the number of DAST genes detected by each method. Since DEXSeq is an exon-based method, for ease of comparison, we consider a gene to be DAST by DEXSeq if at least one virtual exon of the gene was detected by DEXSeq. 89% of the genes detected by PennDiff were detected by at least another method (IUTA, SplicingCompass, or DEXSeq), and the corresponding numbers were 90% for IUTA, 74% for SplicingCompass and 80% for DEXSeq. To have a better understanding on the behavior of DEXSeq and PennDiff when the interest is in virtual exons, we further compared DEXSeq and PennDiff exon-based test when the testing unit is virtual exons. Figure 7A (right panel) shows that PennDiff detected 6675 exons and 60% were also detected by DEXSeq. As a comparison, DEXSeq detected 8303 exons and 48% were detected by PennDiff. Given the high percentage of unique exons detected by DEXSeq, we next examined if the corresponding genes detected by DEXSeq can be detected by other gene-based methods. Our results indicate that only 76% of the DEXSeq detected genes were detected by at least another gene-based method, whereas the corresponding percentage was 87% for PennDiff exon-based test. Supplementary Figure S9 shows that the lack of concordance for DEXSeq with other methods is likely due to its inflated false positive rates.
Fig. 7.
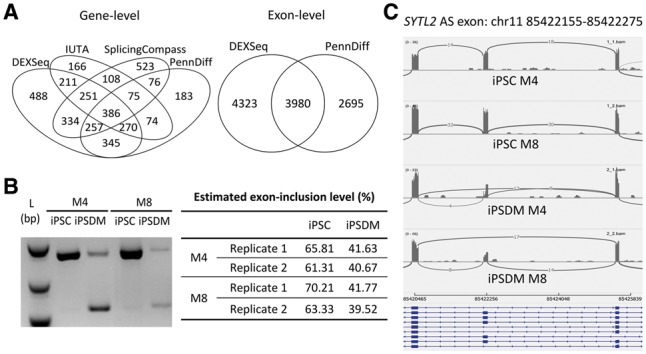
(A) DAST genes detected by different methods for human induced pluripotent stem cells (iPSCs) versus iPSC-derived macrophages (iPSDMs). (B) RT-PCR validation of alternatively spliced exon chr11: 5422155–85422275 in SYTL2 in samples of two human donors we performed the RNA-seq studies. The exon-inclusion levels shown in the table were estimated based on the gel image. (C) IGV sashimi plot of gene SYTL2. M4 and M8 are two study subjects
For genes that were detected by PennDiff but missed by IUTA and SplicingCompass, we searched for empirical evidence in RNA-seq coverage plot. Among the 528 genes detected by PennDiff only, 35.1% have more than three isoforms, which is significantly higher than the corresponding percentage among the remaining 4302 qualified genes (21.5%). We randomly picked 10 genes (ACSL3, CAPS, INTS12, LRRCD8, MGAT1, MYO9B, ST7, SYTL2, TGIF2 and UBA1) among the 528 genes, and generated coverage plots using the sashimi plot feature in IGV to verify our results. For example, ACSL3 and INTS12 both have two annotated isoforms but only one alternatively spliced exon. The boxed areas in Supplementary Figure S9 show visual evidence of DAST between iPSC and iPSDM in these two genes. The P-value from PennDiff was 0.000014 for ACSL3, and was 0.00058 for INTS12, whereas both genes were missed by IUTA and SplicingCompass. For SYTL2, we generated coverage plot for alternatively spliced exon chr11: 85422155–85422275, and further conducted real time polymerase chain reaction (RT-PCR) to validate the differential usage of this cassette exon. Both results were consistent with PennDiff (P = 0.00011, estimated exon-inclusion level difference = 0.23).
To compare the performance of PennDiff and DEXSeq, we next randomly picked a gene ST7 and generated coverage plot for alternatively spliced exons to empirically check the evidence of DAST. The isoform structure of ST7, shown in Supplementary Figure S7C, is relatively simple with only two isoforms and three alternatively spliced exons. The signals of exon 15 and alternate exon 15 have ‘switch-like' pattern, which strongly suggests DAST. However, exon 7 was detected by PennDiff but not by DEXSeq. Since exon 15 and exon 7 share the same exon-inclusion level, and the exon-inclusion level of the alternate exon 15 is complementary to exon 15 and exon 7, these three exons should yield the same result if there is evidence of DAST. PennDiff reported evidence of DAST for all three exons, but DEXSeq failed to detect DAST for exon 7. This example indicates the importance of exon grouping in DAST analysis. We further examined the DAST events detected by PennDiff by their types (Supplementary Fig. S8). Consistent with our simulations, most of the detected DAST events had alternative promoters.
4 Discussion
Detection of genes with mis-regulated alternative splicing or transcription is a critical step in transcriptomics studies. Existing methods either test each exon individually or examine the overall distribution of isoform relative abundances. Exon-based approaches focus on junction reads and those that map exclusively to the exon of interest, but ignore other reads even if they are informative for alternative splicing or transcription. Gene-based methods can prevent information loss, but they often have little power to detect DAST for genes with many isoforms. In this article, we present PennDiff, a statistical approach that can be considered as a hybrid of exon- and gene-based methods. The central idea of PennDiff is to quantify exon-inclusion levels using relative abundances of isoforms sharing the same exon and grouping exons based on their isoform origin. This was motivated by the observation that the distribution of isoform relative abundances offers the most general characterization of splicing pattern because any other features on alternative splicing or transcription can be derived from this distribution.
Compared to existing methods, PennDiff has several advantages. First, collapsing isoforms sharing the same alternative exon leads to more accurate estimation of exon-inclusion levels than methods that only utilize junction reads. Additionally, PennDiff can estimate exon-inclusion levels for both alternative splicing and alternative transcription event, a desirable feature as the majority of alternative exon events in human genes are extremely complex. Indeed, a recent study showed that alternative transcription exceeds alternative splicing in generating transcriptome diversity (Pal et al., 2011). Through simulations and the examination of real data, we found PennDiff to be particularly powerful in detecting alternative promoters, a versatile mechanism for creating diversity and flexibility in gene regulation.
Second, exon-based methods such as DEXSeq test all exons in a gene, regardless whether an exon is alternatively spliced or transcribed. The ignorance of isoform structure may increase the number of tests unnecessarily. For example, we found that among the 105 312 virtual exons from the 7390 human genes with two or more isoforms annotated by RefSeq, 72 460 of the exons are not alternatively spliced or transcribed. However, DEXSeq would perform tests on these non-informative exons, and unnecessarily increases the number of tests.
Third, exon-based methods such as rMATS rely on junction reads mapped to splicing sites to detect differential alternative splicing, which limit their ability to detect splicing events with complex exonic structure and those without enough junction read coverage. Here we showed that PennDiff offers a useful alternative to rMATS. By quantifying exon-inclusion levels using pre-estimated isoform relative abundances, PennDiff cannot only test all alternatively spliced exons but also those alternative exons and this feature makes PennDiff particularly attractive in real studies.
Fourth, by grouping alternative exons that originate from the same isoform(s), PennDiff avoids conducting unnecessary tests, thus reduces the burden of multiple testing. In our simulations, we found that PennDiff was more powerful than DEXSeq, especially when more alternative exons are grouped together. Since PennDiff is performed for each exon group under the assumption that exons within the same group share a common exon-inclusion level, more reads can be utilized to estimate exon-inclusion level when more exons are included and hence lead to more accurate estimation.
The current evaluation of PennDiff used isoform relative abundances estimated from PennSeq as the input. Since PennSeq is robust to non-uniformity of read coverage, it is of interest to evaluate the contribution of PennSeq to the good performance of PennDiff. To this end, we divided genes in our simulated data into two categories based on the degree of non-uniformity of read coverage. Supplementary Figure S5 shows that PennDiff consistently outperforms the other methods for both exon-based and gene-based tests regardless of the degree of non-uniformity, suggesting that the good performance of PennDiff is not completely due to the use of PennSeq. Supplementary Figure S5 also shows that PennDiff performed especially better than the other methods for genes showing higher degrees of non-uniformity, indicating that when read coverage is not uniform, PennDiff would benefit from the use of an isoform expression estimation method that is robust to non-uniformity. Although PennDiff can take isoform relative abundances estimated from other programs as the input, to achieve better performance, we recommend using PennSeq for isoform relative abundance estimation or other programs that can properly handle non-uniformity in read coverage.
We also evaluated the impact of gene expression levels on PennDiff. Specifically, we divided genes in the simulated data into three groups based on the number of mapped reads adjusted by gene length (low: [0, 33%); medium: [33, 66%); high: [66, 100%]). Supplementary Figure S6 shows that all methods had increased power when gene expression level increased, but PennDiff was more robust to the impact of gene expression levels than the other methods.
In summary, we have developed a flexible regression framework to detect DAST at both exon and gene levels. Through extensive simulations and the analysis of a real RNA-seq dataset, we showed that PennDiff outperformed competing methods, particularly when sample size is small or the difference between groups under comparison are small. We believe that PennDiff will be a valuable tool for future transcriptomics studies.
Funding
This research was supported by R01GM108600 and R01HL113147 to M.L., and R01HL113147 to M.P.R. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
Supplementary Material
References
- Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschoff M. et al. (2013) SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics, 29, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Griebel T. et al. (2012) Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res., 40, 10073–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. et al. (2011) Pre-mRNA splicing: where and when in the nucleus. Trends Cell Biol., 21, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. et al. (2012) A Gaussian copula approach for the analysis of secondary phenotypes in case-control genetic association studies. Biostatistics, 13, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper J.E. (2014) A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum. Genomics, 8, 3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. et al. (2013) DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res., 41, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. et al. (2014) PennSeq: accurate isoform-specific gene expression quantification in RNA-Seq by modeling non-uniform read distribution. Nucleic Acids Res., 42, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Wong W.H. (2009) Statistical inferences for isoform expression in RNA-Seq. Bioinformatics, 25, 1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y. et al. (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods, 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz A., Carninci P. (2014) The devil in the details of RNA-seq. Nat. Biotechnol., 32, 882–884. [DOI] [PubMed] [Google Scholar]
- Li B., Dewey C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. (2014) Comprehensive analysis of gene expression in human retina and supporting tissues. Hum. Mol. Genet., 23, 4001–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monlong J. et al. (2014) Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat. Commun, 5, 4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L. et al. (2014) IUTA: a tool for effectively detecting differential isoform usage from RNA-Seq data. BMC Genomics, 15, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S. et al. (2011) Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res., 21, 1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti M.M., Swanson M.S. (2016) RNA mis-splicing in disease. Nat. Rev. Genet., 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. et al. (2012) MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res., 40, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S. et al. (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA, 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Jiang H. (2013) rSeqDiff: detecting differential isoform expression from RNA-Seq data using hierarchical likelihood ratio test. PloS One, 8, e79448.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P.X. et al. (2009) Joint regression analysis of correlated data using Gaussian copulas. Biometrics, 65, 60–68. [DOI] [PubMed] [Google Scholar]
- Trapnell C. et al. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T. et al. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature, 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2013) Identifying differentially spliced genes from two groups of RNA-seq samples. Gene, 518, 164–170. [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. (2015) Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circ. Res., 117, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.